

Abstract
Background
The typical familial form of Alzheimer’s disease (FAD) accounts for about 5% of total Alzheimer’s disease (AD) cases. Presenilins (PSEN1 and PSEN2) and amyloid-β (A4) precursor protein (APP) genes carry all reported FAD-linked mutations. However, other genetic loci may be involved in AD. For instance, seizure-related gene 6 (SEZ6) has been reported in brain development and psychiatric disorders and is differentially expressed in the cerebrospinal fluid of AD cases.
Methods
We describe a targeted exome sequencing analysis of a large Italian kindred with AD, negative for PSEN and APP variants, that indicated the SEZ6 heterozygous mutation R615H is associated with the pathology.
Results
We overexpressed R615H mutation in H4-SW cells, finding a reduction of amyloid peptide Aβ(1–42). Sez6 expression decreased with age in a mouse model of AD (3xTG-AD), but independently from transgene expression.
Conclusions
These results support a role of exome sequencing for disease-associated variant discovery and reinforce available data on SEZ6 in AD models.
Electronic supplementary material
The online version of this article (10.1186/s13195-018-0435-2) contains supplementary material, which is available to authorized users.
Keywords: Alzheimer’s disease, SEZ6, Exome sequencing, Rare variants
Background
Alzheimer’s disease (AD) is a multifactorial neurodegenerative disorder whose onset is mostly sporadic [1]. The genetic background has a major role in AD, and DNA variants may contribute, ranging from predisposing risk factors (having from medium to large effect size, such as the ε4 allele of the APOE gene) [2] to full penetrant causal mutations in a few genes, namely presenilins (PSEN1 and PSEN2) and the amyloid-β (A4) precursor protein (APP) [3, 4]. PSEN1/2 and APP gene mutations have been linked to early-onset, autosomal dominant familial forms of Alzheimer’s disease (FAD) [5, 6]. Recently, large-scale whole-exome sequencing has found rare variants reported to contribute to AD risk, such as in the PLCG2, ABI3, and TREM2 genes [7]. These findings indicate the involvement in familiar forms of AD of variants belonging to genes other than PSEN1/2 and APP, which may have a causal or predisposing role, as recently reported for SORL1 gene [8].
We report an Italian family with several cases of AD (having an onset between 60 and 70 years) negative for PSEN1/2 or APP mutations and whose available affected members were found to bear SEZ6 gene rare missense variant R615H. We describe the genetic, in vitro, and in vivo findings further supporting a role for SEZ6 in AD molecular mechanisms.
Methods
Family and patient description
The family’s pedigree is reported in Fig. 1. We extracted DNA for exome sequencing analysis from the members indicated by the code PR (seven subjects). We had clinical details about three generations after the founder. Ten dementia cases were reported in the whole pedigree, with an additional member having Parkinson’s disease. The age of onset of neurodegenerative disorders ranged from 60 to 70 years. In the first generation, one early-onset dementia case was reported (age at death, 48 years). In the second generation, 8 of 25 siblings (32%) were diagnosed with AD, with an additional case in the third generation (age at onset 64 years). The remaining siblings of this generation were cognitively normal, aged between 35 and 45 years. Apolipoprotein E genotype (APOE) of available patients was in all cases ε3//ε3 apart from PR5 (ε3//ε4). Two siblings of PR5, diagnosed with AD, had dementia too, but they were unavailable for sampling.
Fig. 1.

Pedigree of the Italian family with Alzheimer’s disease. We report clinical information for the last three generations after the founders. Sex, age at sampling, and apolipoprotein E (APOE) genotype of each available family member indicated in the box. The numbers next to subjects with dementia are the age at death. The roman numbers refer to the generation, with the progressive numbers linking to every generation sibling
Sporadic AD cases (n = 9) and cognitively normal elderly control subjects (n = 191) were included for independent evaluation of the SEZ6(R615H) variant frequency by digital droplet PCR (ddPCR).
Patients and healthy control subjects were recruited by the same clinical center, and AD was diagnosed according to international criteria. Healthy control subjects were spouses of patients coming to clinical attention, and they had no sign of neurodegenerative disorders and Mini Mental State Examination (MMSE) scores in the normal range [9].
Exome sequencing and APOE genotyping
The full-exome sequencing of 4811 disease-associated genes (clinical exome) was done starting from 50 ng of DNA diluted in Tris-HCl 10 mM, pH 8.5 (TruSight One Sequencing Panel; Illumina, San Diego, CA, USA), following the manufacturer’s instructions. Briefly, capture-based libraries were prepared by pooling three samples per time. The libraries’ concentrations were calculated using a Qubit® dsDNA High-Sensitivity Assay Kit (Invitrogen, Carlsbad, CA, USA), and the distribution of DNA fragments for each library was evaluated using a high-sensitivity DNA kit and a 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA). Each library was run on a MiSeq platform (Illumina) using a 2 × 150-bp (300 cycles) configuration on a V3 sequencing flow cell.
Data analysis was performed according to best practice from the bioinformatics community. Raw sequence fragments (reads) were aligned to the reference genome (human, build hg19) with the Burrows-Wheeler alignment tool [10], followed by post-processing to recalibrate base call quality scores. Variants were called with the Genome Analysis Toolkit [11–13], using the HaplotypeCaller method, then annotated with the Variant Effect Predictor [14] and loaded into a specialized database [15] for further analysis. In silico mutation impact predictions were extracted from the dbNSFP database [16]. For computation, we used the “bcbio” pipeline (https://github.com/chapmanb/bcbio-nextgen) running on a high-performance computing platform as part of the Cloud4CaRE project. Data files were uploaded to the European Nucleotide Archive with accession number pending.
Selection of candidate variants used the following criteria: (a) depth at least 30×; (b) low frequency in the general population (< 1% in the 1000 Genomes Project); (c) at least a damaging predicted effect as reported from the dbNSFP; and (d) present in all family members affected by AD or their offspring. The APOE genotype was assessed by restriction fragment length polymorphism using the CfoI (Roche, Basel, Switzerland) restriction enzyme, as previously described [17].
Exome sequencing validation by digital droplet PCR
ddPCR experiments were done with the Bio-Rad QX200TM ddPCR system (Bio-Rad Laboratories, Hercules, CA, USA). The mutational assay for SEZ6 R615H was carried out according to the manufacturer’s instructions. Briefly, the TaqMan™ reaction mix, composed of 2× ddPCR Supermix for probes (no deoxyuridine triphosphate), 20× custom target probes for mut SEZ6 (probe sequence: CTACGGTCATGGGCAG-FAM), and 20× reference probes for wild-type SEZ6 (probe sequence: CTACGGTCGTGGGCA-HEX), was assembled at a final concentration of 450 nM and 20 ng of DNA in a volume of 20 μl. This reaction mix was added to a DG8 cartridge together with 60 μl of droplet generation oil for probe and used for droplet generation (QX200 droplet generator; Bio-Rad Laboratories). Droplets were then manually transferred to 96-well PCR plates and placed on a thermal cycler (T100 Thermal Cycler; Bio-Rad Laboratories) for the PCR amplification (thermal cycling conditions: 95 °C for 5 min, 95 °C for 30 s, and 55 °C for 1 min, 40 cycles; 98 °C for 10 min and 4 °C infinite; ramping rate 2 °C/s). The PCR plate was then transferred into the QX100 Droplet Reader for the fluorescence measurement of FAM and HEX probes. The numbers of positive and negative droplets were used to calculate the concentrations (copies/μl) of the target and the reference SEZ6 DNA sequence and their Poisson-based 95% CIs, excluding reactions with fewer than 10,000 total events (positive and negative) (QuantaSoft Analysis pro software 1.0.596; Bio-Rad Laboratories).
For family members and patients with sporadic AD, experiments were run in duplicate; the assay on the healthy population was run once.
Cloning and overexpression of SEZ6(R615H) in H4-SW cells
pSEZ6(R615H) cloning
Synthetic SEZ6(R615H) complementary DNA was provided by GenScript® in pCDNA3.1(+) vector and expanded in competent Escherichia coli cells (strain JM109; Promega, Madison, WI, USA). After purification, pSEZ6(R615H) was verified through the unique enzymatic restriction site PmeI (New England Biolabs, Hitchin, UK) and agarose gel electrophoresis.
Cell culture
H4-SW neuroglioma cells overexpressing human APP gene harboring the Swedish (SW) mutation [18] were grown in DMEM supplemented with 10% FBS, 2 mM l-glutamine, and antibiotics (100 U/ml penicillin, 100 μg/ml streptomycin, 300 μg/ml hygromycin B, 10 μg/ml blasticidin-S).
Transient transfection was done using FuGENE® HD Transfection Reagent (Promega), and cells were selected with G418 (1200 μg/ml) after 48 h. For clonal selection of SEZ6(R615H) mutants, we picked colonies and analyzed DNA and protein extracts by PCR and Western blotting. Finally, a single-point mutation (G→A) leading to R615H substitution was checked by Sanger sequencing.
PCR for SEZ6(R615H) expression in H4-SW cells
PCR was run in a 20-μl mixture containing 50 ng of DNA, 0.5 mM each of forward primer 5′-CTACGGTCATGGGCAGGATTG-3′, which contains the single-point mutation (G→A), and the reverse oligonucleotide primer 5′- ATCATGGCAGGTGAGGATGGACT-3′ (metabion, Planegg, Germany); 1× PCR buffer 200 mM Tris-HCl, 500 mM KCl (Thermo Fisher Scientific, Waltham, MA, USA); 2.5 mM deoxynucleotide triphosphate (Thermo Fisher Scientific); 25 mM MgCl2 (Thermo Fisher Scientific); and 1 unit of Taq polymerase (Thermo Fisher Scientific). Amplification was done with an initial denaturation at 95 °C for 2 min, followed by 30 cycles of denaturation at 95 °C for 30 s, annealing at 61.7 °C for 30 s, extension at 72 °C for 70 s, and a final 5-min extension at 72 °C. The resulting PCR fragments were resolved by 1% agarose gel electrophoresis (Sigma-Aldrich, St. Louis, MO, USA).
Western blotting for SEZ6 overexpression in H4-SW cells
To assess protein overexpression of SEZ6 in H4-SW, protein extracts (18 μg) were separated on 8% SDS-PAGE gel and transferred to a nitrocellulose membrane. Blots were developed using horseradish peroxidase-conjugated secondary antibodies and the ECL chemiluminescence system (MerckMillipore, Burlington, MA, USA). All blots were normalized to α-tubulin and quantified using ImageJ software (National Institutes of Health, Bethesda, MD, USA). The following antibodies were used: anti-α-tubulin (1:7500; Abcam, Cambridge, UK) and anti-SEZ6 (1:1000; Aviva Systems Biology, San Diego, CA, USA).
DNA sequencing
To verify the presence of the single point mutation, we amplified the region containing the mutated base by PCR with forward primer 5′- GAGATCACAGACTCGGCTG-3′ and the reverse primer 5′- ATCATGGCAGGTGAGGATGGACT-3′ (metabion). The total amount of the generated PCR product was purified using the Wizard SV Gel PCR Clean-Up System (Promega) and sent to a Sanger sequencing service (Eurofins Genomics, Ebersberg, Germany). Output data were analyzed using Chromas Lite 2.01 software.
Aβ(1–42) and Aβ(1–40) in H4-SW cells expressing SEZ6(R615H)
A specific sandwich enzyme-linked immunosorbent assay (ELISA) (Immuno-Biological Laboratories Co., Gunma, Japan) was used to measure Aβ(1–42) and Aβ(1–40) concentrations in conditioned media from cultured H4-SW cells. A total of 150 × 103 cells were seeded in a six-well plate and grown overnight. The next day, the medium was changed, and after 48 h it was collected and immediately frozen after the addition of a broad-spectrum protease inhibitor (Sigma-Aldrich). An aliquot of 100 μl was used for ELISA to assess each value in triplicate.
Western blot analysis for Sez6 brain expression in 3xTG-AD mice
For Sez6 brain expression analysis, we used 3xTG-AD mice at 3, 9, and 19 months of age. This triple-transgenic model harbors human PS1(M146 V), APP(SW), and MAPT(P301L) transgenes, and starting from around 9 months of age, mice develop at brain level amyloid plaques and protein tau tangles. They also show early signs of synaptic dysfunction (starting from around 3 months of age), including long-term potentiation alteration [19]. Strain, age, and sex-matched nontransgenic animals were used as controls. Mice were housed at 23 °C room temperature with food and water ad libitum and a 12-h/12-h light/dark cycle. To obtain brain protein extract, the cortex was dissected from a single brain hemisphere and homogenized with ice-cold lysis buffer (pH 7.4) containing 1% Triton X-100 and a broad-range protease inhibitor cocktail. Cortex protein extract (20 μg) was analyzed as described above.
Statistics
Data analysis was done using Prism® version 6.0 software (GraphPad Software, La Jolla, CA, USA). In vitro and in vivo data were compared using one-way analysis of variance followed by Tukey’s post hoc test. Two-tailed levels of significance were used, and p < 0.05 was considered significant.
Results
Exome sequencing and APOE genotyping
To identify variants linked to dementia phenotype, we sequenced DNA samples from family members (healthy and AD cases) and unrelated patients with sporadic AD for a set of over 4000 genes reported as implicated in rare and genetic diseases. Our initial analysis identified 15,745 variants passing our quality control filters (variant depth 30× or more). Many of these were common polymorphisms present in the general population, so we selected only those rare in the European population (< 1% frequency), lowering the count to 612 (Additional file 1: Table S1).
To further narrow the search for variants of interest, we used in silico analysis to restrict our findings to those predicted as damaging for protein, finding 138 variants (Additional file 1: Table S1). The majority (96.4%) of possible damaging variants were common between both familial and sporadic AD samples. On the contrary, five variants (3.6%) were exclusive to the family samples (Table 1). In particular, a missense variant in the SEZ6 neuronal gene (c.1844G>A, R615H) was present only in the two available AD cases (PR1 and PR5) and in a first-degree relative (PR2, son of PR5). This variant was localized on one of the extracellular CUB domains of the protein [20, 21] and was predicted to have a high damaging potential (Combined Annotation Dependent Domain [CADD] score = 23). This prompted us to further focus on this variant.
Table 1.
Variants exclusive of family members and satisfying the filtering criteria
| Chr | Position | Gene | Variant | Amino acid change | (%) | dbSNP ID |
Found in (family code) |
|---|---|---|---|---|---|---|---|
| chr8 | 144,589,984 | ZC3H3 | c.1646 C > T |
p.Ser549 Leu |
0.5% | rs 149,025,999 |
PR 1, PR 2, PR 3, PR 4, PR 5, PR 7, PR 9 |
| chr9 | 738,341 | KANK1 | c.3391 G > C |
p.Ala1131Pro | 0.1% | rs 180,816,986 |
PR1, PR3, PR5 |
| chr17 | 27,286,417 | SEZ6 | c.1844 G > A |
p.Arg615 His |
0.01% | rs 371,753,097 |
PR1, PR2, PR5 |
| chr20 | 57,598,807 | TUBB1 | c.326 G > A |
p.Gly109 Glu |
0.2% | rs 41,303,899 |
PR1, PR2, PR3, PR5 |
| chr22 | 24,717,509 | SPECC1L | c.562 C > T |
p.Leu188 Phe |
0.9% | rs 56,168,869 |
PR1, PR2, PR3, PR5, PR9 |
Chr Chromosome number
Percentage population frequency refers to data of the European population frequency derived from the 1000 Genomes Project at the time the manuscript was written. See the “Methods” section of text for further details. Chromosome positions refer to the hg19 assembly. The gene of interest (SEZ6) is highlighted in bold, and members affected with AD are Italic
Validation of exome sequencing SEZ6(R615H) data and variant screening in sporadic AD cases and healthy control subjects
Because the clinical exome results indicated a mutation in SEZ6 gene (c.1844G>A) as unique to the available family members with AD, we performed an independent validation to confirm the result. Using ddPCR, we tested for the SEZ6 variant in exome sequencing-positive family members (n = 3) and in sporadic AD cases (n = 9). To exclude the possibility that the polymorphic variant of SEZ6 identified could be detected at low frequency in the healthy population, too, the mutational assay was also done in a control group of 191 cognitively healthy people.
Figure 2 shows SEZ6 mutational analysis of three family members (PR1, PR2, and PR5) and a representative case of sporadic AD (PR11). Wild-type SEZ6 (green droplets) was detected in all samples, whereas mutated SEZ6 (blue) was detected only in the PR1, PR2 and PR5 samples. A single event with both wild-type and mutated SEZ6 was detected in PR11, probably a polymerase artifact.
Fig. 2.

Digital droplet PCR validation of the exome sequencing data. For each patient, a 2D dot plot is shown, reporting the distribution of fluorescence (on the y-axis FAM amplitude, and on the x-axis HEX amplitude). FAM and HEX are the fluorescent dyes for the SEZ6 mutant and SEZ6 wild type, respectively. On the basis of the fluorescence measurements and the droplet distributions, thresholds (pink lines) were set to 5000 for the FAM channel (y-axis) and 3000 for the HEX channel (x-axis). Negative droplets (gray), FAM-positive (blue), HEX-positive (green), and FAM/HEX double-positive (orange) droplets are reported for the four cases and no-template control (NTC) analyzed. Each case represents the sum of independent reactions
Regarding a quantitative measure of the SEZ6 variant, Table 2 reports the concentration as the number of target molecules/μl of wild-type and mutant SEZ6 in all sporadic cases (n = 9), in family members (n = 3), and in healthy individuals (n = 191). Wild-type SEZ6 copies were detected in all groups. The means of wild-type SEZ6 copies/μl were 564, 258, and 130 in the healthy control group, sporadic AD cases, and family members, respectively. A high concentration of mutant SEZ6 was detected in family member samples. The simultaneous presence of the wild-type and the mutated form of SEZ6, with ratios (mutated SEZ6 to wild-type SEZ6) ranging from 0.95 to 1.1, confirmed the heterozygous nature of the SEZ6 C>T 27,286,417–27,186,418 substitution.
Table 2.
Mutant SEZ6 assay by digital droplet PCR in healthy control subjects, patients with sporadic Alzheimer’s disease, and family members
| Healthy population (n = 191) | Sporadic AD cases (n = 9) | Family members (n = 3) | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Sample | Target | Concentration (copies/μl) |
Target | Concentration (copies/μl) |
Sample | Target | Concentration (copies/μl) | Target | Concentration (copies/μl) | Sample | Target | Concentration (copies/μl) |
Target | Concentration (copies/μl) | RATIO (mut/wt) |
| 6 | MUT SEZ6 | N.D. | WT SEZ6 | 17 | PR3 | MUT SEZ6 | N.D. | WT SEZ6 | 239 | PR1 | MUT SEZ6 | 116 | WT SEZ6 | 103 | 1.13 |
| 7 | MUT SEZ6 | N.D. | WT SEZ6 | 14 | MUT SEZ6 | N.D. | WT SEZ6 | 226 | MUT SEZ6 | 114 | WT SEZ6 | 120 | 0.95 | ||
| 8 | MUT SEZ6 | N.D. | WT SEZ6 | 11 | PR4 | MUT SEZ6 | N.D. | WT SEZ6 | 220 | PR2 | MUT SEZ6 | 130 | WT SEZ6 | 130 | 1.00 |
| 9 | MUT SEZ6 | N.D. | WT SEZ6 | 59 | MUT SEZ6 | N.D. | WT SEZ6 | 224 | MUT SEZ6 | 131 | WT SEZ6 | 127 | 1.03 | ||
| 11 | MUT SEZ6 | N.D. | WT SEZ6 | 41 | PR6 | MUT SEZ6 | N.D. | WT SEZ6 | 267 | PR5 | MUT SEZ6 | 149 | WT SEZ6 | 153 | 0.97 |
| 12 | MUT SEZ6 | N.D. | WT SEZ6 | 49 | MUT SEZ6 | N.D. | WT SEZ6 | 261 | MUT SEZ6 | 154 | WT SEZ6 | 152 | 1.01 | ||
| 13 | MUT SEZ6 | N.D. | WT SEZ6 | 36 | PR7 | MUT SEZ6 | N.D. | WT SEZ6 | 331 | ||||||
| 14 | MUT SEZ6 | N.D. | WT SEZ6 | 29 | MUT SEZ6 | N.D. | WT SEZ6 | 371 | |||||||
| 16 | MUT SEZ6 | N.D. | WT SEZ6 | 29 | PR8 | MUT SEZ6 | N.D. | WT SEZ6 | 307 | ||||||
| 17 | MUT SEZ6 | N.D. | WT SEZ6 | 27 | MUT SEZ6 | N.D. | WT SEZ6 | 303 | |||||||
| 18 | MUT SEZ6 | N.D. | WT SEZ6 | 43 | PR9 | MUT SEZ6 | N.D. | WT SEZ6 | 254 | ||||||
| 19 | MUT SEZ6 | N.D. | WT SEZ6 | 30 | MUT SEZ6 | N.D. | WT SEZ6 | 266 | |||||||
| 21 | MUT SEZ6 | N.D. | WT SEZ6 | 35 | PR10 | MUT SEZ6 | N.D. | WT SEZ6 | 239 | ||||||
| 22 | MUT SEZ6 | N.D. | WT SEZ6 | 53 | MUT SEZ6 | N.D. | WT SEZ6 | 273 | |||||||
| 23 | MUT SEZ6 | N.D. | WT SEZ6 | 37 | PR11 | MUT SEZ6 | N.D. | WT SEZ6 | 233 | ||||||
| 24 | MUT SEZ6 | N.D. | WT SEZ6 | 45 | MUT SEZ6 | N.D. | WT SEZ6 | 228 | |||||||
| 25 | MUT SEZ6 | N.D. | WT SEZ6 | 32 | PR12 | MUT SEZ6 | N.D. | WT SEZ6 | 212 | ||||||
| 27 | MUT SEZ6 | N.D. | WT SEZ6 | 26 | MUT SEZ6 | N.D. | WT SEZ6 | 190 | |||||||
| 28 | MUT SEZ6 | N.D. | WT SEZ6 | 47 | |||||||||||
| 29 | MUT SEZ6 | N.D. | WT SEZ6 | 48 | |||||||||||
| 30 | MUT SEZ6 | N.D. | WT SEZ6 | 30 | |||||||||||
| 34 | MUT SEZ6 | N.D. | WT SEZ6 | 30 | |||||||||||
| 36 | MUT SEZ6 | N.D. | WT SEZ6 | 49 | |||||||||||
| 38 | MUT SEZ6 | N.D. | WT SEZ6 | 32 | |||||||||||
| 39 | MUT SEZ6 | N.D. | WT SEZ6 | 34 | |||||||||||
| 41 | MUT SEZ6 | N.D. | WT SEZ6 | 74 | |||||||||||
| 42 | MUT SEZ6 | N.D. | WT SEZ6 | 43 | |||||||||||
| 44 | MUT SEZ6 | N.D. | WT SEZ6 | 53 | |||||||||||
| 46 | MUT SEZ6 | N.D. | WT SEZ6 | 64 | |||||||||||
| 51 | MUT SEZ6 | N.D. | WT SEZ6 | 55 | |||||||||||
| 52 | MUT SEZ6 | N.D. | WT SEZ6 | 19 | |||||||||||
| 53 | MUT SEZ6 | N.D. | WT SEZ6 | 32 | |||||||||||
| 60 | MUT SEZ6 | N.D. | WT SEZ6 | 46 | |||||||||||
| 61 | MUT SEZ6 | N.D. | WT SEZ6 | 44 | |||||||||||
| 62 | MUT SEZ6 | N.D. | WT SEZ6 | 64 | |||||||||||
| 64 | MUT SEZ6 | N.D. | WT SEZ6 | 55 | |||||||||||
| 66 | MUT SEZ6 | N.D. | WT SEZ6 | 45 | |||||||||||
| 67 | MUT SEZ6 | N.D. | WT SEZ6 | 46 | |||||||||||
| 69 | MUT SEZ6 | N.D. | WT SEZ6 | 48 | |||||||||||
| 70 | MUT SEZ6 | N.D. | WT SEZ6 | 45 | |||||||||||
| 71 | MUT SEZ6 | N.D. | WT SEZ6 | 67 | |||||||||||
| 72 | MUT SEZ6 | N.D. | WT SEZ6 | 57 | |||||||||||
| 74 | MUT SEZ6 | N.D. | WT SEZ6 | 54 | |||||||||||
| 89 | MUT SEZ6 | N.D. | WT SEZ6 | 47 | |||||||||||
| 90 | MUT SEZ6 | N.D. | WT SEZ6 | 78 | |||||||||||
| 91 | MUT SEZ6 | N.D. | WT SEZ6 | 64.3 | |||||||||||
| 101 | MUT SEZ6 | N.D. | WT SEZ6 | 283 | |||||||||||
| 112 | MUT SEZ6 | N.D. | WT SEZ6 | 524 | |||||||||||
| 113 | MUT SEZ6 | N.D. | WT SEZ6 | 1451 | |||||||||||
| 114 | MUT SEZ6 | N.D. | WT SEZ6 | 962 | |||||||||||
| 115 | MUT SEZ6 | N.D. | WT SEZ6 | 534 | |||||||||||
| 118 | MUT SEZ6 | N.D. | WT SEZ6 | 527 | |||||||||||
| 119 | MUT SEZ6 | N.D. | WT SEZ6 | 1691 | |||||||||||
| 120 | MUT SEZ6 | N.D. | WT SEZ6 | 359 | |||||||||||
| 129 | MUT SEZ6 | N.D. | WT SEZ6 | 186 | |||||||||||
| 130 | MUT SEZ6 | N.D. | WT SEZ6 | 258 | |||||||||||
| 133 | MUT SEZ6 | N.D. | WT SEZ6 | 232 | |||||||||||
| 135 | MUT SEZ6 | N.D. | WT SEZ6 | 373 | |||||||||||
| 137 | MUT SEZ6 | N.D. | WT SEZ6 | 319 | |||||||||||
| 144 | MUT SEZ6 | N.D. | WT SEZ6 | 310 | |||||||||||
| 151 | MUT SEZ6 | N.D. | WT SEZ6 | 396 | |||||||||||
| 152 | MUT SEZ6 | N.D. | WT SEZ6 | 180 | |||||||||||
| 160 | MUT SEZ6 | N.D. | WT SEZ6 | 574 | |||||||||||
| 162 | MUT SEZ6 | N.D. | WT SEZ6 | 400 | |||||||||||
| 163 | MUT SEZ6 | N.D. | WT SEZ6 | 142 | |||||||||||
| 164 | MUT SEZ6 | N.D. | WT SEZ6 | 39 | |||||||||||
| 170 | MUT SEZ6 | N.D. | WT SEZ6 | 96 | |||||||||||
| 179 | MUT SEZ6 | N.D. | WT SEZ6 | 94 | |||||||||||
| 180 | MUT SEZ6 | N.D. | WT SEZ6 | 27 | |||||||||||
| 182 | MUT SEZ6 | N.D. | WT SEZ6 | 1406 | |||||||||||
| 184 | MUT SEZ6 | N.D. | WT SEZ6 | 1994 | |||||||||||
| 185 | MUT SEZ6 | N.D. | WT SEZ6 | 161 | |||||||||||
| 192 | MUT SEZ6 | N.D. | WT SEZ6 | 14.5 | |||||||||||
| 193 | MUT SEZ6 | N.D. | WT SEZ6 | 1740 | |||||||||||
| 197 | MUT SEZ6 | N.D. | WT SEZ6 | 185 | |||||||||||
| 198 | MUT SEZ6 | N.D. | WT SEZ6 | 250 | |||||||||||
| 199 | MUT SEZ6 | N.D. | WT SEZ6 | 145 | |||||||||||
| 200 | MUT SEZ6 | N.D. | WT SEZ6 | 132 | |||||||||||
| 202 | MUT SEZ6 | N.D. | WT SEZ6 | 663 | |||||||||||
| 205 | MUT SEZ6 | N.D. | WT SEZ6 | 658 | |||||||||||
| 206 | MUT SEZ6 | N.D. | WT SEZ6 | 118 | |||||||||||
| 210 | MUT SEZ6 | N.D. | WT SEZ6 | 103 | |||||||||||
| 212 | MUT SEZ6 | N.D. | WT SEZ6 | 23 | |||||||||||
| 214 | MUT SEZ6 | N.D. | WT SEZ6 | 385 | |||||||||||
| 215 | MUT SEZ6 | N.D. | WT SEZ6 | 125 | |||||||||||
| 219 | MUT SEZ6 | N.D. | WT SEZ6 | 223 | |||||||||||
| 223 | MUT SEZ6 | N.D. | WT SEZ6 | 316 | |||||||||||
| 228 | MUT SEZ6 | N.D. | WT SEZ6 | 109 | |||||||||||
| 233 | MUT SEZ6 | N.D. | WT SEZ6 | 385 | |||||||||||
| 237 | MUT SEZ6 | N.D. | WT SEZ6 | 767 | |||||||||||
| 240 | MUT SEZ6 | N.D. | WT SEZ6 | 318 | |||||||||||
| 241 | MUT SEZ6 | N.D. | WT SEZ6 | 15 | |||||||||||
| 243 | MUT SEZ6 | N.D. | WT SEZ6 | 30 | |||||||||||
| 245 | MUT SEZ6 | N.D. | WT SEZ6 | 166 | |||||||||||
| 247 | MUT SEZ6 | N.D. | WT SEZ6 | 161 | |||||||||||
| 251 | MUT SEZ6 | N.D. | WT SEZ6 | 164 | |||||||||||
| 253 | MUT SEZ6 | N.D. | WT SEZ6 | 491 | |||||||||||
| 254 | MUT SEZ6 | N.D. | WT SEZ6 | 772 | |||||||||||
| 255 | MUT SEZ6 | N.D. | WT SEZ6 | 771 | |||||||||||
| 257 | MUT SEZ6 | N.D. | WT SEZ6 | 148 | |||||||||||
| 261 | MUT SEZ6 | N.D. | WT SEZ6 | 875 | |||||||||||
| 263 | MUT SEZ6 | N.D. | WT SEZ6 | 381 | |||||||||||
| 267 | MUT SEZ6 | N.D. | WT SEZ6 | 442 | |||||||||||
| 270 | MUT SEZ6 | N.D. | WT SEZ6 | 368 | |||||||||||
| 275 | MUT SEZ6 | N.D. | WT SEZ6 | 317 | |||||||||||
| 276 | MUT SEZ6 | N.D. | WT SEZ6 | 368 | |||||||||||
| 277 | MUT SEZ6 | N.D. | WT SEZ6 | 186 | |||||||||||
| 278 | MUT SEZ6 | N.D. | WT SEZ6 | 63 | |||||||||||
| 279 | MUT SEZ6 | N.D. | WT SEZ6 | 234 | |||||||||||
| 287 | MUT SEZ6 | N.D. | WT SEZ6 | 99 | |||||||||||
| 293 | MUT SEZ6 | N.D. | WT SEZ6 | 125 | |||||||||||
| 324 | MUT SEZ6 | N.D. | WT SEZ6 | 605 | |||||||||||
| 325 | MUT SEZ6 | N.D. | WT SEZ6 | 153 | |||||||||||
| 326 | MUT SEZ6 | N.D. | WT SEZ6 | 692 | |||||||||||
| 327 | MUT SEZ6 | N.D. | WT SEZ6 | 713 | |||||||||||
| 328 | MUT SEZ6 | N.D. | WT SEZ6 | 391 | |||||||||||
| 332 | MUT SEZ6 | N.D. | WT SEZ6 | 759 | |||||||||||
| 333 | MUT SEZ6 | N.D. | WT SEZ6 | 661 | |||||||||||
| 337 | MUT SEZ6 | N.D. | WT SEZ6 | 798 | |||||||||||
| 338 | MUT SEZ6 | N.D. | WT SEZ6 | 903 | |||||||||||
| 340 | MUT SEZ6 | N.D. | WT SEZ6 | 40 | |||||||||||
| 341 | MUT SEZ6 | N.D. | WT SEZ6 | 274 | |||||||||||
| 342 | MUT SEZ6 | N.D. | WT SEZ6 | 240 | |||||||||||
| 344 | MUT SEZ6 | N.D. | WT SEZ6 | 209 | |||||||||||
| 345 | MUT SEZ6 | N.D. | WT SEZ6 | 873 | |||||||||||
| 348 | MUT SEZ6 | N.D. | WT SEZ6 | 2330 | |||||||||||
| 350 | MUT SEZ6 | N.D. | WT SEZ6 | 387 | |||||||||||
| 351 | MUT SEZ6 | N.D. | WT SEZ6 | 430 | |||||||||||
| 353 | MUT SEZ6 | N.D. | WT SEZ6 | 360 | |||||||||||
| 360 | MUT SEZ6 | N.D. | WT SEZ6 | 473 | |||||||||||
| 361 | MUT SEZ6 | N.D. | WT SEZ6 | 553 | |||||||||||
| 362 | MUT SEZ6 | N.D. | WT SEZ6 | 2470 | |||||||||||
| 363 | MUT SEZ6 | N.D. | WT SEZ6 | 889 | |||||||||||
| 366 | MUT SEZ6 | N.D. | WT SEZ6 | 1990 | |||||||||||
| 367 | MUT SEZ6 | N.D. | WT SEZ6 | 452 | |||||||||||
| 368 | MUT SEZ6 | N.D. | WT SEZ6 | 1736 | |||||||||||
| 369 | MUT SEZ6 | N.D. | WT SEZ6 | 1436 | |||||||||||
| 375 | MUT SEZ6 | N.D. | WT SEZ6 | 588 | |||||||||||
| 376 | MUT SEZ6 | N.D. | WT SEZ6 | 544 | |||||||||||
| 377 | MUT SEZ6 | N.D. | WT SEZ6 | 623 | |||||||||||
| 401 | MUT SEZ6 | N.D. | WT SEZ6 | 803 | |||||||||||
| 404 | MUT SEZ6 | N.D. | WT SEZ6 | 494 | |||||||||||
| 406 | MUT SEZ6 | N.D. | WT SEZ6 | 200 | |||||||||||
| 407 | MUT SEZ6 | N.D. | WT SEZ6 | 482 | |||||||||||
| 408 | MUT SEZ6 | N.D. | WT SEZ6 | 105 | |||||||||||
| 409 | MUT SEZ6 | N.D. | WT SEZ6 | 3260 | |||||||||||
| 418 | MUT SEZ6 | N.D. | WT SEZ6 | 190 | |||||||||||
| 422 | MUT SEZ6 | N.D. | WT SEZ6 | 1325 | |||||||||||
| 430 | MUT SEZ6 | N.D. | WT SEZ6 | 772 | |||||||||||
| 434 | MUT SEZ6 | N.D. | WT SEZ6 | 1233 | |||||||||||
| 435 | MUT SEZ6 | N.D. | WT SEZ6 | 1844 | |||||||||||
| 440 | MUT SEZ6 | N.D. | WT SEZ6 | 90 | |||||||||||
| 446 | MUT SEZ6 | N.D. | WT SEZ6 | 745 | |||||||||||
| 451 | MUT SEZ6 | N.D. | WT SEZ6 | 1366 | |||||||||||
| 453 | MUT SEZ6 | N.D. | WT SEZ6 | 1185 | |||||||||||
| 454 | MUT SEZ6 | N.D. | WT SEZ6 | 2950 | |||||||||||
| 466 | MUT SEZ6 | N.D. | WT SEZ6 | 329 | |||||||||||
| 468 | MUT SEZ6 | N.D. | WT SEZ6 | 681 | |||||||||||
| 493 | MUT SEZ6 | N.D. | WT SEZ6 | 80 | |||||||||||
| 497 | MUT SEZ6 | N.D. | WT SEZ6 | 154 | |||||||||||
| 499 | MUT SEZ6 | N.D. | WT SEZ6 | 128 | |||||||||||
| 501 | MUT SEZ6 | N.D. | WT SEZ6 | 1814 | |||||||||||
| 511 | MUT SEZ6 | N.D. | WT SEZ6 | 547 | |||||||||||
| 512 | MUT SEZ6 | N.D. | WT SEZ6 | 48.2 | |||||||||||
| 513 | MUT SEZ6 | N.D. | WT SEZ6 | 40.8 | |||||||||||
| 514 | MUT SEZ6 | N.D. | WT SEZ6 | 1019 | |||||||||||
| 519 | MUT SEZ6 | N.D. | WT SEZ6 | 1382 | |||||||||||
| 520 | MUT SEZ6 | N.D. | WT SEZ6 | 791 | |||||||||||
| 521 | MUT SEZ6 | N.D. | WT SEZ6 | 1858 | |||||||||||
| 522 | MUT SEZ6 | N.D. | WT SEZ6 | 2180 | |||||||||||
| 523 | MUT SEZ6 | N.D. | WT SEZ6 | 1849 | |||||||||||
| 531 | MUT SEZ6 | N.D. | WT SEZ6 | 2110 | |||||||||||
| 532 | MUT SEZ6 | N.D. | WT SEZ6 | 3030 | |||||||||||
| 535 | MUT SEZ6 | N.D. | WT SEZ6 | 1096 | |||||||||||
| 537 | MUT SEZ6 | N.D. | WT SEZ6 | 1941 | |||||||||||
| 538 | MUT SEZ6 | N.D. | WT SEZ6 | 78 | |||||||||||
| 539 | MUT SEZ6 | N.D. | WT SEZ6 | 917 | |||||||||||
| 542 | MUT SEZ6 | N.D. | WT SEZ6 | 1650 | |||||||||||
| 543 | MUT SEZ6 | N.D. | WT SEZ6 | 937 | |||||||||||
| 545 | MUT SEZ6 | N.D. | WT SEZ6 | 1423 | |||||||||||
| 546 | MUT SEZ6 | N.D. | WT SEZ6 | 818 | |||||||||||
| 549 | MUT SEZ6 | N.D. | WT SEZ6 | 1196 | |||||||||||
| 550 | MUT SEZ6 | N.D. | WT SEZ6 | 716 | |||||||||||
| 558 | MUT SEZ6 | N.D. | WT SEZ6 | 845 | |||||||||||
| 567 | MUT SEZ6 | N.D. | WT SEZ6 | 724 | |||||||||||
| 570 | MUT SEZ6 | N.D. | WT SEZ6 | 765 | |||||||||||
| 571 | MUT SEZ6 | N.D. | WT SEZ6 | 2290 | |||||||||||
| 574 | MUT SEZ6 | N.D. | WT SEZ6 | 790 | |||||||||||
| 575 | MUT SEZ6 | N.D. | WT SEZ6 | 1399 | |||||||||||
| 578 | MUT SEZ6 | N.D. | WT SEZ6 | 1293 | |||||||||||
| 580 | MUT SEZ6 | N.D. | WT SEZ6 | 947 | |||||||||||
ND Not detectable
Alzheimer’s disease cases are underlined. For each group, patient code, digital droplet PCR target, and the calculated concentration (copies/μl) are reported. For the last group, the ratio, defined as concentration mutant SEZ6/concentration wild-type SEZ6, is also reported
Aβ peptide generation in H4-SW cells
Three different H4-SW stable clonal lines (C3, C4, and C13) transfected with a pCDNA3.1 plasmid coding for SEZ6(R615H) mutant were selected, and the presence of the variant at DNA level was confirmed by allele-specific PCR and sequencing (data not shown). The effect of the R615H substitution on Aβ(1–42) and Aβ(1–40) production by H4-SW cells was assessed in conditioned media from cultured H4-SW(R615H) in comparison to H4-SW cells (untransfected or mock-transfected with an empty pCDNA3.1 vector) (Fig. 3a). The mean concentration of released Aβ(1–42), normalized to cell total protein content, was significantly lower in C4 and C13 than in controls, whereas for the C3 line, there was a trend in the same direction (p = 0.07). The Aβ(1–40) assay showed no differences (Fig. 3b).
Fig. 3.
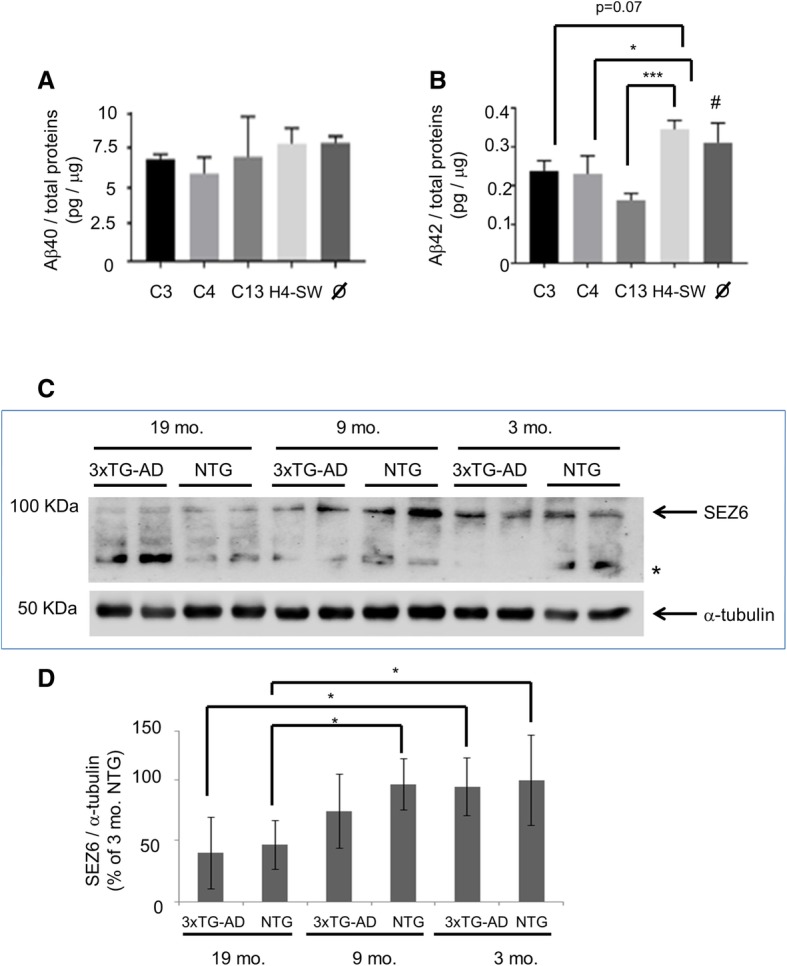
Evaluation of SEZ6 relevance for Alzheimer’s disease (AD) mechanisms in in vitro and in vivo models. a Quantification by enzyme-linked immunosorbent assay of soluble amyloid-β 1–40 (Aβ1–40) in conditioned media from H4-SW clonal lines (C3, C4, and C13) overexpressing SEZ6(R615H). The amyloid peptide concentration was normalized to the total protein content of the producing cells of each replicate. Measures are the mean ± SD of three independent wells. H4-SW Untransfected control; Ø H4-SW control transfected with pCDNA3.1 empty vector. b Same as in (a) except for the assessment of Aβ1–42 soluble form. * p < 0.05; *** p < 0.001, one-way analysis of variance (ANOVA) and post hoc test; # p < 0.05 vs. C4 and p < 0.01 vs. C13, one-way ANOVA and post hoc test. c Representative Western blotting for Sez6 protein detection in brain cortical extract from 3xTG-AD mice. Mice were killed at ages 3, 9, or 19 months, and Sez6 expression was assessed in transgenic and matched nontransgenic (NTG) animals. Each group was composed of three mice, and every animal was loaded in duplicate in the SDS-PAGE experiment. * Unspecific signal. d Densitometric quantification of all Western blot analysis data for Sez6 protein cortical expression (n = 3 mice/group) using ImageJ software. Each signal was normalized to the corresponding α-tubulin band to control for unequal protein loading. Results are expressed as a percentage of the youngest group (3 months) * p < 0.05, one-way ANOVA and post hoc test. mo. Months from birth
Sez6 brain expression in 3xTG-AD mice
Given that few experimental data linked SEZ6 to AD, we also examined murine Sez6 expression in a transgenic line model of AD (3xTG-AD), in comparison with age-matched nontransgenic controls (NTG) (Fig. 3c and d). Mice were killed at ages ranging from 3 to 19 months, and Sez6 protein expression was assessed at brain cortical level. Sez6 protein markedly decreased with age, particularly between 3 and 19 months. However, this reduction was common to both the 3xTG-AD and NTG lines and thus not unique to the AD model.
Discussion
Pathogenic mutations in APP, PSEN1, or PSEN2 genes are linked to FAD [3, 4]. PSEN1 mutations are responsible for about 60% of the genetic cases of AD, and 286 pathogenic variants have been described in the three above-cited genes [22]. We report an Italian family with AD that we previously screened by denaturing high-performance liquid chromatography (data not shown) for APP, PSEN1, or PSEN2 mutations with no results. Considering that rare variants in other genes have been associated with AD [7], we decided to perform targeted exome-sequencing analysis that yielded a large number of variants; in order to identify those closely related to the disease, we employed a recursive filtering strategy. This strategy was based on the removal of high-frequency (> 1%) variants using a public database (1000 Genomes Project) with in silico prediction software (SIFT, PolyPhen2, CADD) to exclude potentially harmless mutations and focus on variants present in FAD but not sporadic AD samples. We gave priority to the SEZ6(R615H) variant among those reported in Table 1, considering that SEZ6 has already been reported as relevant for molecular mechanisms involved in AD pathogenesis, because it is a substrate of the BACE-1 enzyme (β-secretase), affects synapse formation, and is reduced in the cerebrospinal fluid of patients with AD, as revealed by a proteomic study [23–25]. SEZ6 gene mutations have been also reported in association with febrile seizures, and SEZ6 was proposed as a candidate gene for epilepsy [26, 27]. Moreover, SEZ6 mutations were found in cases of childhood-onset schizophrenia [28]. The rare variant R615H (rs371753097, C/T) was reported in the 1000 Genomes Project as absent in Toscani in Italy (TSI) population and had a frequency in the whole project of 0.0002 [29]. Another interesting genetic variant we found by exome sequencing that is deserving of attention is A1131P in the KANK1 gene [30], which was present in the two AD cases (PR1 and PR5) and in PR3, sibling of PR1. However, PR3 did not have dementia at sampling (age 67 years), and her clinical state is currently unchanged, even though we are not able to exclude a possible later onset. The human KANK1 gene (alias ANKRD15) was originally described to be a tumor suppressor in renal cell carcinoma, and it encodes an ankyrin repeat domain-containing protein (Kank). It belongs to a family of four homologous members that have a role in actin stress fiber formation and renal pathophysiology [31, 32]. There is no reported interaction of KANK1 with SEZ6 or AD-related genes. However, a role of KANK1 mutation or deletion was reported in cerebral palsy spastic quadriplegic type 2, a central nervous system developmental disorder [33]. Moreover, to the best of our knowledge, no data associate KANK1 with AD.
In our study’s family, we were able to correlate the AD pathology to R615H presence, which was found in the two available AD-affected members and one first-degree relative of an AD case, whose age at sampling in 2003 (PR2, 37 years) was far below the family age of onset (range, 60–70 years) to expect clinical signs. The current clinical diagnosis of PR2 (51 years) is unchanged. We also confirmed that R615H frequency is very low (< 1%) in the Italian population, because we were unable to detect the variant in 200 family-unrelated subjects.
Because it is a common finding that AD pathogenic mutations increase Aβ(1–42) peptide generation [34], we examined the effect of the R615H variant in a cell model in this respect. In the H4-SW line, we noticed a decrease in Aβ(1–42), whereas Aβ(1–40) was unchanged. However, the increase of Aβ(1–42) in association with FAD-linked mutations is not always replicated. In fact, some presenilin mutants with proved pathologic action did not increase Aβ(1–42) but acted on other Aβ peptide generation or even had no impact on this proteolytic cleavage. In the latter case, the hypothesis is that the mutation affects important functions of presenilin other than the γ-secretase activity [35, 36]. It is worth underlining that we found a peculiar biochemical effect of the PSEN1 mutation E318G that increased Aβ(1–40) only in cultured skin primary fibroblasts [17]. Our failure to detect an increase of Aβ(1–42) might depend on the reported role of SEZ6 protein as substrate for BACE-1 [23], so its overexpression may be competitive for APP in the cell model tested. We need further experiments to clarify the role of the R615H variant in this context.
Finally, we followed SEZ6 cortical expression in a mouse model of AD (3xTG-AD). Considering that it changed similarly in 3xTG-AD and control mice, we were unable to link this result to AD-specific patterns, but we did notice a decrease of SEZ6 protein with age, in agreement with this gene’s reported role in brain development [37, 38]. A damaging mutation (as R615H is predicted to be) may have an impact on the protein activity from birth, with possible neuropathologic outcomes, likely in combination with other triggering factors, also considering the reported role of SEZ6 in dendritic spine dynamics and cognition [39].
This study has limitations, mainly linked to the unavailability of genomic DNA from all the family’s AD-affected members alive at sampling. Moreover, we decided to use a targeted exome-sequencing strategy that, on one hand, gave us clinical data supporting a rational choice of candidate variants to be prioritized, but on the other hand, prevented us from ruling out that additional coding mutations in genes not included in our panel may be linked to AD phenotype, thus acting in synergy with SEZ6 (R615H).
Conclusions
In summary, by using a targeted exome-sequencing approach, we discovered a rare SEZ6 variant exclusive to AD members of a large Italian family carrying no typical FAD-linked mutations that might have a role in disease onset, in particular taking into account the already described involvement of SEZ6 in AD pathogenic mechanisms linked to amyloid-β (A4) precursor protein (APP) and brain physiology, even though the exact molecular pathway linking SEZ6 to AD is still unclear.
Additional file
Table reporting the sequencing results of DNA from PR family members and unrelated sporadic AD cases, including only rare variants in the European population (frequency less than 1%) [low frequency page]. The same reults were further filtered to show variants with predicted damaging action [predicted damage page]. (XLSX 85 kb)
Acknowledgements
We are grateful to the family that kindly participated in this study. We thank Judith Baggott for English-language editing.
Funding
This work was supported by Fondazione Italo Monzino (Milan, Italy).
Availability of data and materials
Exome sequencing data files were uploaded to the European Nucleotide Archive (https://www.ebi.ac.uk/ena) with accession numbers pending.
Authors’ contributions
LP performed the digital droplet PCR assay and exome sequencing. LBe alanyzed genomic data and produced bioinformatics output. FF and LBo prepared the H4-SW clonal lines and measured SEZ6 gene expression in transgenic mice. PC recruited the families with sporadic Alzheimer’s disease and control subjects. DA, SM, and GF drafted the manuscript. All authors critically revised the manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to participate
All patients or their relatives gave informed consent for participation in this study, which was approved by the local ethics committee at Istituto di Neurologia, Università di Parma (PC), under the responsibility of Prof. Paolo Caffarra. Animal studies were run in compliance with national laws, regulations, and policies governing the care and use of laboratory animals: Italian Governing Law (D.lgs 26/2014; authorization no. 19/2008-A, issued March 6, 2008, by the Ministry of Health); Mario Negri institutional regulations and policies providing internal authorization for people conducting animal experiments (Quality Management System Certificate UNI EN ISO 9001:2008 registration no. 6121); the National Institutes of Health (NIH) Guide for the Care and Use of Laboratory Animals (2011 edition), and European Union directives and guidelines (EEC Council Directive 2010/63/UE). The Statement of Compliance (Assurance) with the Public Health Service (PHS) Policy on Human Care and Use of Laboratory Animals was recently reviewed (September 9, 2014) and will expire on September 30, 2019 (Animal Welfare Assurance no. A5023-01, DL. vo 116/1992, Gazzetta Ufficiale, Suppl. 40, Feb.18, 1992; Circolare nr. 8, Gazzetta Ufficiale, July 14, 1994) and international laws and policies (EEC Council Directive 86/609, OJL 358, 1, Dec.12, 1987; NIH Guide for the Care and Use of Laboratory Animals 8th edition, 2011).
Consent for publication
Not applicable, because this article does not contain any individual person’s data, images, or videos.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Lara Paracchini, Email: lara.paracchini@marionegri.it.
Luca Beltrame, Email: luca.beltrame@marionegri.it.
Lucia Boeri, Email: lucia.boeri@polimi.it.
Federica Fusco, Email: federica.fusco@marionegri.it.
Paolo Caffarra, Email: caffarra@unipr.it.
Sergio Marchini, Email: sergio.marchini@marionegri.it.
Diego Albani, Phone: +39 02 39014594, Email: diego.albani@marionegri.it.
Gianluigi Forloni, Email: gianluigi.forloni@marionegri.it.
References
- 1.Chandra V, Schoenberg BS. Inheritance of Alzheimer’s disease: epidemiologic evidence. Neuroepidemiology. 1989;8:165–174. doi: 10.1159/000110179. [DOI] [PubMed] [Google Scholar]
- 2.Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science. 1993;261:921–923. doi: 10.1126/science.8346443. [DOI] [PubMed] [Google Scholar]
- 3.Brunkan AL, Goate AM. Presenilin function and gamma-secretase activity. J Neurochem. 2005;93:769–792. doi: 10.1111/j.1471-4159.2005.03099.x. [DOI] [PubMed] [Google Scholar]
- 4.Bertram L, Tanzi RE. The current status of Alzheimer’s disease genetics: what do we tell the patients? Pharmacol Res. 2004;50:385–396. doi: 10.1016/j.phrs.2003.11.018. [DOI] [PubMed] [Google Scholar]
- 5.Sherrington R, Rogaev EI, Liang Y, Rogaeva EA, Levesque G, Ikeda M, et al. Cloning of a gene bearing missense mutations in early-onset familial Alzheimer’s disease. Nature. 1995;375:754–760. doi: 10.1038/375754a0. [DOI] [PubMed] [Google Scholar]
- 6.Goate A, Chartier-Harlin MC, Mullan M, Brown J, Crawford F, Fidani L, et al. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer’s disease. Nature. 1991;349:704–706. doi: 10.1038/349704a0. [DOI] [PubMed] [Google Scholar]
- 7.Sims R, van der Lee SJ, Naj AC, Bellenguez C, Badarinarayan N, Jakobsdottir J, et al. Rare coding variants in PLCG2, ABI3, and TREM2 implicate microglial-mediated innate immunity in Alzheimer’s disease. Nat Genet. 2017;49:1373–1384. doi: 10.1038/ng.3916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Thonberg H, Chiang HH, Lilius L, Forsell C, Lindström AK, Johansson C, et al. Identification and description of three families with familial Alzheimer disease that segregate variants in the SORL1 gene. Acta Neuropathol Commun. 2017;5:43. doi: 10.1186/s40478-017-0441-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer’s disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology. 1984;34:939–944. doi: 10.1212/WNL.34.7.939. [DOI] [PubMed] [Google Scholar]
- 10.Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010;26:589–595. doi: 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43:491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, Del Angel G, Levy-Moonshine A, et al. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr Protoc Bioinformatics. 2013;43:11.10.1–11.1033. doi: 10.1002/0471250953.bi1110s43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McLaren W, Gil L, Hunt SE, Riat HS, Ritchie GR, Thormann A, et al. The Ensembl Variant Effect Predictor. Genome Biol. 2016;17:122. doi: 10.1186/s13059-016-0974-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Paila U, Chapman BA, Kirchner R, Quinlan AR. GEMINI: integrative exploration of genetic variation and genome annotations. PLoS Comput Biol. 2013;9(7):e1003153. doi: 10.1371/journal.pcbi.1003153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu X, Jian X, Boerwinkle E. dbNSFP v2.0: a database of human non-synonymous SNVs and their functional predictions and annotations. Hum Mutat. 2013;34:E2393–E2402. doi: 10.1002/humu.22376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Albani D, Roiter I, Artuso V, Batelli S, Prato F, Pesaresi M, et al. Presenilin-1 mutation E318G and familial Alzheimer’s disease in the Italian population. Neurobiol Aging. 2007;28:1682–1688. doi: 10.1016/j.neurobiolaging.2006.07.003. [DOI] [PubMed] [Google Scholar]
- 18.Haugabook SJ, Yager DM, Eckman EA, Golde TE, Younkin SG, Eckman CB. High throughput screens for the identification of compounds that alter the accumulation of the Alzheimer’s amyloid β peptide (Aβ) J Neurosci Methods. 2001;108:171–179. doi: 10.1016/S0165-0270(01)00388-0. [DOI] [PubMed] [Google Scholar]
- 19.Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, et al. Triple-transgenic model of Alzheimer’s disease with plaques and tangles: intracellular Aβ and synaptic dysfunction. Neuron. 2003;39:409–421. doi: 10.1016/S0896-6273(03)00434-3. [DOI] [PubMed] [Google Scholar]
- 20.Shimizu-Nishikawa K, Kajiwara K, Sugaya E. Cloning and characterization of seizure-related gene, SEZ-6. Biochem Biophys Res Commun. 1995;216:382–389. doi: 10.1006/bbrc.1995.2635. [DOI] [PubMed] [Google Scholar]
- 21.Bork P, Beckmann G. The CUB domain: a widespread module in developmentally regulated proteins. J Mol Biol. 1993;231:539–545. doi: 10.1006/jmbi.1993.1305. [DOI] [PubMed] [Google Scholar]
- 22.Alzheimer Disease & Frontotemporal Dementia Mutation Database (AD&FTDMDB). http://www.molgen.ua.ac.be/ADmutations/. Accessed 21 May 2018.
- 23.Pigoni M, Wanngren J, Kuhn PH, Munro KM, Gunnersen JM, Takeshima H, et al. Seizure protein 6 and its homolog seizure 6-like protein are physiological substrates of BACE1 in neurons. Mol Neurodegener. 2016;11:67. doi: 10.1186/s13024-016-0134-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhu K, Xiang X, Filser S, Marinković P, Dorostkar MM, Crux S, et al. Β-site amyloid precursor protein cleaving enzyme 1 inhibition impairs synaptic plasticity via seizure protein 6. Biol Psychiatry. 2018;83:428–437. doi: 10.1016/j.biopsych.2016.12.023. [DOI] [PubMed] [Google Scholar]
- 25.Khoonsari PE, Häggmark A, Lönnberg M, Mikus M, Kilander L, Lannfelt L, et al. Analysis of the cerebrospinal fluid proteome in Alzheimer’s disease. PLoS One. 2016;11:e0150672. doi: 10.1371/journal.pone.0150672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yu ZL, Jiang JM, Wu DH, Xie HJ, Jiang JJ, Zhou L, et al. Febrile seizures are associated with mutation of seizure-related (SEZ) 6, a brain-specific gene. J Neurosci Res. 2007;85:166–172. doi: 10.1002/jnr.21103. [DOI] [PubMed] [Google Scholar]
- 27.Mulley JC, Iona X, Hodgson B, Heron SE, Berkovic SF, Scheffer IE, et al. The role of seizure-related SEZ6 as a susceptibility gene in febrile seizures. Neurol Res Int. 2011;2011:917565. doi: 10.1155/2011/917565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ambalavanan A, Girard SL, Ahn K, Zhou S, Dionne-Laporte A, Spiegelman D, et al. De novo variants in sporadic cases of childhood onset schizophrenia. Eur J Hum Genet. 2016;24:944–948. doi: 10.1038/ejhg.2015.218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.1000 Genomes Browser. https://www.ncbi.nlm.nih.gov/variation/tools/1000genomes/. Accessed 18 July 2017.
- 30.Sarkar S, Roy BC, Hatano N, Aoyagi T, Gohji K, Kiyama R. A novel ankyrin repeat-containing gene (Kank) located at 9p24 is a growth suppressor of renal cell carcinoma. J Biol Chem. 2002;277:36585–36591. doi: 10.1074/jbc.M204244200. [DOI] [PubMed] [Google Scholar]
- 31.Zhu Y, Kakinuma N, Wang Y, Kiyama R. Kank proteins: a new family of ankyrin-repeat domain-containing proteins. Biochim Biophys Acta. 2008;1780:128–133. doi: 10.1016/j.bbagen.2007.09.017. [DOI] [PubMed] [Google Scholar]
- 32.Gee HY, Zhang F, Ashraf S, Kohl S, Sadowski CE, Vega-Warner V, et al. KANK deficiency leads to podocyte dysfunction and nephrotic syndrome. J Clin Invest. 2015;125:2375–2384. doi: 10.1172/JCI79504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lerer I, Sagi M, Meiner V, Cohen T, Zlotogora J, Abeliovich D. Deletion of the ANKRD15 gene at 9p24.3 causes parent-of-origin-dependent inheritance of familial cerebral palsy. Hum Mol Genet. 2005;14:3911–3920. doi: 10.1093/hmg/ddi415. [DOI] [PubMed] [Google Scholar]
- 34.Suzuki N, Cheung TT, Cai XD, Odaka A, Otvos L, Jr, Eckman C, et al. An increased percentage of long amyloid β protein secreted by familial amyloid β protein precursor (β APP717) mutants. Science. 1994;264:1336–1340. doi: 10.1126/science.8191290. [DOI] [PubMed] [Google Scholar]
- 35.Bentahir M, Nyabi O, Verhamme J, Tolia A, Horré K, Wiltfang J, et al. Presenilin clinical mutations can affect gamma-secretase activity by different mechanisms. J Neurochem. 2006;96:732–742. doi: 10.1111/j.1471-4159.2005.03578.x. [DOI] [PubMed] [Google Scholar]
- 36.Szaruga M, Veugelen S, Benurwar M, Lismont S, Sepulveda-Falla D, Lleo A, et al. Qualitative changes in human γ-secretase underlie familial Alzheimer’s disease. J Exp Med. 2015;212:2003–2013. doi: 10.1084/jem.20150892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kim MH, Gunnersen JM, Tan SS. Localized expression of the seizure-related gene SEZ-6 in developing and adult forebrains. Mech Dev. 2002;118:171–174. doi: 10.1016/S0925-4773(02)00238-1. [DOI] [PubMed] [Google Scholar]
- 38.Osaki G, Mitsui S, Yuri K. The distribution of the seizure-related gene 6 (Sez-6) protein during postnatal development of the mouse forebrain suggests multiple functions for this protein: an analysis using a new antibody. Brain Res. 2011;1386:58–69. doi: 10.1016/j.brainres.2011.02.025. [DOI] [PubMed] [Google Scholar]
- 39.Gunnersen JM, Kim MH, Fuller SJ, De Silva M, Britto JM, Hammond VE, et al. Sez-6 proteins affect dendritic arborization patterns and excitability of cortical pyramidal neurons. Neuron. 2007;56:621–639. doi: 10.1016/j.neuron.2007.09.018. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table reporting the sequencing results of DNA from PR family members and unrelated sporadic AD cases, including only rare variants in the European population (frequency less than 1%) [low frequency page]. The same reults were further filtered to show variants with predicted damaging action [predicted damage page]. (XLSX 85 kb)
Data Availability Statement
Exome sequencing data files were uploaded to the European Nucleotide Archive (https://www.ebi.ac.uk/ena) with accession numbers pending.