

Abstract
Background
A decision tree model was built to estimate the economic impact of introducing screening for X-linked adrenoleukodystrophy (X-ALD) into an existing tandem mass spectrometry based newborn screening programme. The model was based upon the UK National Health Service (NHS) Newborn Blood Spot Screening Programme and a public service perspective was used with a lifetime horizon. The model structure and parameterisation were based upon literature reviews and expert clinical judgment. Outcomes included health, social care and education costs and quality adjusted life years (QALYs). The model assessed screening of boys only and evaluated the impact of improved outcomes from hematopoietic stem cell transplantation in patients with cerebral childhood X-ALD (CCALD). Threshold analyses were used to examine the potential impact of utility decrements for non-CCALD patients identified by screening.
Results
It is estimated that screening 780,000 newborns annually will identify 18 (95%CI 12, 27) boys with X-ALD, of whom 10 (95% CI 6, 15) will develop CCALD. It is estimated that screening may detect 7 (95% CI 3, 12) children with other peroxisomal disorders who may also have arisen symptomatically. If results for girls are returned an additional 17 (95% CI 12, 25) cases of X-ALD will be identified. The programme is estimated to cost an additional £402,000 (95% CI £399–407,000) with savings in lifetime health, social care and education costs leading to an overall discounted cost saving of £3.04 (95% CI £5.69, £1.19) million per year. Patients with CCALD are estimated to gain 8.5 discounted QALYs each giving an overall programme benefit of 82 (95% CI 43, 139) QALYs.
Conclusion
Including screening of boys for X-ALD into an existing tandem mass spectrometry based newborn screening programme is projected to reduce lifetime costs and improve outcomes for those with CCALD. The potential disbenefit to those identified with non-CCALD conditions would need to be substantial in order to outweigh the benefit to those with CCALD. Further evidence is required on the potential QALY impact of early diagnosis both for non-CCALD X-ALD and other peroxisomal disorders. The favourable economic results are driven by estimated reductions in the social care and education costs.
Electronic supplementary material
The online version of this article (10.1186/s13023-018-0921-4) contains supplementary material, which is available to authorized users.
Keywords: Adrenoleukodystrophy, Neonatal screening, Newborn screening, Cost-benefit analysis, Cost-effectiveness analysis, Economic analysis, Hematopoietic stem cell transplantation, Decision trees
Background
X-linked adrenoleukodystrophy (X-ALD) is a rare genetic disorder caused by a defect in the ABCD1 gene. The disorder interrupts peroxisomal fatty acid beta oxidation resulting in the accumulation of very long chain fatty acids with consequent damage to tissue throughout the body and brain. X-ALD demonstrates X-linked recessive inheritance, with reports of incidence varying between 0.8 and 4.76 people affected per 100,000 births [1–3].
Males with X-ALD can present with adrenal insufficiency, the cerebral form of X-ALD, or progressive myelopathy (adrenomyeloneuropathy (AMN)). The majority of men with X-ALD will go on to develop AMN. Most women with X-ALD will also develop symptomatic AMN over their lifetime, but women do not appear to be affected by cerebral deterioration or adrenal insufficiency [4–7]. Cerebral X-ALD is the most severe phenotype and without treatment patients may experience neurodegenerative decline leading to a vegetative state and death. Studies have shown that haematopoietic stem cell transplantation (HSCT) and more recently gene therapy can be successful in preventing long term deterioration in patients with cerebral X-ALD presenting in childhood or adolescence (CCALD) [8–11]. However, this benefit is dependent on being transplanted at the first signs of neurological development with little or no benefit to patients transplanted after this point [9–11]. There is also emerging evidence of similar benefits in early transplanted adults [12].
Patients currently undergoing HSCT before the onset of significant neurological symptoms, referred to as early HSCT are most frequently identified due to family history or through presenting with adrenal insufficiency. While screening of the extended family of X-ALD patients is currently offered it is estimated that between 5 and 18% of new patients present with a spontaneous mutation [13, 14]. A population screening approach using existing newborn blood spots and high throughput tandem mass spectrometry has been shown to accurately diagnose affected individuals and to have a high sensitivity and specificity [13, 15]. The aim of screening is to identify patients before they become symptomatic to enable them to be monitored for the initial signs of CCALD and transplanted at an optimal time. This method of screening has been in use in New York State since 2014 and has been found to identify patients with other peroxisomal disorders as well as X-ALD [16, 17].
Screening programmes use criteria, often based on the Wilson and Jungner criteria, to decide which conditions should be screened for. These criteria often incorporate an economic component, for example, the Wilson and Jungner criteria state that the cost of case-finding, including diagnosis and treatment of all patients diagnosed should be economically balanced in relation to possible expenditure on medical care as a whole [18]. In the United Kingdom (UK), the National Screening Committee (NSC) criteria describe this economic requirement in terms of the potential cost-effectiveness of a screening technology [19]. The aim of this study was to address this criterion and estimate the potential cost-effectiveness of including screening for X-ALD in the UK National Health Service (NHS) Newborn Blood Spot Screening Programme.
Methods
A decision analytic model was built to estimate the economic impact of screening for X-ALD in the NHS Newborn Blood Spot Screening Programme. The model took an NHS and personal social services (PSS) perspective, included a lifetime horizon and discounted costs and benefits at 3.5% [20]. The model estimated life years and quality adjusted life years (QALYs) gained, health, social care and special education costs. Costs were estimated for 2014/15 with uplifting according to hospital and community health services indices [21]. The incremental cost effectiveness ratio (ICER) is defined as the cost per QALY gained. The model used a decision tree structure, shown in Fig. 1 to estimate the number of newborns that might be identified with a positive screening result, the distribution across the peroxisomal disorders and the number developing CCALD and undergoing HSCT with and without screening. It was assumed that the outcomes of early HSCT in patients identified through screening would be equivalent to outcomes in those identified early without screening. Lifetime costs and QALYs for the different outcomes with and without screening were estimated using lifetables. The annual number of births for the UK was estimated based on a 10 year average [22–24]. Patients identified with other peroxisomal disorders were assumed to incur incremental costs of screening and confirmatory diagnosis, but no health benefits or disbenefits were associated with early diagnosis from screening. Details of all model parameters are given in Table 1 and further details on the model distribution used in the probabilistic sensitivity analysis are given in Additional file 1.
Fig. 1.

X-ALD Screening Decision Tree
Table 1.
Parameters Table
| Parameter | Mean (95% Confidence Interval) | Reference |
|---|---|---|
| Base case parameters | ||
| Number of births per year | 400,308 | [22–24] |
| X-ALD incidence | 1 in 22,361 (15,083, 33,153) | [3, 29–32] |
| Proportion CCALD, AMN, Addison’s/Asymptomatic |
0.53 (0.36, 0.69), 0.32 (0.18, 0.49), 0.15 (0.05, 0.28) | [14] |
| Non-X-ALD peroxisomal incidence | 1 in 63,000 (33,897, 117,090) | [32] |
| Age at presentation CCALD | 7 (6.76, 7.24) | [11] |
| Survival from onset CCALD Weibull distribution - shape parameter - scale parameter - correlation |
ᅟ ᅟ −2.970 0.162 −0.8994 |
[11] |
| Time to CCALD progression (years) | 1.6 (1.34, 1.86) | [11] |
| Mortality risk HSCT | 0.08 (0.01, 0.21) | [10] |
| Proportion of CCALD currently undergoing early transplant (Family history) | 0.33 (0.23, 0.43) | [10] |
| Proportion ALD-DRS 0, ALD-DRS1, ALD-DRS2, ALD-DRS 3–4, after HSCT | 0.62 (0.35, 0.85), 0.23 (0.05, 0.48), 0.08 (0.002, 0.26), 0.08 (0.002, 0.26) | [10] |
| Proportion successful HSCT develop AMN | 0.6 (0.19, 0.93) | [65] |
| Sensitivity | 0.995 | |
| Specificity | 1 | |
| Proportion of AMN mild | 0.51 (0.38, 0.64) | [44] |
| Proportion of AMN developing adult onset cerebral X-ALD | 0.63 (0.44, 0.8) | [40] |
| Age at presentation AMN (years) | 35.3 (26.7, 43.9) | [40] |
| Time to development of adult onset cerebral X-ALD (years) | 10.2 (3.3, 17.1) | [40] |
| Survival adult onset cerebral X-ALD (years) | 3.4 (0.5, 6.3) | [40] |
| QALYs | See Additional file 2 | |
| Costs | See Additional file 3 | |
A comprehensive, systematic search of bibliographic databases was conducted to identify literature on X-ALD to inform model structure and parameterisation. Information requirements were defined prospectively, however data searching and data extraction remained dynamic in order to reflect additional information needs identified during model development [25]. Full details of evidence searches and reviews are provided in the Additional file 2.
The NHS Newborn Blood Spot Screening Programme already uses tandem mass spectrometry hence the incremental cost of including testing for X-ALD is small and was estimated at £0.50 per baby based on a previous economic evaluation [26]. The review identified four studies that reported the sensitivity and specificity of newborn blood spot tandem mass spectrometry for X-ALD. All studies showed either 100% sensitivity or 100% specificity or both [13, 15, 27, 28]. However, as false negatives are likely in a population based screening programme we assumed a sensitivity of 99.5%.
Incidence of X-ALD from four studies identified in the review and an additional study identified after the review that included data on incidence from the New York screening programme were synthesised using a random effects model in WinBugs [3, 29–32]. Studies were included that gave both the number of cases and relevant population figure, retrospective studies that included both sexes fatty acid measurements were excluded due to historic underreporting of X-ALD in women and studies that included cases from before the adoption of very long-chain fatty acid measurements were also excluded as they are likely to only report a minimum estimates [33]. The Moser et al. study [32] was included as it provided an important estimate of the incidence of X-ALD once a screening programme has been implemented of both X-ALD and the other peroxisomal conditions that are identified. The incidence values from the five studies [3, 29–32] and the synthesised incidence of 1 in 22,000 (95%Confidence Interval (CI) 1 in 33,000, 1 in 15,000) used in the model are shown in a forest plot in Fig. 2. There is no direct evidence concerning the incidence of the other peroxisomal conditions that might be identified by screening in the UK, we therefore used the incidence of 1 in 63,000 (95% CI 1 in 117,000, 1 in 34,000) from the New York Screening Programme [32].
Fig. 2.

Forest Plot of Incidence Values. The black horizontal line represents the mean incidence and the black vertical line the 95% confidence interval
The review identified 10 studies that estimated the phenotype breakdown of X-ALD [2, 14, 29, 31, 34–39]. The studies were heterogeneous and differed in terms of how the phenotypes were classified. Due to this no attempt was made to synthesise evidence, rather the Horn et al. [14] study of X-ALD in Norway was chosen on the basis of study quality and relative generalisability to the UK population. This assumption was tested in a sensitivity analysis. The study of AMN by de Beer et al. [40] was used to estimate the proportion of AMN patients that go on to develop cerebral involvement.
The review identified 26 studies on either the natural history or outcome following transplantation [6, 9–11, 35, 40–62]. For patients with CCALD, there is no direct comparative evidence on survival with and without HSCT. Survival and time to progression without transplantation were estimated from the Mahmood et al. study [11], selected from the 26 studies [6, 9–11, 35, 40–62] as it presented data on a large cohort, 283 patients, with follow-up of up to 30 years. The model assumed that CCALD patients start off with mild to moderate disease before progressing to severe CCALD indicated by developing two or more neurological deficits with a mean time to progression of 1.6 years. The mean survival in these patients of 19 years (16.8–21) was estimated using simulated patient level data from the Mahmood study [11, 63] with parametric extrapolation of long term survival according to the methods recommended by the National Institute for Health and Care Excellence (NICE) Decision Support Unit [64].
For those undergoing HSCT the Peters et al. [10] study was selected from the 26 studies identified [6, 9–11, 35, 40–62] as it provided detailed outcomes following HSCT, including the ALD-Disability rating scale (ALD-DRS) and provided survival and outcomes data based on the severity of the disease at transplant. The model assumed that only patients with a Loes score of < 10 undergo HSCT and that those who survive and have a good outcome following HSCT have a normal life expectancy as no deaths occurred post 1.5 years for those undergoing early HSCT [9, 10]. The proportion of patients identified through a family history without screening in the Peters et al. [10] study was used to estimate the number of those diagnosed early enough to undergo HSCT. In the screen arm it was assumed that all CCALD patients undergo HSCT. This assumption was explored in a sensitivity analysis.
Estimates of patient outcomes following HSCT were based on their functional level assessed by the ALD-DRS before and after HSCT [10]. The outcomes of patients with an ALD-DRS rating of 0 before HSCT were used to model outcomes following successful HSCT [10]. Yearly costs and QALYs were applied to the different disability levels to estimate long term outcomes. A normal life expectancy was assumed for those with an outcome of ALD-DRS level 0–2 and life expectancy of non-transplanted CCALD for those with an outcome of ALD-DRS of 3–4.
HSCT does not prevent myelopathy in adulthood. In a small study of five patients who had undergone successful HSCT for CCALD [65], age range 18–25, three patients developed myelopathy. This rate of development of AMN type symptoms was applied to those who had undergone transplantation and whose outcomes were either ALD-DRS 0 or 1. A lower age of symptom onset (20) was used for these patients in line with the study. Additional costs and QALY decrements were incurred in line with AMN patients.
The model assumed normal survival for AMN and progression per se is not modelled but patients are split between a mild or moderate/severe form from the onset of the disease. The model parameters were based on a study with a cohort of 60 men that provided expanded disability status scale (EDSS) used to estimate quality of life and costs [44]. A second study by de Beer et al. [40] was used to estimate a number of parameters for AMN and adult onset cerebral X-ALD that were not provided in the Keller et al. study [44] (see Table 1). The model assumed that the same proportion of both the mild and moderate/severe AMN patients go on to develop adult onset cerebral X-ALD.
Asymptomatic and Addison’s only cases were assumed to have normal life expectancy and morbidity. It is also assumed that the Addison’s/Asymptomatic cases are monitored from birth in both arms but that all AMN cases are diagnosed symptomatically in the no-screen arm.
No studies were identified providing direct evidence on quality of life utilities for X-ALD patients; furthermore no suitable proxy condition was identified for CCALD. Age specific general UK population QALYs were used for pre-symptomatic patients, those with Addison’s only, and for those with ALD-DRS of 0 following transplant [66]. Based on the description of the ALD-DRS each state was assigned an equivalent EuroQol Five Dimension Five Level (EQ-5D-5 L) health state, shown in Additional file 3 [67]. QALYs for mild/moderate CCALD were calculated as the average of ALD-DRS1 and 2 and the QALYs for moderate/severe CCALD were calculated as the average of ALD-DRS 3 and 4.
For patients with AMN and women with X-ALD multiple sclerosis (MS) was used as a proxy as the Expanded Disability Status Scale (EDSS) has been used in both patient populations [40, 44]. The difference between the mean value for the EDSS state 3 (mild AMN) and EDSS state 6.5 (moderate/severe AMN) and the general population norms were calculated and proportionate differences were then applied to the age specific general population norms to give the utility decrements for each age group [66, 68]. Further detail on the calculations of the QALYs is provided in Additional file 3.
The costs of monitoring, diagnosis and a yearly cost of management were estimated for each phenotype. This was an iterative process that involved developing a resource use profile based on published guidelines and guidance for patients and families with X-ALD [5, 16]. The resource use profiles were sent out for consultation by ALD Life. The feedback from this process was used to create final resource use descriptions presented in Additional file 4 and were costed using appropriate sources [21, 69–71].
The cost of diagnosis in symptomatic patients included GP and specialist appointments associated with the increased diagnostic journey over and above the standard tests and consultations for all diagnosed patients as outlined in the Additional file 4. Untreated CCALD results in substantial disability with consequent high social care costs and education costs. In the experiences of the users of ALD Life all CCALD patients require education support often at the 1:1 or 2:1 level. Special education costs have been estimated based on the costs of education of children with autism who attended, in line with patients with CCALD, mainstream, special, and residential schools. The uplifted yearly cost is around £24,000 [72]. Social and education costs are not separated in the model as care packages can include funding for special education and social care. Social care costs included respite care and equipment but not the costs of home adaptation. For those over 18 years just the social care package and no education costs were included. Non-medical costs such as aids and home help and transportations for AMN and women with X-ALD were taken from a study on MS which provided costs for EDSS state 2 and EDSS 6.5 which were used for mild AMN and moderate/severe AMN respectively. The costs for EDSS state 2 were also used for the women with X-ALD in the corresponding age group of 40+ [6, 44, 68, 73]. Details of the costs are provided in Additional file 4.
Input parameters were characterised probabilistically, see Additional file 1, and uncertainty was propagated with Monte Carlo sampling with 100,000 replicates in the base case and 10,000 replicates in the sensitivity analyses. A number of one-way sensitivity analyses were undertaken to explore the impact of assumptions and structural uncertainties in the model.
The proportion of patients who develop CCALD may be overestimated as they are the cases most classically associated with X-ALD and there were differences between the identified studies [2, 14, 29, 31, 34–39]. The proportion of X-ALD patients that develop CCALD was decreased to 10%, 15%, and 20%. In each case it was estimated, based on the base case inputs, that 69% of non-CCALD X-ALD was AMN, and 31% was Addison’s only/Asymptomatic.
The proportion of CCALD patients in the screen arm who undergo HSCT was reduced from 100 to 60%
Scenario analyses were conducted that varied both the proportion of patients who developed CCALD and the proportion of CCALD patients in the screen arm who undergo HSCT
Both sexes were screened for. It is assumed based on data from the New York Screening Programme that the incidence in females is the same as that in males [7, 32]. For this sensitivity analysis the study by Engelen et al. [6] was used to model progression of the disease in women. It was assumed that the disease progressed with age and that all women would become affected. EDSS scores were converted into EQ-5D quality of life scores and the average score for three age groups 18–39, 40–59, and 60+ years were used in the model [6, 68, 73].
NICE specifies that a lower discount rate of 1.5% can be used for public health interventions [20]. A sensitivity analysis using the lower discount rate was also conducted.
As there is some uncertainty as to the incidence rate in the UK the incidence rate was doubled and halved in order to explore the impact on the results.
Patients or parents of patients with non-CCALD X-ALD and those with other non-X-ALD disorders may experience a disbenefit from a positive screen results. For those with non-CCALD they or their parents may experience anxiety about the potential for developing CCLAD or developing the non-treatable AMN. Patients and parents of those with who test positive for other non X-ALD peroxisomal disorders may also experience anxiety or distress from being diagnosed before they become symptomatic. It is unclear how these disbenefits would present and if they would be limited to anxiety or if they would present in other behavioural changes. In order to explore and try to quantify this uncertainty an exploratory threshold analysis was undertaken that explored the maximum disbenefit, expressed in QALYs, per patient per year that non-CCALD identified patients would need to experience in order to cancel out the benefits that accrue to CCALD patients due to screening. The number of cases is multiplied by the length of time they would be expected to be asymptomatic. For non CCALD X-ALD this is assumed to be the age of onset of AMN (35 years), for the non-X-ALD cases we have assumed that is will be 5 years, and for woman with X-ALD we have assumed that is will be 50 years.
Results
The results of including screening for X-ALD in the NHS Newborn Blood Spot Screening Programme are presented in Tables 2 and 3. It is estimated that screening an annual UK birth cohort of approximately 780,000 newborns would identify 18 (95%CI 12, 27) males with X-ALD. It is expected that 6 (95% CI 3, 10) of these newborns will develop AMN, with 10 (95% CI 6, 15) progressing to CCALD, and approximately 3 (95% CI 0.9, 6) having Addison’s only or being asymptomatic. The model also estimates that screening will detect 7 (95% CI 3, 12) cases of other peroxisomal disorders each year. If girls are also screened it will result in an additional 17 (95% CI 12, 25) cases of X-ALD and around 13 (95%CI 7, 23) cases of other peroxisomal disorders in total.
Table 2.
Model estimated number of X-ALD and CCALD cases per year
| Sensitivity Analyses | Number of cases | |||
|---|---|---|---|---|
| X-ALD Cases | 95% Confidence Interval | CCALD Cases | 95% Confidence Interval | |
| Base case | 18.3 | (12.1,26.6) | 9.7 | (5.5,15.3) |
| Incidence rate doubled | 36.5 | (24.,53.2) | 19.3 | (11.1,31.0) |
| Incidence rate halved | 9.1 | (6.,13.2) | 4.8 | (2.7,7.6) |
| CCALD 20% of total X-ALD | – | – | 3.6 | (2.4,5.3) |
| CCALD 15% of total X-ALD | – | – | 2.7 | (1.8,4.) |
| CCALD 10% of total X-ALD | – | – | 1.8 | (1.2,2.7) |
| Both girls and boys screened | 35.6 | (23.7,51.9) | – | – |
– Number of cases are the same as the base case
Table 3.
Cost-effectiveness results
| Sensitivity analyses | Screening | No screening | Incremental | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Total Costs (m) | Total QALYs | Total Costs (m) | Total QALYs | Costs (m) | 95% Confidence Interval (m) | QALYs | 95% Confidence Interval | ICER | |
| Base case | £3.01 | 390 | £6.44 | 307 | -£3.04 | (−£5.69, −£1.19) | 82 | (43, 139) | Dominates |
| Incidence rate doubled | £5.97 | 778 | £12.88 | 614 | -£6.50 | (−£11.74, −£2.80) | 164 | (86, 277) | Dominates |
| Incidence rate halved | £1.51 | 194 | £3.21 | 153 | -£1.30 | (−£2.62, −£0.39) | 41 | (22, 69) | Dominates |
| CCALD 10% of total X-ALD | £2.02 | 412 | £2.21 | 397 | £0.21 | (−£0.06, £0.46) | 16 | (9, 24) | £13,600 |
| CCALD 15% of total X-ALD | £2.13 | 409 | £2.70 | 386 | -£0.17 | (−£0.63, £0.22) | 23 | (14, 36) | Dominates |
| CCALD 60% HSCT rate | £5.20 | 340 | £6.41 | 307 | -£0.81 | (−£2.01, £0.02) | 33 | (15, 60) | Dominates |
| CCALD 10% of total X-ALD and 80% HSCT rate | £2.23 | 407 | £2.21 | 396 | £0.42 | (£0.24, £0.62) | 11 | (6, 17) | £38,701 |
| CCALD 15% of total X-ALD and 80% HSCT rate | £2.43 | 401 | £2.69 | 385 | £0.15 | (−£0.18, £0.42) | 16 | (9, 26) | £8927 |
| CCALD 20% of total X-ALD and 80% HSCT rate | £2.65 | 396 | £3.18 | 375 | -£0.12 | (−£0.59, £0.24) | 22 | (13, 34) | Dominates |
| 1.5% Discount Rate | £4.59 | 611 | £9.35 | 455 | -£4.36 | (−£7.97, −£1.81) | 156 | (83, 260) | Dominates |
| Both girls and boys screened | £3.27 | 800 | £6.96 | 718 | -£3.27 | (−£5.97, −£1.36) | 82 | (43, 139) | Dominates |
m million
Adding X-ALD to the screening programme as a whole results in an increase in total discounted QALYs and life years per year of 82 (95% CI 43, 139) and 79 (95% CI 42, 131) respectively. The increase in QALYs and life years is due to improvements in the outcomes of patients with CCALD only who on average have a gain of 8.5 QALYS per CCALD patient and a life year gain of just over 8 years per CCALD patient.
The screening programme is estimated to cost an additional £402,000 (95% CI £399–407,000) per year with discounted marginal lifetime health and social care / education costs of £256,000 (95% CI £12,000, £527,000) and -£3.69 (95% CI -£6.27, −£1.92) million respectively, leading to an overall discounted cost saving of £3.04 (95% CI £5.69, £1.19) million per year of screening.
As screening is estimated to result in more QALYs and fewer costs, screening is said to dominate no screening. The results of the probabilistic sensitivity analysis are shown in the cost-effectiveness plane in Fig. 3. In the base case screening dominates no screening. This means that adding X-ALD screening into the existing screening programme results in additional QALYs gained and lower total discounted costs.. Across the sensitivity analyses screening dominated no screening except when the proportion of X-ALD that developed CCALD was reduced to 10%. The ICER went above the threshold of £30,000 per QALY when both the proportion of X-ALD developing CCALD was reduced to 10% and the proportion of CCALD patients receiving an HSCT was reduced to 80%.
Fig. 3.
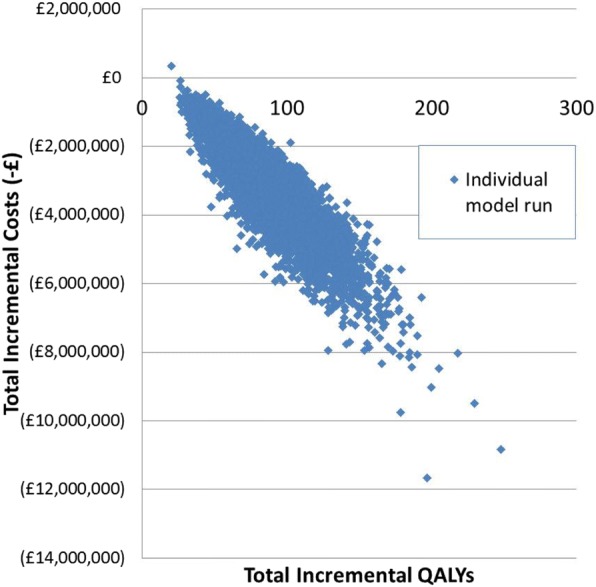
Cost-Effectiveness Plane. Each of the blue diamonds represents one of the 100,000 model runs. Costs in () represent model runs where screening is estimated to be cost saving
The results of the disbenefit analysis are shown in Table 4. Results are shown for both undiscounted QALYs and a discounted analysis which used the total discounted incremental QALYs and discounted age at symptomatic presentation. In the male babies only screened analysis the disbenefit to the non CCALD patients would have to be a relatively substantial 0.84 or 0.40 per case per year in the undiscounted or discounted analysis respectively in order to wipe out the benefit CCALD patients derive from screening. If both sexes were screened the disbenefit to the non CCALD patients that would wipe out the benefit to the CCALD patients would need to be 0.13 in the discounted analysis.
Table 4.
Results of the Disbenefit Analysis
| Male babies only screened | Both sexes screened | |||
|---|---|---|---|---|
| Undiscounted | Discounted | Undiscounted | Discounted | |
| Non CCALD X-ALD | 8.61 | 8.61 | 8.61 | 8.61 |
| Age of symptomatic presentation | 35.00 | 20.55 | 35.00 | 20.55 |
| Person years without diagnosis | 301.34 | 176.95 | 301.34 | 176.95 |
| Non X-ALD disorders | 6.68 | 6.68 | 13.04 | 13.04 |
| Age of symptomatic presentation | 5.00 | 4.66 | 5.00 | 4.66 |
| Person years without diagnosis | 33.39 | 31.11 | 65.21 | 60.76 |
| Number of cases of X-ALD in females | – | – | 17.36 | 17.36 |
| Age of symptomatic presentation | – | – | 50.00 | 24.11 |
| Person years without diagnosis | – | – | 867.96 | 418.60 |
| Total person years without diagnosis | 334.73 | 208.06 | 1234.52 | 656.32 |
| Total incremental QALYs from screening | 280.15 | 82.42 | 279.78 | 82.30 |
| Maximum QALY decrement per non CCALD case per year | 0.84 | 0.40 | 0.23 | 0.13 |
– No cases in females when only male babies are screened
Discussion
This study has attempted to address whether screening for X-ALD is cost-effective and so fulfils the economic criterion for a screening programme. The study suggests that screening for X-ALD is cost saving, however problems with measurement and valuation of some of the key benefits and harms of screening mean that it is difficult to capture the full scope of these within an economic model. These benefits and harms also relate to other screening criteria, in particular the evidence of treatment benefit and the evidence on benefits and harms from over diagnosis, overtreatment and uncertain findings [19].
Evidence for treatment benefit for CCALD in screening comes from small observational studies in a non-screening setting. Long term evidence on morbidity is difficult to interpret because of the range of different outcome measures that have been used, together with little evidence on quality of life utilities in both transplanted and non-transplanted patients [9–11]. The approach taken here of mapping the ALD-DRS onto the EQ-5D-5 L is an imperfect solution to the quality of life issue but due to methodological and practical issues with valuing health states in children and in those with cognitive disabilities no suitable proxy conditions were found [74–76]. The QALY estimates produced are in line with recent studies have shown that neurological quality of life outcomes are similar in early transplanted CCALD patients to the general population [77, 78]. There is also a lack of evidence concerning the impact of early X-ALD diagnosis on patients who might go on to develop Addison’s disease.
The evidence demonstrates that transplanting CCALD patients at the first signs of cerebral involvement offers the best outcomes in terms of both survival and morbidity [9–11]. However, there currently exists no way of identifying which X-ALD patients will go on to develop cerebral involvement in childhood [4]. The need to intervene before there are significant symptoms and the lack of other treatment options brings with it the potential for over or under-treatment if there is variation in the implementation of clinical guidelines [79]. This could also be exacerbated if CCALD patients or families do not follow monitoring protocols or do not consent to transplantation, a procedure with potentially severe complications.
The quality of life and behavioural impacts of receiving an early diagnosis of X-ALD particularly for those who do not develop CCALD are also not well understood or valued. This is also the case for those diagnosed with other peroxisomal disorders. This is particularly relevant as despite recent studies investigating transplantation in adult onset cerebral ALD, there are no established treatment options for non-CCALD patients and therefore screening is currently unlikely to improve clinical outcomes in these patients outside of improved adrenal monitoring [12, 16, 79]. The issue is also particularly relevant if both sexes are screened as this increases the number of non-CCALD patients identified.
Where possible the impact of these issues has been explored through sensitivity, probabilistic sensitivity and threshold analyses. The results of which suggest that the results are robust to the assumptions made in the model. In addition some of these evidence gaps, such as the types and number of other peroxisomal disorders identified, will be addressed by the results coming out of the New York X-ALD screening programme and other implementing sites [16, 79]. In the longer term the existing X-ALD screening programmes will also be able to address other evidence gaps such as the feasibility and efficacy of monitoring, transplantation protocols and disease natural history. Specific studies may also need to be undertaken in order to fully understand the impact of identifying those with non-CCALD, improve the evidence on the quality of life and resource use of those currently living with CCALD and understand the quality of life and resource impact of the diagnostic journey in symptomatic patients.
There are methodological issues associated with measuring and valuing many of the potential benefits and harms of newborn screening. For instance, impacts on families and carers, including future family planning, measuring and valuing quality of life in children, especially those with cognitive disabilities, and estimating the impact on families of incidental findings arising through screening [74–76, 80]. Not all of these issues are covered by current guidelines on economic evaluation of newborn screening interventions [81] and further methodological work is required to improve the quality and scope of future economic evaluations.
Conclusion
This study estimates that including screening for X-ALD in a tandem MS based screening programme such as the UK NHS Newborn Blood Spot Screening Programme would result in an increase in QALYs and a decrease in total discounted health, social care and education costs. The results are driven by the reduction in social care costs and the increase in QALYs for CCALD patients. Sensitivity analyses suggest that the results are sensitive to the proportion of patients with X-ALD that go on to develop CCALD. Threshold analyses suggest that any potential disbenefits arising for those with non-CCALD conditions would need to be substantial in order to outweigh the benefit to those with CCALD. However the uncertainties associated with measuring and valuing the benefits and harms of screening in the X-ALD population need to be addressed in order to fully demonstrate that the economic criteria can be fulfilled.
Additional files
Full parameters table. This file contains the full parameters table including the distributions used in the probabilistic sensitivity analysis. (DOCX 30 kb)
Systematic review of ALD for model parameters. This file contains the full methods and results of the systematic review that was carried out to inform the model parameters presented in this manuscript. (DOCX 315 kb)
Calculation of the Quality Adjusted Life Years (QALYs). This file presents more detail on the calculations that were used to calculate the QALYs in the model presented in this manuscript. (DOCX 27 kb)
Calculation of the costs and their sources. This file contains a table that outlines the included costs for the model presented in this manuscript and their sources. (DOCX 48 kb)
Acknowledgements
Acknowledgments are due to Sara Hunt and Karen Harrison of ALD Life and Professor Colin Steward of the University of Bristol. The views and opinions expressed are those of the authors.
Funding
The study was supported by the UK charity ALD Life (www.aldlife.org) and Bluebird Bio (www.bluebirdbio.com). The funding source had no involvement in study design, in the collection, analysis and interpretation of data, in the writing of the report or in the decision to submit an article for publication.
Availability of data and materials
All data generated or analysed during this study are included in this published article and its supplementary information file.
Abbreviations
- ALD-DRS
Adrenoleukodystrophy Disability Rating Scale
- AMN
Adrenomyeloneuropathy
- CCALD
Childhood cerebral adrenoleukodystrophy
- CI
Confidence interval
- EDSS
Expanded Disability Status Scale
- EQ-5D-5 L
EuroQol Five Dimension Five Level
- HSCT
Haematopoietic stem cell transplantation
- ICER
Incremental cost effectiveness ratio
- MS
Multiple sclerosis
- NHS
National Health Service
- NICE
National Institute for Health and Care Excellence
- NSC
National Screening Committee
- PSS
Personal social services
- QALY
Quality adjusted life years
- UK
United Kingdom
- X-ALD
X-linked adrenoleukodystrophy
Authors’ contributions
All authors were involved in the design and execution of the study and in the preparation of the manuscript. AB lead the study, JC and AB undertook the health economic modelling, JL led the evidence review and AS provided information specialist expertise. All authors read and approved the final manuscript.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The submitted work was supported by ALD Life and Bluebird Bio. AB has received reimbursement of travel expenses from ALD Life, JC has received reimbursement of conference and travel expenses from Bluebird Bio. None of the authors has non-financial interests that may be relevant to the submitted work.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Alice Bessey, Email: a.r.bessey@sheffield.ac.uk.
James B Chilcott, Email: j.b.chilcott@sheffield.ac.uk.
Joanna Leaviss, Email: j.leaviss@sheffield.ac.uk.
Anthea Sutton, Email: a.sutton@sheffield.ac.uk.
References
- 1.Bezman L, Moser HW. Incidence of X-linked adrenoleukodystrophy and the relative frequency of its phenotypes. Am J Med Genet. 1998;76:415–419. doi: 10.1002/(SICI)1096-8628(19980413)76:5<415::AID-AJMG9>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 2.Heim P, Claussen M, Hoffmann B, Conzelmann E, Gartner J, Harzer K, et al. Leukodystrophy incidence in Germany. Am J Med Genet. 1997;71:475–478. doi: 10.1002/(SICI)1096-8628(19970905)71:4<475::AID-AJMG20>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 3.Bezman L, Moser AB, Raymond GV, Rinaldo P, Watkins PA, Smith KD, et al. Adrenoleukodystrophy: incidence, new mutation rate, and results of extended family screening. Ann Neurol. 2001;49:512–517. doi: 10.1002/ana.101. [DOI] [PubMed] [Google Scholar]
- 4.Kemp S, Berger J, Aubourg P. X-linked adrenoleukodystrophy: clinical, metabolic, genetic and pathophysiological aspects. Biochim Biophys Acta Mol basis Dis. 2012;1822:1465–1474. doi: 10.1016/j.bbadis.2012.03.012. [DOI] [PubMed] [Google Scholar]
- 5.Engelen M, Kemp S, de Visser M, van Geel BM, Wanders RJ, Aubourg P, et al. X-linked adrenoleukodystrophy (X-ALD): clinical presentation and guidelines for diagnosis, follow-up and management. Orphanet J Rare Dis. 2012;7:51. doi: 10.1186/1750-1172-7-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Engelen M, Barbier M, Dijkstra IME, Schur R, De Bie RMA, Verhamme C, et al. X-linked adrenoleukodystrophy in women: a cross-sectional cohort study. Brain. 2014;137:693–706. doi: 10.1093/brain/awt361. [DOI] [PubMed] [Google Scholar]
- 7.Turk BR, Moser AB, Fatemi A. Therapeutic strategies in adrenoleukodystrophy. Wien Med Wochenschr. 2017;167:219–226. doi: 10.1007/s10354-016-0534-2. [DOI] [PubMed] [Google Scholar]
- 8.Eichler F, Duncan C, Musolino PL, Orchard PJ, De Oliveira S, Thrasher AJ, et al. Hematopoietic stem-cell gene therapy for cerebral Adrenoleukodystrophy. N Engl J Med. 2017;377:1630–1638. doi: 10.1056/NEJMoa1700554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Miller W, Rothman S, Nascene D, Kivisto T, DeFor T, Ziegler R, et al. Outcomes after allogenic hematopoetic cell transplantation for childhood cerebral adrenoleukodystrophy: the largest single-institution cohort report. Blood. 2011;118:1971–1978. doi: 10.1182/blood-2011-01-329235. [DOI] [PubMed] [Google Scholar]
- 10.Peters C, Charnas LR, Tan Y, Ziegler RS, Shapiro EG, DeFor T, et al. Cerebral X-linked adrenoleukodystrophy: the international hematopoietic cell transplantation experience from 1982 to 1999. Blood. 2004;104:881–888. doi: 10.1182/blood-2003-10-3402. [DOI] [PubMed] [Google Scholar]
- 11.Mahmood A, Raymond GV, Dubey P, Peters C, Moser HW. Survival analysis of haematopoietic cell transplantation for childhood cerebral X-linked adrenoleukodystrophy: a comparison study. Lancet Neurol. 2007;6:687–692. doi: 10.1016/S1474-4422(07)70177-1. [DOI] [PubMed] [Google Scholar]
- 12.Kühl JS, Suarez F, Gillet FT, Hemmati PG, Snowden JA, Stadler M, et al. Long-term outcomes of allogeneic haematopoietic stem cell transplantation for adult cerebral X-linked adrenoleukodystrophy. Brain. 2017;140:953–966. doi: 10.1093/brain/awx016. [DOI] [PubMed] [Google Scholar]
- 13.Theda C, Gibbons K, DeFor TE, Donohue PK, Golden WC, Kline AD, et al. Newborn screening for X-linked adrenoleukodystrophy: further evidence high throughput screening is feasible. Mol Genet Metab. 2014;111:55–57. doi: 10.1016/j.ymgme.2013.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Horn MA, Retterstol L, Abdelnoor M, Skjeldal OH, Tallaksen CM. Adrenoleukodystrophy in Norway: high rate of de novo mutations and age-dependent penetrance. Pediatr Neurol. 2013;48:212–219. doi: 10.1016/j.pediatrneurol.2012.12.007. [DOI] [PubMed] [Google Scholar]
- 15.Hubbard WC, Moser AB, Liu AC, Jones RO, Steinberg SJ, Lorey F, et al. Newborn screening for X-linked adrenoleukodystrophy (X-ALD): validation of a combined liquid chromatography-tandem mass spectrometric (LC-MS/MS) method. Mol Genet Metab. 2009;97:212–220. doi: 10.1016/j.ymgme.2009.03.010. [DOI] [PubMed] [Google Scholar]
- 16.Vogel B, Bradley S, Adams D, D'Aco K, Erbe R, Fong C, et al. Newborn screening for X-linked adrnoluekodystophy in New York state: diagnostic protocol, surveillance protocol and treatment guidelines. Mol Gen Metab. 2015;114:599–603. doi: 10.1016/j.ymgme.2015.02.002. [DOI] [PubMed] [Google Scholar]
- 17.Braverman NE, Raymond GV, Rizzo WB, Moser AB, Wilkinson ME, Stone EM, et al. Peroxisome biogenesis disorders in the Zellweger spectrum: an overview of current diagnosis, clinical manifestations, and treatment guidelines. Mol Genet Metab. 2016;117:313–321. doi: 10.1016/j.ymgme.2015.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wilson JM, Jungner G. Principles and practice of screening for disease. Public health paper 34. Geneva: WHO; 1968. [Google Scholar]
- 19.Public Health England . Effectiveness and appropriateness of a screening programme. 2015. Criteria for appraising the viability. [Google Scholar]
- 20.National Institute of Health and Care Excellence. Guide to the methods of technology appraisal. London: National Institute of Health and Care Excellence; 2013. [PubMed]
- 21.Curtis L. Unit costs of health and social care 2014. Canterbury: Personal Social Services Research Unit, University of Kent; 2014. [Google Scholar]
- 22.Office for National Statistics . Birth Summary Tables - England and Wales 2014. 2015. [Google Scholar]
- 23.National Records of Scotland . Vital Events Reference Tables 2014. 2015. [Google Scholar]
- 24.Northern Ireland Statistics and Research Agency . Live births 1887 to 2014. 2015. [Google Scholar]
- 25.Kaltenthaler E, Tappenden P, Paisley S, Squires H. NICE DSU technical support document 13: identifying and reviewing evidence to inform the conceptualisation and population of cost-effectiveness models. 2011. [PubMed] [Google Scholar]
- 26.Bessey A, Chilcott J, Pandor A, Paisley S. The cost-effectiveness of expanding the NHS newborn bloodspot screening Programme to include homocystinuria (HCU), maple syrup urine disease (MSUD), glutaric aciduria type 1 (GA1), isovaleric acidaemia (IVA), and long-chain hydroxyacyl-coa dehydrogenase deficiency (LCHADD) Value Health. 2014;17:A531. doi: 10.1016/j.jval.2014.08.1685. [DOI] [PubMed] [Google Scholar]
- 27.Hubbard WC, Moser AB, Tortorelli S, Liu A, Jones D, Moser H. Combined liquid chromatography-tandem mass spectrometry as an analytical method for high throughput screening for X-linked adrenoleukodystrophy and other peroxisomal disorders: preliminary findings. Mol Genet Metab. 2006;89:185–118. doi: 10.1016/j.ymgme.2006.05.001. [DOI] [PubMed] [Google Scholar]
- 28.Turgeon CT, Moser AB, Mørkrid L, Magera MJ, Gavrilov DK, Oglesbee D, et al. Streamlined determination of lysophosphatidylcholines in dried blood spots for newborn screening of X-linked adrenoleukodystrophy. Mol Genet Metab. 2015;114:46–50. doi: 10.1016/j.ymgme.2014.11.013. [DOI] [PubMed] [Google Scholar]
- 29.Kirk EP, Fletcher JM, Sharp P, Carey B, Poulos A. X-linked adrenoleukodystrophy: the Australasian experience. Am J Med Genet. 1998;76:420–423. doi: 10.1002/(SICI)1096-8628(19980413)76:5<420::AID-AJMG10>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 30.Di Biase A, Salvati S, Avellino C, Cappa M, Bertini E, Moroni I, et al. X-linked adrenoleukodystrophy: first report of the Italian study group. Ital J Neurol Sci. 1998;19:315–319. doi: 10.1007/BF00713859. [DOI] [PubMed] [Google Scholar]
- 31.Jardim LB, da Silva AC, Blank D, Villanueva MM, Renck L, Costa ML, et al. X-linked adrenoleukodystrophy: clinical course and minimal incidence in South Brazil. Brain Dev. 2010;32:180–190. doi: 10.1016/j.braindev.2009.02.002. [DOI] [PubMed] [Google Scholar]
- 32.Moser BA, Jones OR, Hubbard CW, Tortorelli S, Orsini JJ, Caggana M, et al. Newborn screening for X-linked Adrenoleukodystrophy. Int J Neonatal Screen. 2016;2:15. doi: 10.3390/ijns2040015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wiesinger C, Eichler FS, Berger J. The genetic landscape of X-linked adrenoleukodystrophy: inheritance, mutations, modifier genes, and diagnosis. Appl Clin Genet. 2015;8:109–121. doi: 10.2147/TACG.S49590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Moser HW. Adrenoleukodystrophy: phenotype, genetics, pathogenesis and therapy. Brain. 1997;120:1485–1508. doi: 10.1093/brain/120.8.1485. [DOI] [PubMed] [Google Scholar]
- 35.Pereira FS, Matte U, Habekost CT, de Castilhos RM, El Husny AS, Lourenco CM, et al. Mutations, clinical findings and survival estimates in south American patients with X-linked adrenoleukodystrophy. PLoS One. 2012;7:e34195. doi: 10.1371/journal.pone.0034195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ruiz M, Coll MJ, Pampols T, Giros M. X-linked adrenoleukodystrophy: phenotype distribution and expression of ALDP in Spanish kindreds. Am J Med Genet. 1998;76:424–427. doi: 10.1002/(SICI)1096-8628(19980413)76:5<424::AID-AJMG11>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 37.Sereni C, Paturneau-Jouas M, Aubourg P, Baumann N, Feingold J. Adrenoleukodystrophy in France: an epidemiological study. Neuroepidemiology. 1993;12:229–233. doi: 10.1159/000110321. [DOI] [PubMed] [Google Scholar]
- 38.Takemoto Y, Suzuki Y, Tamakoshi A, Onodera O, Tsuji S, Hashimoto T, et al. Epidemiology of X-linked adrenoleukodystrophy in Japan. J Hum Genet. 2002;47:590–593. doi: 10.1007/s100380200090. [DOI] [PubMed] [Google Scholar]
- 39.Van Geel BM, Assies J, Weverling GJ, Barth PG. Predominance of the adrenomyeloneuropathy phenotype of X-linked adrenoleukodystrophy in the Netherlands: a survey of 30 kindreds. Neurology. 1994;44:2343–2346. doi: 10.1212/WNL.44.12.2343. [DOI] [PubMed] [Google Scholar]
- 40.De Beer M, Engelen M, Van Geel BM. Frequent occurrence of cerebral demyelination in adrenomyeloneuropathy. Neurology. 2014;83:2227–2231. doi: 10.1212/WNL.0000000000001074. [DOI] [PubMed] [Google Scholar]
- 41.Cox CS, Dubey P, Raymond GV, Mahmood A, Moser AB, Moser HW. Cognitive evaluation of neurologically asymptomatic boys with X-linked adrenoleukodystrophy. Arch Neurol. 2006;63:69–73. doi: 10.1001/archneur.63.1.69. [DOI] [PubMed] [Google Scholar]
- 42.Dubey P, Raymond GV, Moser AB, Kharkar S, Bezman L, Moser HW. Adrenal insufficiency in asymptomatic adrenoleukodystrophy patients identified by very long-chain fatty acid screening. J Pediatr. 2005;146:528–532. doi: 10.1016/j.jpeds.2004.10.067. [DOI] [PubMed] [Google Scholar]
- 43.Kaga M, Furushima W, Inagaki M, Nakamura M. Early neuropsychological signs of childhood adrenoleukodystrophy (ALD) Brain Dev. 2009;31:558–561. doi: 10.1016/j.braindev.2009.05.001. [DOI] [PubMed] [Google Scholar]
- 44.Keller JL, Wang JI, Kang JY, Hanson JA, Kamath P, Swain JO, et al. Strength: a relevant link to functional performance in the neurodegenerative disease of adrenomyeloneuropathy. Neurorehabil Neural Repair. 2012;26:1080–1088. doi: 10.1177/1545968312441682. [DOI] [PubMed] [Google Scholar]
- 45.Liang JS, Lee WT, Hwu WL, Peng SS, Chu LW, Wang PJ, et al. Adrenoleukodystrophy: clinical analysis of 9 Taiwanese children. Acta Paediatr Taiwan. 2004;45:272–277. [PubMed] [Google Scholar]
- 46.Moser HW, Bezman L, Lu SE, Raymond GV. Therapy of X-linked adrenoleukodystrophy: prognosis based upon age and MRI abnormality and plans for placebo-controlled trials. J Inherit Metab Dis. 2000;23:273–277. doi: 10.1023/A:1005688130338. [DOI] [PubMed] [Google Scholar]
- 47.Stephenson DJ, Bezman L, Raymond GV. Acute presentation of childhood adrenoleukodystrophy. Neuropediatrics. 2000;31:293–297. doi: 10.1055/s-2000-12952. [DOI] [PubMed] [Google Scholar]
- 48.Suzuki Y, Takemoto Y, Shimozawa N, Imanaka T, Kato S, Furuya H, et al. Natural history of X-linked adrenoleukodystrophy in Japan. Brain Dev. 2005;27:353–357. doi: 10.1016/j.braindev.2004.09.008. [DOI] [PubMed] [Google Scholar]
- 49.Walterfang MA, O'Donovan J, Fahey MC, Velakoulis D. The neuropsychiatry of adrenomyeloneuropathy. Cns Spectrums. 2007;12:696–701. doi: 10.1017/S1092852900021532. [DOI] [PubMed] [Google Scholar]
- 50.Winstone A, Stellitano L, Verity C. The presentation and diagnosis of UK children with X-lined adrenoluekodystrophy ascertained over 17 years. Dev Med Child Neurol. 2015;57:2. [Google Scholar]
- 51.Zachowski K, Dubey P, Raymond G, Mori S, Bastian A. Sensorimotor function and axonal integrity in adrenalmyeloneuropathy. Arch Neurol. 2006;63:74–80. doi: 10.1001/archneur.63.1.74. [DOI] [PubMed] [Google Scholar]
- 52.Baumann M, Korenke GC, Weddige-Diedrichs A, Wilichowski E, Hunneman DH, Wilken B, et al. Haematopoietic stem cell transplantation in 12 patients with cerebral X-linked adrenoleukodystrophy. Eur J Pediatr. 2003;162:6–14. doi: 10.1007/s00431-002-1097-3. [DOI] [PubMed] [Google Scholar]
- 53.Beam D, Poe MD, Provenzale JM, Szabolcs P, Martin PL, Prasad V, et al. Outcomes of unrelated umbilical cord blood transplantation for X-linked adrenoleukodystrophy. Biol Blood Marrow Transplant. 2007;13:665–674. doi: 10.1016/j.bbmt.2007.01.082. [DOI] [PubMed] [Google Scholar]
- 54.Gassas A, Raiman J, White L, Schechter T, Clarke J, Doyle J. Long-term adaptive functioning outcomes of children with inherited metabolic and genetic diseases treated with hematopoietic stem cell transplantation in a single large pediatric center: parents’ perspective. J Pediatr Hematol Oncol. 2011;33:216–220. doi: 10.1097/MPH.0b013e3182050945. [DOI] [PubMed] [Google Scholar]
- 55.Gess A, Christiansen SP, Pond D, Peters C. Predictive factors for vision loss after hematopoietic cell transplant for X-linked adrenoleukodystrophy. J AAPOS. 2008;12:273–276. doi: 10.1016/j.jaapos.2007.11.012. [DOI] [PubMed] [Google Scholar]
- 56.Loes DJ, Stillman AE, Hite S, Shapiro E, Lockman L, Latchaw RE, et al. Childhood cerebral form of adrenoleukodystrophy: short-term effect of bone marrow transplantation on brain MR observations. AJNR Am J Neuroradiol. 1994;15:1767–1771. [PMC free article] [PubMed] [Google Scholar]
- 57.Martin PL, Carter SL, Kernan NA, Sahdev I, Wall D, Pietryga D, et al. Results of the cord blood transplantation study (COBLT): outcomes of unrelated donor umbilical cord blood transplantation in pediatric patients with lysosomal and peroxisomal storage diseases. Biol Blood Marrow Transplant. 2006;12:184–194. doi: 10.1016/j.bbmt.2005.09.016. [DOI] [PubMed] [Google Scholar]
- 58.Petryk A, Polgreen LE, Chahla S, Miller W, Orchard PJ. No evidence for the reversal of adrenal failure after hematopoietic cell transplantation in X-linked adrenoleukodystrophy. Bone Marrow Transplant. 2012;47:1377–1378. doi: 10.1038/bmt.2012.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Prasad V, Mendizabal A, arikh SH, Szabolcs P, Driscoll T, Page K, et al. Unrelated donor umbilical cord blood transplantation for inherited metabolic disorders in 159 pediatric patients from a single center: influence of cellular composition of the graft on transplantation outcomes. Blood. 2008;112:2979–2989. doi: 10.1182/blood-2008-03-140830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sakata N, Kawa K, Kato K, Yabe H, Yabe M, Nagasawa M, et al. Unrelated donor marrow transplantation for congenital immunodeficiency and metabolic disease: an update of the experience of the Japan marrow donor program. Int J Hematol. 2004;80:174–182. doi: 10.1532/IJH97.04055. [DOI] [PubMed] [Google Scholar]
- 61.Shapiro E, Krivit W, Lockman L, Jambaque I, Peters C, Cowan M, et al. Long-term effect of bone-marrow transplantation for childhood-onset cerebral X-linked adrenoleukodystrophy. Lancet. 2000;356:713–718. doi: 10.1016/S0140-6736(00)02629-5. [DOI] [PubMed] [Google Scholar]
- 62.Wilken B, Dechent P, Brockmann K, Finsterbusch J, Baumann M, Ebell W, et al. Quantitative proton magnetic resonance spectroscopy of children with adrenoleukodystrophy before and after hematopoietic stem cell transplantation. Neuropediatrics. 2003;34:237–246. doi: 10.1055/s-2003-43254. [DOI] [PubMed] [Google Scholar]
- 63.Guyot P, Ades AE, Ouwens MJNM, Welton NJ. Enhanced secondary analysis of survival data: reconstructing the data from published Kaplan-Meier survival curves. BMC Med Res Methodol. 2012;12:9. doi: 10.1186/1471-2288-12-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Latimer N. NICE DSU Technical Support Document 14: Undertaking survival analysis for economic evaluations alongside clinical trials - extrapolation with patient-level data. 2011. [PubMed] [Google Scholar]
- 65.Van Geel BM, Poll-The BT, Verrips A, Boelens JJ, Kemp S, Engelen M. Hematopoietic cell transplantation does not prevent myelopathy in X-linked adrenoleukodystrophy: a retrospective study. J Inherit Metab Dis. 2015;38:359–361. doi: 10.1007/s10545-014-9797-1. [DOI] [PubMed] [Google Scholar]
- 66.Kind P, Dolan P, Gudex C, Williams A. Variations in population health status: results from a United Kingdom national questionnaire survey. BMJ. 1998;316:736. doi: 10.1136/bmj.316.7133.736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Devlin N, Van Hout B. An EQ-5D-5L value set for England: Office of Health Economics. https://www.ohe.org/publications/valuing-health-related-quality-life-eq-5d-5l-value-set-england. Accessed on 23 Mar 2015
- 68.Kobelt G, Berg J, Lindgren P, Fredrikson S, Jönsson B. Costs and quality of life of patients with multiple sclerosis in Europe. J Neurol Neurosurg Psychiatry. 2006;77:918. doi: 10.1136/jnnp.2006.090365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Department of Health . NHS Reference Costs 2014 to 2015. 2015. [Google Scholar]
- 70.NHS UK Genetic Testing Network . Find a Test. 2015. [Google Scholar]
- 71.Paediatric Formulary Committee. British National Formulary for Children 2014–2015. London: BMJ Group, Pharmaceutical Press, and RCPCH Publications; 2015.
- 72.Barrett B, Mosweu I, Jones CRG, Charman T, Baird G, Simonoff E, et al. Comparing service use and costs among adolescents with autism spectum disorders, special needs and typical development. Autism. 2015;19:5. doi: 10.1177/1362361314536626. [DOI] [PubMed] [Google Scholar]
- 73.Kobelt G, Berg J, Lindgren P, Kerrigan J, Russel N, Nixon R. Costs and quality of life of multiple sclerosis in the United Kingdom. Eur J Health Econ. 2006;7(Suppl 5):S96–S104. doi: 10.1007/s10198-006-0380-z. [DOI] [PubMed] [Google Scholar]
- 74.Stevens K. Developing a descriptive system for a new preference-based measure of health-related quality of life for children. Qual Life Res. 2009;18:1105–1113. doi: 10.1007/s11136-009-9524-9. [DOI] [PubMed] [Google Scholar]
- 75.Adlard N, Kinghorn P, Frew E. Is the UK NICE reference case influencing the practice of pediatric quality-adjusted life-year measurement within economic evaluations? Value Health. 2014;17:454–461. doi: 10.1016/j.jval.2014.02.007. [DOI] [PubMed] [Google Scholar]
- 76.Lamsal R, Zwicker JD. Economic evaluation of interventions for children with neurodevelopmental disorders: opportunities and challenges. Appl Health Econ Health Policy. 2017;15:763–772. doi: 10.1007/s40258-017-0343-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Beckmann NB, Miller WP, Dietrich MS, Orchard PJ. Quality of life among boys with adrenoleukodystrophy following hematopoietic stem cell transplant. Child Neuropsychol. 2018;24:986–998. doi: 10.1080/09297049.2017.1380176. [DOI] [PubMed] [Google Scholar]
- 78.Pierpont EI, Eisengart JB, Shanley R. Neurocognitive trajectory of boys who received a hematopoietic stem cell transplant at an early stage of childhood cerebral adrenoleukodystrophy. JAMA Neurol. 2017;74:710–717. doi: 10.1001/jamaneurol.2017.0013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kemper AR, Brosco J, Comeau AM, Green NS, Grosse SD, Jones E, et al. Newborn screening for X-linked adrenoleukodystrophy: evidence summary and advisory committee recommendation. Genet Med. 2016;9:12. doi: 10.1038/gim.2016.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Grosse SD, Rogowski WH, Ross LF, Cornel MC, Dondorp WJ, Khoury MJ. Population screening for genetic disorders in the 21st century: evidence, economics, and ethics. Public Health Genomics. 2010;13:106–115. doi: 10.1159/000226594. [DOI] [PubMed] [Google Scholar]
- 81.Langer A, Holle R, John J. Specific guidelines for assessing and improving the methodological quality of economic evaluations of newborn screening. BMC Health Serv Res. 2012;12:300. doi: 10.1186/1472-6963-12-300. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Full parameters table. This file contains the full parameters table including the distributions used in the probabilistic sensitivity analysis. (DOCX 30 kb)
Systematic review of ALD for model parameters. This file contains the full methods and results of the systematic review that was carried out to inform the model parameters presented in this manuscript. (DOCX 315 kb)
Calculation of the Quality Adjusted Life Years (QALYs). This file presents more detail on the calculations that were used to calculate the QALYs in the model presented in this manuscript. (DOCX 27 kb)
Calculation of the costs and their sources. This file contains a table that outlines the included costs for the model presented in this manuscript and their sources. (DOCX 48 kb)
Data Availability Statement
All data generated or analysed during this study are included in this published article and its supplementary information file.