

Abstract
Introduction
The distinct properties of the centrally-acting analgesic tapentadol derive from the combined contributions of an opioid component and a nonopioid component. However, the opioid component’s relative contribution to analgesic and adverse effects has not previously been elucidated. Tapentadol’s analgesic effect derives from the combined contribution of an opioid mechanism and a nonopioid mechanism, the extent of which can vary for different pains. Likewise, the interaction can vary for various adverse effects. Hence, the contribution of each mechanism to adverse effects can be different from the contribution to analgesia. We here estimate the percent contribution of each component of the mechanism of action to analgesia and to adverse effects.
Areas Covered
Several approaches to in vitro and in vivo data to estimate the contribution of tapentadol’s opioid component to analgesia and to the two important opioid adverse effects, respiratory depression and constipation. The results are then compared with clinical data.
Expert Opinion
Traditional opioids, such as morphine, oxycodone, and others, produce their analgesic effects primarily through a single mechanism—the activation of µ-opioid receptors (MOR). Therefore, the contribution of the opioid component to adverse effects is 100%. In contrast, the newer strong analgesic tapentadol produces its analgesic effect via two separate and complementary analgesic mechanisms, only one of which is µ-opioid. We applied standard drug–receptor theory and novel techniques to in vitro and in vivo data to estimate by several different ways the μ-load of tapentadol (the % contribution of the opioid component to the adverse effect magnitude relative to a pure/classical µ-opioid at equianalgesia) in respiratory depression and constipation, and we compared the results to clinical evidence. The estimate is remarkably consistent over the various approaches and indicates that the μ-load of tapentadol is ≤ 40% (relative to pure MOR agonists, which have, by definition, a µ-load of 100%).
Funding
Grünenthal GmbH.
Keywords: Adverse effects, Analgesia, MOR-NRI, Tapentadol, µ-load
Introduction
The first opioids were plant-derived opiates, including morphine and codeine (which undergoes Phase 1 cytochrome P450-catalyzed metabolism to morphine). Their therapeutic pain-relieving effects were discovered serendipitously, and they were used for centuries without much rational scientific understanding of how they produced their beneficial effects. However, in addition to pain relief, the opiates unfortunately produce a spectrum of adverse effects such as constipation and respiratory depression that are inexorably linked to the analgesic effect, and they have abuse potential related to their activation of MOR (μ-opioid receptor) [1, 2]. The explanation for the lack of dissociation between therapeutic and adverse effects is now understood—their major analgesic mechanism is the same as their adverse effect mechanism, namely MOR agonism, so they are in this regard in essence mono-modal or one-dimensional (albeit differences in pharmacology or pharmacokinetics can result in nuances in their clinical profile).
In the absence of a molecular target, subsequent semi-synthetic and synthetic opioids were discovered by structure–activity relationships in which the dependent variable was obtained from in vivo screening using animal models (such as abdominal constriction, hot plate, tail flick, etc., tests). Such screening led to a great number of useful opioids with individual differences [3, 4], but, due to the way they were discovered, they are essentially pharmacologic ‘clones’, that is, they display fundamentally the same pharmacology as the predecessors that were used as the template and standards.
The discovery in the early 1970s that opioids produce their effects via binding to 7-transmembrane G protein-coupled opioid receptors [2] and activating their 2nd-messenger transduction systems transformed the initial stages of drug discovery from screening through animal models to ‘directed’ in vitro high-throughput screening in selective assays for affinity for, and intrinsic activity at, opioid receptors and receptor subtypes expressed in specialized cells or modeled on computers using computational techniques [5, 6].
Based on the discovery of opioid receptors, the logical hypothesis was reached that there must be endogenous ligands for these receptors, and endogenous opioid peptides were soon identified. The existence of opioid receptors and endogenous ligands for these receptors revealed for the first time a cellular-level mechanistic explanation for how the exogenous opioids produced both their analgesic, and their other receptor-mediated (side-) effects [2]. It also helped to delineate what is now recognized as a major afferent nociceptive (pain-detecting/transmitting) pathway. For years, opioid pharmacology and drug discovery was directed toward compounds that had ever-greater selectivity and binding affinity for opioid receptors. There was thus a race to a more mono-modal mechanism of analgesic action. And with that came the more mono-modal opioid receptor-mediated adverse effects. As a result, with pure opioid drugs, the MOR contribution to adverse effects (e.g., constipation, respiratory depression, etc.) is 100%.
During the course of such drug discovery, however, it was inevitable that drugs with more than one pharmacologic mechanism of action were discovered [7]. For such drugs, four outcomes are possible: the mechanisms contribute either positively or negatively to the analgesic effect, and positively or to a less extent to adverse effects [8]. The latter category—drugs that have mechanisms of action that are either additive or synergistic on the analgesic endpoint, but less than additive on adverse effect endpoints—would possess clinically advantageous properties [9, 10]. Two such drugs (buprenorphine [11–15] and tramadol [16, 17]) were recognized after discovery. One (tapentadol) was designed and discovered with a specific dual mechanism as the goal [18–20].
Concurrent with the identification of analgesics with favorable clinical features was the discovery of pain modulatory pathways. Thus, in addition to long-known afferent (‘ascending’) pain-sensation transmitting pathways, there are ‘descending’ pain-sensation modulating pathways in the brain and spinal cord [21]. Now collectively known as DNIC (diffuse noxious inhibitory control), activity in these pathways can modify the amplitude or temporal characteristics of pain signals [22]. The monoamines, norepinephrine and serotonin, play prominent roles in the descending pathways, with variable contributions in different types of pain, at different anatomical sites, and at different periods in the progression or time course of pain. Furthermore, experience has shown that many pains involve more than one (patho)physiological process [23, 24], having, for example, both a neuropathic component and a nociceptive component. Therefore, treatment of such pains with mono-mechanistic analgesics usually yields sub-optimal results (either insufficient pain relief or excess adverse effects). In such cases, better separation of therapeutic from adverse effects can be achieved by using analgesics with multiple mechanisms of action that match the multiple mechanisms of pain (patho)physiology [25, 26].
Buprenorphine and tramadol are examples of analgesics that were found to have multiple mechanisms of analgesic action after their synthesis. Buprenorphine has very high affinity for MOR, which is a major mechanism of its analgesic action [27]. It has recently been shown to have an additional supraspinal naloxone-, PTX- (pertussis toxin), and NOP (nociception/orphanin FQ peptide)-insensitive, Gz- and Ser/Thr-sensitive mechanism, and possibly other contributory mechanisms [28]. Tramadol produces its analgesic effect by the combined action of the enantiomers of the parent drug and enantiomers of its O-desmethyl (M1) metabolite. Tramadol has at least three mechanisms: affinity for MOR, inhibition of neuronal reuptake of norepinephrine (NRI), and inhibition of neuronal reuptake of serotonin (SRI).
Tapentadol is the only approved centrally-acting analgesic that was chemically engineered from the beginning to possess strong analgesic efficacy by combining two specific synergistic mechanisms of analgesic action (‘directed polypharmacology’) [29, 30]. The two mechanisms are: activation of MOR and the inhibition of neuronal reuptake of NRI (MOR-NRI) [18–20, 31–33]. The outcome of this approach is that tapentadol is more potent in a variety of animal models, and in clinical trials has been shown to have comparable efficacy to oxycodone, with more favorable tolerability [34–40]. It is a single molecule, has no analgesically active metabolite [41], and is metabolized primarily by glucuronidation (rather than CYP450) [42].
Although tapentadol is a strong analgesic in animal models [43] and humans [44–46], it binds to recombinant human MOR with an affinity of 0.16 μM (Ki value). For comparison, that is about 10- to 20-fold lower affinity than morphine or oxycodone for MOR [18, 47, 48]. How does one account for a lower receptor affinity yet greater analgesic potency and efficacy of a molecule? In the case of tapentadol, the answer came from an animal study that demonstrated that tapentadol’s two mechanisms of analgesic action (MOR-NRI) independently contribute to the analgesic effect, and in fact produce a synergistic (greater than the expected additive)–analgesic effect [49, 50]. The synergistic interaction can account for its greater functional (therapeutic effect) activity.
Importantly, though, the two mechanisms do not interact synergistically on an adverse effect endpoint (constipation) [51]. There is thus a mechanistic explanation for a favorable separation between the analgesic effect and adverse effects [51]. Nevertheless, such a separation needs to be demonstrated clinically. In fact, data from clinical trials demonstrate that tapentadol produces pain relief comparable to oxycodone, but that it has a better tolerability profile, i.e., a better balance of benefit (effective analgesia) and risk (adverse effects) [52–55]. There is therefore compelling evidence that each component of tapentadol, opioid and non-opioid, independently contributes significantly to its analgesic effect; that is, the overall effect is a sort of hybrid of a polypharmacological ligand. It is also suggested by the data that the contribution of the opioid component to adverse effects might be less than its contribution to analgesia.
Two questions thus arise. First, what is the relative contribution of tapentadol’s opioid component to its analgesic effect? Second, what is the relative contribution of tapentadol’s opioid component to adverse effects? To date, there has been no formal attempt to answer these questions. In the case of a drug that has enantiomers, for example, tramadol, the assessment is facilitated by the ability to administer each enantiomer separately, and observing the effect of each independently, then together. In the case of a single molecule such as tapentadol, the analysis is less obvious or direct. We use available data to provide the first attempt at a quantitative answer to the question of the relative contribution of tapentadol’s opioid component to analgesia and to adverse effects. Since we are not aware of any prior attempt to answer such questions for any other drug, there is no definitive way to do so. We thus decided to take a variety of different approaches, and analyzed whether the answers would converge to an agreeably consistent answer. We selected approaches that we loosely label ‘in vitro and PK’, ‘in vivo’, and ‘clinical’. The ‘in vitro’ approach will be recognized as emanating directly from affinity, intrinsic activity, and pharmacokinetic data. The ‘in vivo’ approach involves novel analysis, but is essentially a type of component factor analysis. And the ‘clinical’ approach utilizes available clinical data. Both clinical and preclinical data are taken from previously conducted studies, and no studies with human participants or animals were performed by any of the authors for this analysis.
The remainder of the manuscript is designed so that the reader uninterested in the mathematical details can read the explanations and conclusions without loss of continuity.
Estimation of the Contribution of Tapentadol’s Opioid Component to Adverse Effects Relative to Analgesia
‘In vitro and PK’ Approach
This approach proceeded as follows. It is first recognized that the published values of 88% intrinsic efficacy for tapentadol versus 100% intrinsic efficacy for morphine (from a GTPγS assay) are not helpful, and in fact are misleading in this context. Such values are obtained in a situation in which receptors are completely saturated with high concentrations of the respective compounds, and in which only one out of potentially several intracellular effector systems is considered. That is not the case under clinical conditions. In the intrinsic efficacy studies, the EC50 at the hMOR of tapentadol was 0.67 μM and the EC50 of morphine was 0.022 μM [18], i.e., morphine was 30 times more potent than tapentadol in producing a given effect at the MOR. In other words, to obtain a given effect via MOR, 30 times more tapentadol than morphine would be needed in terms of exposure/molar concentration in the target areas.
In clinical practice, the nominal equianalgesic dose of tapentadol is about 2.5- to 3-fold higher than the equivalent morphine dose. Based on measured plasma concentrations of morphine and tapentadol at steady state after treatment with presumed equianalgesic (prolonged/sustained release) doses, and assuming that plasma concentrations can be taken as a proxy for brain concentrations, it follows that the local concentration of tapentadol is approximately 5-fold higher than the local concentration of morphine.
Taken together with the 30-fold difference in EC50, this would mean that the μ-load of tapentadol is only 5/30 = 17% of that of morphine.
A similar calculation can be based on the Ki values of tapentadol (0.16 μM) and morphine (0.009 μM) at the hMOR [18]. In that case, there is an 18-fold difference with respect to receptor affinity versus the 5-fold difference in exposure, yielding a tapentadol μ-load of 28% of that of morphine, or about 1/4th that of morphine.
‘In vivo’ (Animal Models) Approach
A model based on conventional drug–receptor-effect concepts was developed and used with available data (rat in vitro assay and in vivo rat pain models) in order to estimate the percent contribution of tapentadol’s opioid component to therapeutic analgesic (antinociceptive) effect and adverse effect [respiratory depression (in terms of inhibition of the respiratory stimulatory effect of CO2), and constipation (in terms of inhibition of gastrointestinal transit)].
Analgesic Response
Tapentadol’s total in vivo effect results from the combined contributions of its opioid component, its non-opioid component, and the positive synergistic interaction between them. Since the interaction derives from, but is separate from, the opioid component, the total effect is given by:
| 1 |
where μ represents the opioid component, and β represents the combined non-opioid and synergistic interaction components. From conventional drug–receptor modeling,
| 2 |
where α is a parameter that relates drug–receptor binding to an in vivo effect. Conc is (free brain) concentration, and Kμ is the dissociation constant of tapentadol at MOR (= 0.096 × 10−6 M) [18]. It should be noted that α is a measure of effect efficacy (functional efficacy) for a particular in vivo endpoint. It is not intrinsic activity or intrinsic efficacy, and therefore is not the same as the property measured by 35S]GTPγS binding assay. The two unknowns α and β can be determined using two separate equations. It is advantageous for practical and interpretative reasons to use data from the rat low-intensity tail-flick test (LITF-r)—which is a model of acute nociceptive pain—and the rat spinal-nerve ligation test (SNL-r)—which is a model of chronic neuropathic pain—at 71.7% and 54.3% effect levels, respectively. The tapentadol doses that produced these effect levels are 4.64 and 2.15 mg/kg, respectively [from Table 1 in Schröder et al. [50]]. The effect-site concentrations can be obtained using the relationship (linear in the range of interest) [from Fig. 5 in Schröder et al. [50]]: tapentadol brain level (ng/g) = 406 × dose (mg/kg), and conversion using rat brain weight of approximately 2 g and volume approximately 1 mL. The estimated tapentadol brain concentrations for doses of 2.15 and 4.64 mg/kg are then 8 × 10−9 and 17 × 10−9 M, respectively. The two equations are thus:
| 3 |
| 4 |
Which yields: α = 257. This value can be substituted into equations (3) and (4) to yield:
| 5 |
| 6 |
Therefore: in the LITF-r test, tapentadol’s μ-opioid component contributes approximately 38.6/71.7 54% to the total effect; and in the SNL-r test, tapentadol’s μ-opioid component contributes approximately 19.8/54.3 36% to the total effect. The result of this analysis is very well in line with results from a study using naloxone and yohimbine as opioid and noradrenergic antagonists, respectively. While the antinociceptive ED50 value of tapentadol in LITF-r was shifted to the right 6.4-fold by naloxone and only 1.7-fold by yohimbine, the antihypersensitive ED50 value in SNL-r was shifted to the right 4.7-fold by yohimbine and only 2.7-fold by naloxone [33]. Taken together, these data show that the relative contribution of the two mechanisms of action of tapentadol to analgesia is dependent on the particular pain indication, as μ-opioid receptor agonism predominantly mediates tapentadol’s antinociceptive effects, whereas noradrenaline reuptake inhibition predominantly mediates its antihypersensitive effects. Note that we do not consider the 36% and 54% opioid contributions to analgesia as a measure of the µ-load of tapentadol, as this refers, by our definition, to the opioid contribution relative to a pure MOR opioid to an effect (other than analgesia) at equianalgesic doses.
Inhibition of Respiratory Stimulatory Effect of CO2 (IRSE-CO2)
From the graph for tapentadol in Fig. 1, tapentadol produces a dose-related inhibition of the respiratory stimulatory effect of CO2. Unlike the analgesic endpoint, where both the opioid and the NRI components are known to contribute, individually and in combination, to the analgesic effect, there is a particular challenge to elucidating the possible effect of NRI on the endpoint IRSE-CO2. In fact, the effect of the NRI component of tapentadol on IRSE-CO2 could be in opposition to that of the opioid component (i.e., stimulation vs. inhibition of respiration). Unfortunately, the data that are needed to model this are not available. Therefore, an alternative approach must be taken. Toward this end, a way is sought to isolate one or the other component in the effect of tapentadol on IRSE-CO2. The opioid component is the easier to isolate. Unfortunately, no naloxone data are available for this endpoint, so a more indirect approach is necessary. The following assumption is made: that the inhibition of IRSE-CO2 is due to the opioid component of action; the NRI component is either neutral or stimulatory.
Fig. 1.
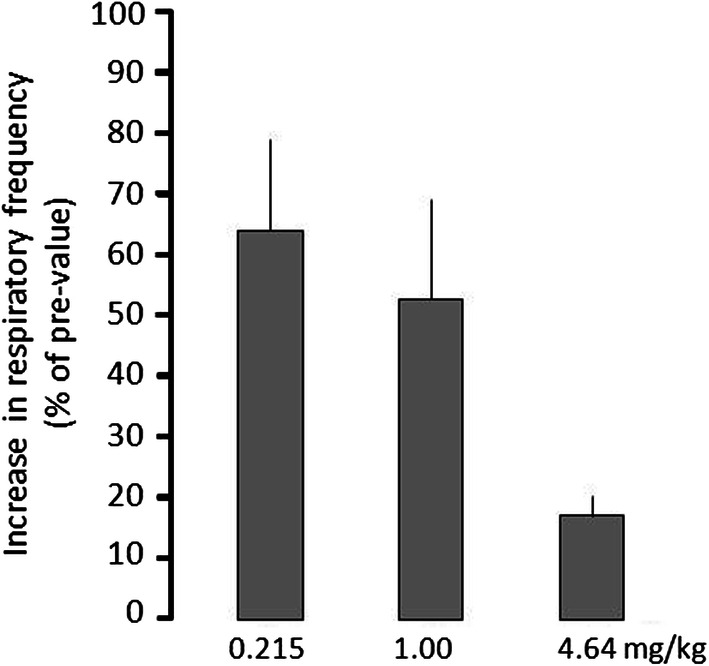
Effect of tapentadol on CO2-induced stimulation of the respiratory frequency in conscious rats. After an equilibration period of 30 min, pre-values of respiratory rate were recorded before and at the end of a 5-min CO2-stimulation period (8% CO2). Tapentadol was given i.v. 5 min later. After 30 min, a baseline value was recorded; then, a 5-min CO2-stimulation was performed again and the respiration rate was recorded at the end of the stimulation period. Drug-mediated inhibition of CO2-induced increase is expressed as the percentage of the increase without drug (assigned as 100%)
Unpublished data, courtesy of T. Christoph, Grünenthal GmbH
This does not give any insight at intermediate points along the IRSE-CO2 dose–response curve, where the effect is possibly the net effect of two possibly competing influences. In contrast, at near 100% inhibition, the NRI component is overcome, and hence negated. Using a similar approach, as shown in equation (2),
| 7 |
where ρ+ β are analogous to α and β, but for IRSE-CO2 instead of analgesia. Tapentadol produces an approximate 84% effect (IRSE-CO2) at the highest dose tested (Fig. 1). There are two possibilities: (1) the 16% ‘residual’ effect is due to the NRI component, or (2) the 84% is due to the opioid component, with negligible influence by NRI (the < 100% is due merely to the dose, i.e., a higher dose would produce a 100% effect). These two possibilities allow the calculation of a range of values for:
| 8 |
which yields ρ = 665, and
| 9 |
which yields ρ= 558. The μ-load can then be calculated as the ratio of parameters for the therapeutic effect (analgesia) and adverse effect (IRSE-CO2). (Note that, for morphine and other single-mechanism opioids, this ratio is 1.0.) For tapentadol, two values are obtained, based on the limiting assumptions leading to equations (8) and (9):
| 10 |
| 11 |
where α is from the calculation on analgesia. Therefore, the μ-load of tapentadol (relative to pure/classic opioids) with respect to the inhibition of respiratory activity is approximately 40%.
Constipation (Inhibition of Gastrointestinal Transit)
The inhibition of the gastrointestinal transit of charcoal is a classic animal model and a quantifiable correlate and measure of drug-induced constipation. From published results of studies using inhibition of gastrointestinal transit (GIT) as the endpoint in mice and rats, it is known that tapentadol inhibits GIT to a lesser extent than does morphine at equi-antinociceptive doses. It is also known that the two mechanisms of action of tapentadol interact in the third possible way on this endpoint, namely in an additive manner, in contrast to the antinociceptive (synergistic) and respiratory (presumably counteractive) endpoints. For the estimation, the same antinociceptive dose as used in sections 5.1. and 5.2. is chosen. From Cowan et al. [51, Table 1], tapentadol (5 mg/kg) reduces GIT from 44% (baseline to 18% when administered alone) to 38% when co-administered with naloxone (revealing the magnitude of the contribution of the opioid component), and to 31% when co-administered with yohimbine (revealing the magnitude of the contribution of the non-opioid component). and , analogous to α and β, but for constipation instead of analgesia, are (38–18)/(44–18) = 0.77 and (31–18)/(44–18) = 0.5, respectively.
and can be used to estimate the relative contribution of each component to the overall (100%) effect. Note that the calculation is for the contribution to overall effect, not the measured effect. Using the same doses of tapentadol as for sections 5.1. and 5.2.,
| 12 |
Substituting the values for (0.77) and (0.5) yields ρ = 860. The μ-load is then calculated (the same as in sections 2.1 and 2.2) as the ratio,
| 13 |
Evidence from clinical data
A logical interpretation of the reported clinical study data is that they are consistent with the estimates presented above, based on in vitro data and animal models, that tapentadol’s μ-load is also substantially less than 100% in humans.
Constipation
Although constipation was not the primary endpoint of tapentadol’s clinical trials, Kwong et al. [37] analyzed patient-reported bowel function from two clinical trials (10- and 90-day) involving patients who were given equianalgesic doses of immediate-release (IR) tapentadol or immediate-release oxycodone to treat chronic low back pain or osteoarthritis pain. During the course of the trials, prospective data were collected on the patients’ perspective of their bowel function and constipation symptoms. Bowel function was assessed in several ways in one or both of the trials: the BMQ (Bowel Movement Questionnaire), the PAC-SYM (Patient Assessment of Constipation Symptoms) questionnaire, and the need for use of a laxative. The analysis set included data from 666 randomized patients (518 completions) with end-stage joint disease (10-day trial) and from 848 randomized patients (457 completions) with chronic low back pain or osteoarthritis pain (90-day trial).
The patients given oxycodone experienced a significantly greater number of days with either no or incomplete bowel movements over the course of the 10-day treatment compared to placebo and compared to tapentadol. In contrast, there was no significant difference between tapentadol and placebo in the proportions of treatment days with either no or incomplete bowel movements. Similarly, the mean overall PAC-SYM scores were significantly lower with tapentadol than with oxycodone after 15 days of treatment and over the full 90-day treatment period, and the tapentadol group showed significantly lower constipation symptom scores versus oxycodone for all three PAC-SYM summary domain scores. Likewise, the use of laxatives by the tapentadol patients was similar to placebo and numerically less than the oxycodone patients in the 10-day trial and significantly less than the oxycodone patients in the 90-day trial, while the proportion of patients who did not report constipation as a side effect and/or laxative use was significantly more in the tapentadol group, and fewer of the patients treated with tapentadol than with oxycodone discontinued therapy because of constipation.
Similar results were found in a randomized, double-blind, active-controlled study of patients with moderate to severe chronic malignant tumor-related pain who were given oral extended-release tapentadol or oral controlled-release oxycodone, and TEAEs (treatment-emergent adverse events) were recorded [54]. At equianalgesic (non-inferior) doses, the incidence of gastrointestinal TEAEs was lower in the tapentadol group than in the oxycodone group.
If it were not for the preceding consistent calculations based on the in vitro and animal model data, we would be reluctant to attempt an estimation of tapentadol’s μ-load based solely on clinical trial data alone, i.e., in isolation. However, given the relative tight range of the values found from the estimates from preclinical studies, an estimation based on the clinical trial data was deemed warranted, if sufficiently circumspect in its conclusions.
In the absence of data on tapentadol brain levels in humans, the estimate is necessarily based on the effect domain. We thus define μ-load simply as the ratio of the clinical endpoint, difference from placebo (P), of tapentadol (T) to an equianalgesic dose of mono-mechanistic oxycodone (O), i.e., μ-load = [T–P]/[O–P]), and summarize the findings from three randomized, double-blind, placebo- and oxycodone-comparator phase 3 studies in patients with chronic low back (cLBP) or osteoarthritis (OA) pain (total n = 2974). In the cLBP study [56], the odds of experiencing constipation were significantly lower with tapentadol than with oxycodone. In the OA pain studies, there were substantially lower incidences of gastrointestinal-related TEAEs associated with treatment with tapentadol than with oxycodone [52], and tapentadol was better tolerated than oxycodone, largely due to fewer gastrointestinal side-effects [57].
cLBP (Buynak et al. [56]): P(5%), T(13.8%), O(26.8%); μ-load = 40.4%.
OA pain (Afilalo et al. [52]): P(6.5%), T(18.9%), O(36.8%): μ-load = 40.9%.
OA pain (Serrie et al. [57]): P(9.2%), T(17.9%), O(35.0%); μ-load = 33.7%.
A pooled analysis of the data yielded a similar value:
Pooled analysis (Lange et al. [58]:) P(6.9%), T(16.9%), O(33.0%); μ-load = 38.3%.
Respiratory Depression
A study that has recently been published by van der Schrier et al. [59] directly compared the effect of oral tapentadol 100 mg IR and oral oxycodone 20 mg IR (doses deduced to be equianalgesic from the literature) on isohypercapnic ventilation (the end-tidal PCO2 concentration 7.3 kPa or 55 mmHg; VE55) in healthy adults. It is the first such study that has directly compared the respiratory effects of tapentadol and oxycodone. Significantly greater respiratory depression was observed following exposure to oxycodone than to tapentadol. The data indicate a respiratory inhibition potency ratio of 7.5 for these doses of oxycodone and tapentadol. This ratio exceeds the commonly used ratio for analgesia of 5.0 (i.e., oxycodone is 5-fold more potent than tapentadol), and therefore the results clearly suggest some advantage of tapentadol over oxycodone in this respect. Unfortunately, these data are the least amenable to a quantitative calculation of μ-load. However, since the above previous approaches were rigorous quantitative methods and gave similar results, it seems that the data from this study should not be wasted, and should be assessed in some way, even if only a qualitative approach is all that can be used. With this in mind, a ´ball park´ estimation can be made from the data. Although the effect difference was not quantitatively described in the publication, tapentadol produced approximately half of the respiratory depression produced by an equianalgesic dose of oxycodone (Fig. 1c in van der Schrier et al [59]). And even a 50% higher dose of tapentadol produced a (numerically) weaker effect than did oxycodone. Thus, it seems reasonable to assume that tapentadol’s μ-load in this case is also ≤ 40%, consistent with the estimates from the clinical constipation data and the preclinical animal models of constipation and respiratory depression.
Conclusion
The analyses described here, using a variety of different approaches, and a variety of in vitro, in vivo, and clinical data, lead to the following findings:
Combining principles of drug–receptor binding and signal transduction (in vitro) data with available pharmacokinetic data suggests that the μ-load of tapentadol should theoretically be about 17–28%.
From in vivo animal models, the opioid (MOR) component of tapentadol contributes slightly more than ½ (~ 54%) to analgesia in a nociceptive pain (LITF-r) model and about 1/3rd (~ 36%) in a neuropathic pain (SNL-r) model. This is consistent with available data on the in vivo activity of tapentadol.
The contribution of tapentadol’s μ-opioid component of mechanism of action to the prototypical opioid adverse effect (μ-load) of respiratory depression (as modeled by inhibition of respiratory stimulatory effect of CO2 in rats and humans) is approximately 40% that of morphine and other single-mechanism opioids.
The contribution of tapentadol’s μ-opioid component of action to the adverse effect of constipation (modeled by the inhibition of gastrointestinal transit of charcoal in rats) is approximately 30%, thus about 1/3rd that of morphine and other single-mechanism opioids.
From post hoc analysis of clinical trials in which constipation-related data were obtained, the μ-load of tapentadol for constipation in humans is in the range of 38–41%, very similar to the calculated estimates from the animal studies.
The results are summarized in Tables 1 and 2.
Table 1.
Summary of the calculated estimates of the contribution of tapentadol’s opioid component to its analgesic (antinociceptive) action
| Pain type | Source of data | Estimate (%) |
|---|---|---|
| Nociceptive | Animal model: LITF-r (low-intensity tail-flick test, rat) | 54 |
| Neuropathic | Animal model: SNL-r (spinal nerve ligation test, rat) | 36 |
Table 2.
Summary of the calculated estimates of the contribution of tapentadol’s opioid component to adverse effects relative to analgesia (its μ-load)
| Endpoint | Source of data | Estimated μ-load (%) |
|---|---|---|
| Constipation | Animal model (inhibition of GI transit, rat) | 30 |
| Clinical trial (cLBP) | 40 | |
| Clinical trials (OA pain) | 33–41 | |
| Respiratory depression | Animal model (inhibition of CO2 stimulation) | 39–46 |
| Clinical pharmacology (VE55)–100/150 mg vs. oxycodone | ~ 40 |
Expert Opinion
Prior to the introduction of a select few analgesics that produce their effects by the combined complementary contribution of an opioid and one or more non-opioid components, the class of opioid analgesics was rather monolithic. Despite subtle differences in pharmacodynamics or pharmacokinetics, they primarily produce their therapeutic and adverse effects which are for the most part mediated by the same receptors (MOR) [4, 53]. It thus did not make sense to talk of a μ-load for this class of drugs, because the μ-load of all of the opioids was essentially 100%. However, the introduction of mixed agonists-antagonists (e.g., nalbuphine, pentazocine) and the three multi-mechanistic centrally-acting analgesics (buprenorphine, tramadol, and most recently tapentadol) raises the question of the relative contribution of the opioid component to their analgesia and to their adverse effects. Multimechanistic analgesic drugs can indeed be different, since the mechanisms can interact in an additive or synergistic way to produce analgesia, yet to a less than additive, even counteractive, way to produce adverse effects [7]. Since tapentadol was the only one of the three that was designed (‘engineered’) to have its specific ‘polypharmacologic’ profile [18, 29], it was of particular interest to determine the relative contribution of its opioid component to analgesia and to adverse effects relative to pure MOR agonists at equianalgesic doses (what we term here its μ-load) (see Table 3).
Table 3.
Article Highlights
| Article highlights |
|---|
| In terms of analgesic response, the main relevant receptor for most commonly-used opioids is MOR (μ-opioid receptor). Thus, when considering the balance between analgesia and opioid-typical adverse effects, the opioids are rather mono-mechanistic |
| In contrast, tapentadol’s analgesic effect results from the combined contributions of an opioid and a non-opioid mechanism of action |
| Tapentadol’s dual opioid (MOR) and non-opioid (norepinephrine reuptake inhibition; NRI) mechanisms of action combine in a complementary and synergistic manner to produce an antinociceptive effect (analgesia) in animal models, but in less than a synergistic manner to produce the adverse effect of constipation. The question is: to what extent does the opioid component contribute to analgesia on the one hand, and to adverse effects on the other hand? |
| We here estimate, using drug–receptor theory and several different approaches, the μ-load of tapentadol for two classic opioid adverse effects (constipation and respiratory depression) |
| The calculations confirm that both components of tapentadol’s mechanism of analgesic action contribute to its therapeutic effect |
| However, unlike mono-mechanistic opioids, the μ-load (the MOR-related effect in comparison to the effect of a pure/classical opioid at equianalgesia) of tapentadol is substantially less than 100% (calculated estimates yield values of ≤ 40%) |
| The μ-load varies somewhat by type of pain (neuropathic and nociceptive) and by type of adverse effect (constipation and respiratory depression) that is considered for the estimation |
| Estimates using clinical trial data yield results similar to the estimates using in vitro and in vivo animal data |
| The results of this analysis are consistent with, and help explain, the favorable clinical characteristics of tapentadol with regards to opioid-induced side effects. Because of the synergistic mechanism of action, tapentadol provides 100% of the analgesic efficacy of a pure strong opioid, but at < 40% of the µ-load |
| The results of our analysis also suggest that for a drug to be a strong analgesic, it does not have to be a strong opioid, and that this distinction is particularly important when considering the side effects of strong analgesics |
Based on extensive in vitro and in vivo data in a large variety of assays and in a large variety of animal models, tapentadol (a single entity without an analgesically-active metabolite) produces its antinociceptive (analgesic) effect by the combined contribution of two components: one opioid and the other non-opioid [18–20, 31–33]. The opioid component consists primarily of activation (an agonist action) at μ-opioid receptors, while the non-opioid component consists primarily of inhibition of the neuronal reuptake of norepinephrine. In addition, preclinical studies have demonstrated a (positive) synergistic interaction between the two components on pain relief, but less than synergistic interaction on an adverse effect endpoint (constipation). It is therefore clearly a mechanistic possibility that, unlike for mono-mechanistic opioids, the μ-load of tapentadol can be less than 100% [34–40], as found here.
Since no study has been specifically designed to determine a drug’s μ-load, there was no precedence to follow, so we decided to intentionally use a variety of techniques and assumptions, some techniques more rigorous and quantitative than others, in order to obtain a broad overview of the range of estimates. We also decided to compare the results obtained from in vitro, in vivo (animal model), and clinical trial data. The reasoning was that diverse approaches would give a better estimate than would a single estimate. In fact, the estimates of μ-load obtained here using the diverse approaches and sources of data converged to a surprising degree, and rather consistently at ≤ 40%.
Given the inherent diversity of the sources and usability of the data, and the number of quite disparate approaches employed, it seems remarkable that the estimates fall within such a narrow range. The fact that they do enhances confidence in their estimates. The results also make sense. For example, tapentadol was designed to have dual contributions to analgesia, only one of which is opioid. So the calculations here that the opioid component is not the only contributor to analgesia, but that the non-opioid component also contributes to a significant extent, is consistent with the original design intent. It is therefore also reasonable to find here that the estimates of the μ-load of tapentadol are consistent from test tube to animal to human data, and yield values substantially less than 100%.
The estimates for tapentadol’s μ-load calculated here are consistent, not only with the intent from the inception of the drug’s design but also with the clinical experience with the drug. In several clinical trials, tapentadol has been shown to produce analgesia that is equivalent to oxycodone, but with greater gastrointestinal tolerability (greater separation of adverse from therapeutic effects). The results are also consistent with tapentadol’s activity against different types of pain [34–40]. As mentioned above, preclinical data suggest that the NRI component instills in it a more potent effect against neuropathic pain than against acute nociceptive pain, another fact consistent with the present findings. Interestingly, this is also consistent with clinical evidence suggesting that tapentadol is more potent in chronic low back pain patients whose pain has a neuropathic component as opposed to pain of purely nociceptive origin [60, 61].
Given the significant contribution of tapentadol’s non-opioid mechanism (NRI) of analgesic action, and its low μ-load for adverse effects estimated here, it might be worth consideration that it is more accurately called a ‘strong analgesic’ rather than a ‘strong opioid’. The results of our analysis suggest that for a drug to be a ‘strong analgesic’, it does not have to be a strong opioid, and that this distinction is particularly important when considering the side effects of strong analgesics.
Acknowledgements
This paper is dedicated to the memory of Ronald J. Tallarida, Ph.D., whose approach to quantitative pharmacology was an inspiration.
Funding
Grünenthal GmbH sponsored the analysis, the article processing charges and the open access fee. All authors had full access to all of the data in this study and take complete responsibility for the integrity of the data and accuracy of the data analysis.
Authorship
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Disclosure
Robert B. Raffa was a previous employee of Johnson & Johnson and has received research support and honoraria from several other pharmaceutical companies involved in analgesics research and development, but he receives no remuneration based on sales of any product. He is a cofounder of CaRafe Drug Innovation (involved in the discovery of non-opioid analgesics) and advisor/investor with Chamounix Ventures (a medical marijuana dispensary licensee). Thomas M. Tzschentke is an employee of Grünenthal GmbH. Christian Elling is an employee of Grünenthal GmbH.
Compliance with Ethics Guidelines
This article is based on previously conducted studies and does not contain any new studies with human participants or animals performed by any of the authors.
Open Access
This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
Footnotes
Enhanced digital features
To view enhanced digital features for this article go to 10.6084/m9.figshare.6979343.
Robert B. Raffa, Christian Elling, Thomas M. Tzschentke contributed equally to the manuscript.
References
- 1.Snyder SH, Pasternak GW. Historical review: opioid receptors. Trends Pharmacol Sci. 2003;24(4):198–205. 10.1016/S0165-6147(03)00066-X [DOI] [PubMed] [Google Scholar]
- 2.Pasternak GW, Pan YX. Mu opioids and their receptors: evolution of a concept. Pharmacol Rev. 2013;65(4):1257–317. 10.1124/pr.112.007138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stanley TH. The fentanyl story. J Pain. 2014;15(12):1215–26. 10.1016/j.jpain.2014.08.010 [DOI] [PubMed] [Google Scholar]
- 4.Trescot AM, Datta S, Lee M, Hansen H. Opioid pharmacology. Pain Phys. 2008;11(2 Suppl):S133–53. [PubMed] [Google Scholar]
- 5.Sliwoski G, Kothiwale S, Meiler J, Lowe EW Jr. Computational methods in drug discovery. Pharmacol Rev. 2014;66(1):334–95. 10.1124/pr.112.007336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hu Y, Bajorath J. Polypharmacology directed compound data mining: identification of promiscuous chemotypes with different activity profiles and comparison to approved drugs. J Chem Inf Model. 2010;50(12):2112–8. 10.1021/ci1003637 [DOI] [PubMed] [Google Scholar]
- 7.Raffa RB. On subclasses of opioid analgesics. Curr Med Res Opin. 2014;30(12):2579–84. 10.1185/03007995.2014.952717 [DOI] [PubMed] [Google Scholar]
- 8.Tallarida RJ. Drug synergism and dose-effect data analysis. Boca Raton: Chapman & Hall/CRC; 2000. [Google Scholar]
- 9.Raffa RB, Clark-Vetri R, Tallarida RJ, Wertheimer AI. Combination strategies for pain management. Expert Opin Pharmacother. 2003;4(10):1697–708. 10.1517/14656566.4.10.1697 [DOI] [PubMed] [Google Scholar]
- 10.Zimmermann GR, Lehar J, Keith CT. Multi-target therapeutics: when the whole is greater than the sum of the parts. Drug Discov Today. 2007;12(1–2):34–42. 10.1016/j.drudis.2006.11.008 [DOI] [PubMed] [Google Scholar]
- 11.Dahan A, Yassen A, Romberg R, et al. Buprenorphine induces ceiling in respiratory depression but not in analgesia. Br J Anaesth. 2006;96(5):627–32. 10.1093/bja/ael051 [DOI] [PubMed] [Google Scholar]
- 12.Lutfy K, Cowan A. Buprenorphine: a unique drug with complex pharmacology. Curr Neuropharmacol. 2004;2(4):395–402. 10.2174/1570159043359477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tallarida RJ, Cowan A, Raffa RB. On deriving the dose-effect relation of an unknown second component: an example using buprenorphine preclinical data. Drug Alcohol Depend. 2010;109(1–3):126–9. 10.1016/j.drugalcdep.2009.12.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wheeler-Aceto H, Cowan A. Buprenorphine and morphine cause antinociception by different transduction mechanisms. Eur J Pharmacol. 1991;195(3):411–3. 10.1016/0014-2999(91)90485-9 [DOI] [PubMed] [Google Scholar]
- 15.Cowan A. Buprenorphine: new pharmacological aspects. Int J Clin Pract Suppl. 2003; 133:3–8. (discussion 23–4). [PubMed]
- 16.Raffa RB, Friderichs E. The basic science aspect of tramadol hydrochloride. Pain Revi. 1996;3:249–71. [Google Scholar]
- 17.Raffa RB, Friderichs E, Reimann W, et al. Complementary and synergistic antinociceptive interaction between the enantiomers of tramadol. J Pharmacol Exp Ther. 1993;267(1):331–40. [PubMed] [Google Scholar]
- 18.Tzschentke TM, Christoph T, Kogel B, et al. (-)-(1R,2R)-3-(3-dimethylamino- 1-ethyl-2-methyl-propyl)-phenol hydrochloride (tapentadol HCl): a novel mu-opioid receptor agonist/norepinephrine reuptake inhibitor with broad-spectrum analgesic properties. J Pharmacol Exp Ther. 2007;323(1):265–76. 10.1124/jpet.107.126052 [DOI] [PubMed] [Google Scholar]
- 19.Tzschentke TM, Jahnel U, Kogel B, et al. Tapentadol hydrochloride: a next- generation, centrally acting analgesic with two mechanisms of action in a single molecule. Drugs Today (Barc). 2009;45(7):483–96. [DOI] [PubMed] [Google Scholar]
- 20.Tzschentke TM, de Vry J, Terlinden R, et al. Tapentadol hydrochloride: analgesic mu-opioid receptor agonist noradrenaline reuptake inhibitor. Drugs Future. 2006;31(12):1053–61. 10.1358/dof.2006.031.12.1047744 [DOI] [Google Scholar]
- 21.Millan MJ. Descending control of pain. Prog Neurobiol. 2002;66(6):355–474. 10.1016/S0301-0082(02)00009-6 [DOI] [PubMed] [Google Scholar]
- 22.Ossipov MH, Dussor GO, Porreca F. Central modulation of pain. J Clin Invest. 2010;120(11):3779–87. 10.1172/JCI43766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brennan TJ. Pathophysiology of postoperative pain. Pain. 2011;152(3 Suppl):S33–40. 10.1016/j.pain.2010.11.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bee LA, Dickenson AH. Neuropathic pain: multiple mechanisms at multiple sites. Future Neurol. 2007; 2:661–71. 10.2217/14796708.2.6.661 [DOI] [Google Scholar]
- 25.Morphy R, Rankovic Z. Designed multiple ligands. An emerging drug discovery paradigm. J Med Chem. 2005;48(21):6523–43. 10.1021/jm058225d [DOI] [PubMed] [Google Scholar]
- 26.Pergolizzi J. Chronic pain-moving from symptom control to mechanism- based treatment. Curr Med Res Opin. 2011;27(10):2079–80. 10.1185/03007995.2011.619446 [DOI] [PubMed] [Google Scholar]
- 27.Budd K, Raffa RB. Buprenorphine—the Unique Opioid analgesic. Berlin: Thieme; 2005. [Google Scholar]
- 28.Ding Z, Raffa RB. Identification of an additional supraspinal component to the analgesic mechanism of action of buprenorphine. Br J Pharmacol. 2009;157(5):831–43. 10.1111/j.1476-5381.2009.00209.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Raffa RB, Buschmann H, Christoph T, et al. Mechanistic and functional differentiation of tapentadol and tramadol. Expert Opin Pharmacother. 2012;13(10):1437–49. 10.1517/14656566.2012.696097 [DOI] [PubMed] [Google Scholar]
- 30.Langford RM, Knaggs R, Farquhar-Smith P, Dickenson AH. Is tapentadol different from classical opioids? A review of the evidence. Br J Pain. 2016;10(4):217–21. 10.1177/2049463716657363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Benade V, Nirogi R, Bhyrapuneni G, et al. Mechanistic evaluation of tapentadol in reducing the pain perception using in vivo brain and spinal cord microdialysis in rats. Eur J Pharmacol. 2017;809:224–30. 10.1016/j.ejphar.2017.04.013 [DOI] [PubMed] [Google Scholar]
- 32.Kress HG. Tapentadol and its two mechanisms of action: is there a new pharmacological class of centrally-acting analgesics on the horizon? Eur J Pain. 2010;14(8):781–3. 10.1016/j.ejpain.2010.06.017 [DOI] [PubMed] [Google Scholar]
- 33.Schröder W, Vry JD, Tzschentke TM, et al. Differential contribution of opioid and noradrenergic mechanisms of tapentadol in rat models of nociceptive and neuropathic pain. Eur J Pain. 2010;14(8):814–21. 10.1016/j.ejpain.2010.05.005 [DOI] [PubMed] [Google Scholar]
- 34.Schwartz S, Etropolski MS, Shapiro DY, et al. A pooled analysis evaluating the efficacy and tolerability of tapentadol extended release for chronic, painful diabetic peripheral neuropathy. Clin Drug Investig. 2015;35(2):95–108. 10.1007/s40261-014-0249-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wade WE, Spruill WJ. Tapentadol hydrochloride: a centrally acting oral analgesic. Clin Ther. 2009;31(12):2804–18. 10.1016/j.clinthera.2009.12.003 [DOI] [PubMed] [Google Scholar]
- 36.Stegmann JU, Weber H, Steup A, et al. The efficacy and tolerability of multiple-dose tapentadol immediate release for the relief of acute pain following orthopedic (bunionectomy) surgery. Curr Med Res Opin. 2008;24(11):3185–96. 10.1185/03007990802448056 [DOI] [PubMed] [Google Scholar]
- 37.Kwong WJ, Hammond G, Upmalis D, et al. Bowel function after tapentadol and oxycodone immediate release (IR) treatment in patients with low back or osteoarthritis pain. Clin J Pain. 2013;29(8):664–72. 10.1097/AJP.0b013e318274b695 [DOI] [PubMed] [Google Scholar]
- 38.Hartrick C, Van Hove I, Stegmann JU, et al. Efficacy and tolerability of tapentadol immediate release and oxycodone HCl immediate release in patients awaiting primary joint replacement surgery for end-stage joint disease: a 10-day, phase III, randomized, double-blind, active- and placebo-controlled study. Clin Ther. 2009;31(2):260–71. 10.1016/j.clinthera.2009.02.009 [DOI] [PubMed] [Google Scholar]
- 39.Hale M, Upmalis D, Okamoto A, et al. Tolerability of tapentadol immediate release in patients with lower back pain or osteoarthritis of the hip or knee over 90 days: a randomized, double-blind study. Curr Med Res Opin. 2009;25(5):1095–104. 10.1185/03007990902816970 [DOI] [PubMed] [Google Scholar]
- 40.Daniels SE, Upmalis D, Okamoto A, et al. A randomized, double-blind, phase III study comparing multiple doses of tapentadol IR, oxycodone IR, and placebo for postoperative (bunionectomy) pain. Curr Med Res Opin. 2009;25(3):765–76. 10.1185/03007990902728183 [DOI] [PubMed] [Google Scholar]
- 41.Terlinden R, Kogel BY, Englberger W, Tzschentke TM. In vitro and in vivo characterization of tapentadol metabolites. Methods Find Exp Clin Pharmacol. 2010;32(1):31–8. 10.1358/mf.2010.32.1.1434165 [DOI] [PubMed] [Google Scholar]
- 42.Nagar S, Raffa RB. Looking beyond the administered drug: metabolites of opioid analgesics. J Fam Pract. 2008;57(6 Suppl Looking):s25–32. [PubMed] [Google Scholar]
- 43.Schiene K, De Vry J, Tzschentke TM. Antinociceptive and antihyperalgesic effects of tapentadol in animal models of inflammatory pain. J Pharmacol Exp Ther. 2011;339(2):537–44. 10.1124/jpet.111.181263 [DOI] [PubMed] [Google Scholar]
- 44.Riemsma R, Forbes C, Harker J, et al. Systematic review of tapentadol in chronic severe pain. Curr Med Res Opin. 2011;27(10):1907–30. 10.1185/03007995.2011.611494 [DOI] [PubMed] [Google Scholar]
- 45.Pergolizzi J, Alegre C, Blake D, et al. Current considerations for the treatment of severe chronic pain: the potential for tapentadol. Pain Pract. 2012;12(4):290–306. 10.1111/j.1533-2500.2011.00487.x [DOI] [PubMed] [Google Scholar]
- 46.Pergolizzi J, Alon E, Baron R, et al. Tapentadol in the management of chronic low back pain: a novel approach to a complex condition? J Pain Res. 2011;4:203–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tzschentke TM, Christoph T, Kogel BY. The mu-opioid receptor agonist/noradrenaline reuptake inhibition (MOR-NRI) concept in analgesia: the case of tapentadol. CNS Drugs. 2014;28(4):319–29. 10.1007/s40263-014-0151-9 [DOI] [PubMed] [Google Scholar]
- 48.Lalovic B, Kharasch E, Hoffer C, et al. Pharmacokinetics and pharmacodynamics of oral oxycodone in healthy human subjects: role of circulating active metabolites. Clin Pharmacol Ther. 2006;79(5):461–79. 10.1016/j.clpt.2006.01.009 [DOI] [PubMed] [Google Scholar]
- 49.Christoph T, De Vry J, Tzschentke TM. Tapentadol, but not morphine, selectively inhibits disease-related thermal hyperalgesia in a mouse model of diabetic neuropathic pain. Neurosci Lett. 2010;470(2):91–4. 10.1016/j.neulet.2009.12.020 [DOI] [PubMed] [Google Scholar]
- 50.Schröder W, Tzschentke TM, Terlinden R, et al. Synergistic interaction between the two mechanisms of action of tapentadol in analgesia. J Pharmacol Exp Ther. 2011;337(1):312–20. 10.1124/jpet.110.175042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cowan A, Raffa RB, Tallarida CS, et al. Lack of synergistic interaction between the two mechanisms of action of tapentadol in gastrointestinal transit. Eur J Pain. 2014;18(8):1148–56. 10.1002/j.1532-2149.2014.00461.x [DOI] [PubMed] [Google Scholar]
- 52.Afilalo M, Etropolski MS, Kuperwasser B, et al. Efficacy and safety of Tapentadol extended release compared with oxycodone controlled release for the management of moderate to severe chronic pain related to osteoarthritis of the knee: a randomized, double-blind, placebo- and active-controlled phase III study. Clin Drug Investig. 2010;30(8):489–505. 10.2165/11533440-000000000-00000 [DOI] [PubMed] [Google Scholar]
- 53.Xiao JP, Li AL, Feng BM, et al. Efficacy and safety of tapentadol immediate release assessment in treatment of moderate to severe pain: a systematic review and meta-analysis. Pain Med. 2017;18(1):14–24. 10.1093/pm/pnw154 [DOI] [PubMed] [Google Scholar]
- 54.Imanaka K, Tominaga Y, Etropolski M, et al. Efficacy and safety of oral tapentadol extended release in Japanese and Korean patients with moderate to severe, chronic malignant tumor-related pain. Curr Med Res Opin. 2013;29(10):1399–409. 10.1185/03007995.2013.831816 [DOI] [PubMed] [Google Scholar]
- 55.Wild JE, Grond S, Kuperwasser B, et al. Long-term safety and tolerability of tapentadol extended release for the management of chronic low back pain or osteoarthritis pain. Pain Pract. 2010;10(5):416–27. 10.1111/j.1533-2500.2010.00397.x [DOI] [PubMed] [Google Scholar]
- 56.Buynak R, Shapiro DY, Okamoto A, et al. Efficacy and safety of tapentadol extended release for the management of chronic low back pain: results of a prospective, randomized, double-blind, placebo- and active-controlled Phase III study. Expert Opin Pharmacother. 2010;11(11):1787–804. 10.1517/14656566.2010.497720 [DOI] [PubMed] [Google Scholar]
- 57.Serrie A, Lange B, Steup A. Tapentadol prolonged-release for moderate-to- severe chronic osteoarthritis knee pain: a double-blind, randomized, placebo- and oxycodone controlled release-controlled study. Curr Med Res Opin. 2017;33(8):1423–32. 10.1080/03007995.2017.1335189 [DOI] [PubMed] [Google Scholar]
- 58.Lange B, Kuperwasser B, Okamoto A, et al. Efficacy and safety of tapentadol prolonged release for chronic osteoarthritis pain and low back pain. Adv Ther. 2010;27(6):381–99. 10.1007/s12325-010-0036-3 [DOI] [PubMed] [Google Scholar]
- 59.van der Schrier R, Jonkman K, van Velzen M, et al. An experimental study comparing the respiratory effects of tapentadol and oxycodone in healthy volunteers. Br J Anaesth. 2017;119(6):1169–77. 10.1093/bja/aex295 [DOI] [PubMed] [Google Scholar]
- 60.Steigerwald I, Muller M, Davies A, et al. Effectiveness and safety of tapentadol prolonged release for severe, chronic low back pain with or without a neuropathic pain component: results of an open-label, phase 3b study. Curr Med Res Opin. 2012;28(6):911–36. 10.1185/03007995.2012.679254 [DOI] [PubMed] [Google Scholar]
- 61.Reimer M, Hullemann P, Hukauf M, et al. Prediction of response to tapentadol in chronic low back pain. Eur J Pain. 2017;21(2):322–33. 10.1002/ejp.926 [DOI] [PMC free article] [PubMed] [Google Scholar]