

Engineered aquatic systems such as aquaria and aquaculture facilities have large societal value. Ensuring the health of animals in these systems requires understanding how microorganisms contribute to chemical cycling and waste removal. Focusing on the largest seawater aquarium in the United States, we explore the microbial communities in specialized reactors designed to remove excess nitrogen through the metabolic activity of sulfur-consuming microbes. We show that the diversity of microbes in these reactors is both high and highly variable, with distinct community types associated with significant differences in nitrogen removal rate. We also show that the genes encoding the metabolic steps of nitrogen removal are distributed broadly throughout community members, suggesting that the chemical transformations in this system are likely a result of microbes relying on other microbes. These results provide a framework for future studies exploring the contributions of different community members, both in waste removal and in structuring microbial biodiversity.
KEYWORDS: Sulfurimonas, aquarium, denitrification, microbial ecology, sulfur oxidation
ABSTRACT
Denitrification by sulfur-oxidizing bacteria is an effective nitrate removal strategy in engineered aquatic systems. However, the community taxonomic and metabolic diversity of sulfur-driven denitrification (SDN) systems, as well as the relationship between nitrate removal and SDN community structure, remains underexplored. This is particularly true for SDN reactors applied to marine aquaria, despite the increasing use of this technology to supplement filtration. We applied 16S rRNA gene, metagenomic, and metatranscriptomic analyses to explore the microbial basis of SDN reactors operating on Georgia Aquarium's Ocean Voyager, the largest indoor closed-system seawater exhibit in the United States. The exhibit's two SDN systems vary in water retention time and nitrate removal efficiency. The systems also support significantly different microbial communities. These communities contain canonical SDN bacteria, including a strain related to Thiobacillus thioparus that dominates the system with the higher water retention time and nitrate removal but is effectively absent from the other system. Both systems contain a wide diversity of other microbes whose metagenome-assembled genomes contain genes of SDN metabolism. These include hundreds of strains of the epsilonproteobacterium Sulfurimonas, as well as gammaproteobacterial sulfur oxidizers of the Thiotrichales and Chromatiales, and a relative of Sedimenticola thiotaurini with complete denitrification potential. The SDN genes are transcribed and the taxonomic richness of the transcript pool varies markedly among the enzymatic steps, with some steps dominated by transcripts from noncanonical SDN taxa. These results indicate complex and variable SDN communities that may involve chemical dependencies among taxa as well as the potential for altering community structure to optimize nitrate removal.
IMPORTANCE Engineered aquatic systems such as aquaria and aquaculture facilities have large societal value. Ensuring the health of animals in these systems requires understanding how microorganisms contribute to chemical cycling and waste removal. Focusing on the largest seawater aquarium in the United States, we explore the microbial communities in specialized reactors designed to remove excess nitrogen through the metabolic activity of sulfur-consuming microbes. We show that the diversity of microbes in these reactors is both high and highly variable, with distinct community types associated with significant differences in nitrogen removal rate. We also show that the genes encoding the metabolic steps of nitrogen removal are distributed broadly throughout community members, suggesting that the chemical transformations in this system are likely a result of microbes relying on other microbes. These results provide a framework for future studies exploring the contributions of different community members, both in waste removal and in structuring microbial biodiversity.
INTRODUCTION
Denitrification is a vital component of the nitrogen cycle and can be harnessed in engineered systems to promote nitrogen removal. In this anaerobic process, microbes reduce dissolved nitrate (NO3−) to gaseous nitrous oxide (N2O) or dinitrogen (N2) through a series of intermediates. The enzymes catalyzing this process are found broadly in diverse Bacteria and Archaea (1, 2). Whereas some microbes carry out all denitrification steps, others conduct partial denitrification, which suggests that in certain environments, complete nitrate reduction to gaseous nitrogen may involve a community of diverse members (3). This community may use a range of chemical substrates. While many denitrifiers use organic compounds as both an electron donor and a carbon source for growth, others are chemolithoautotrophs that fix CO2 and couple denitrification to the oxidation of inorganic compounds such as hydrogen sulfide. This diversity suggests that the compositions of denitrifying communities can be highly variable, potentially with certain communities dominated by a small number of taxa and others supporting a consortium of interacting species. Characterizing this variation can help microbial ecologists understand the processes of community assembly as well as inform efforts to engineer nitrate removal, potentially by suggesting community types associated with high removal efficiency.
Nitrate removal is a particular challenge for closed aquatic systems with high biomass, such as the Ocean Voyager marine exhibit at Georgia Aquarium in Atlanta. Ocean Voyager holds 23,814,000 liters of recirculating artificial seawater that supports thousands of animals, including large whale sharks, manta rays, and a sea turtle. Nitrate is produced continuously in this system through the activity of nitrifying microbes that oxidize the nitrite and ammonia originating from animal waste. If not removed, nitrate may accumulate to levels that threaten animal health (4–6), for example, by inhibiting cellular oxygen transport (7). Lowering nitrate by replacing exhibit water is not cost effective, as Georgia Aquarium lacks access to natural seawater. Moreover, water replacement and other nonbiological nitrate removal strategies (e.g., ion exchange) may destabilize system parameters such as pH, temperature, and salinity.
Rather, nitrate is removed from Ocean Voyager in custom sulfur-driven denitrification (SDN) reactors that promote the denitrifying activity of sulfur-oxidizing chemolithoautotrophs. SDN reactors are widespread in wastewater treatment (8) and are increasingly applied to supplement filtration in commercial and home aquariums and aquaculture systems (9, 10). To remove nitrate from Ocean Voyager, exhibit water is diverted to two sets (pads) of SDN vessels. Each vessel contains prills of elemental sulfur (S0) for use as an energy source by denitrifiers. The flow is reduced through the vessels, enabling microbial respiration to deplete dissolved oxygen and an anaerobic denitrifying community to develop. Nitrate removal is monitored daily. While both SDN pads remove nitrate, the pads can differ significantly in removal efficiency, raising the possibility that microbial communities could vary substantially, with this variation potentially affecting removal efficiency.
While nitrate removal efficiency has been examined in SDN systems (11, 12), the taxonomic variation in the underlying microbial community is rarely characterized. Many reactor communities are composed of microbes that colonize from the influent water, as is the case for the Ocean Voyager systems. Alternatively, reactors may be seeded with a sample from an established SDN system or a culture of a known chemoautotrophic denitrifier, such as Thiobacillus denitrificans or Sulfurimonas denitrificans (13–15). These bacteria have been repeatedly isolated from SDN systems, and their biochemistry and kinetics have been extensively described (8, 16–21). However, studies that have assessed community composition indicate that SDN systems contain a diverse range of other denitrifying species. A clone library analysis identified both autotrophic and heterotrophic denitrifiers in a sulfide-driven reactor fed with raw sewage (22) and in an aquaculture SDN system (9). Similar results were recently obtained in a study using deep-coverage 16S rRNA gene sequencing to characterize an SDN reactor, which in that case, was seeded with wastewater sludge (23). While quintessential SDN taxa such as Thiobacillus and Sulfurimonas were detected in these studies, they were typically less abundant than other putative denitrifiers, including heterotrophs of the genera Azoarcus and Thauera (9) or potential sulfur-utilizing members of the genera Chlorobaculum and Dechloromonas (23). These studies suggest that the SDN community is complex and may support a high diversity of functional niches.
The extent to which this diversity varies among systems and in relationship to nitrate removal efficiency is not known for SDN reactors applied to marine aquaria. On the basis, presumably, of studies of wastewater reactors, marine aquarium SDN systems are generally assumed to be dominated by the obligate chemolithoautotroph T. denitrificans (24, 25). However, to our knowledge, community-level characterizations of marine aquarium SDN reactors have not been described. Additionally, it remains unknown how protein-coding genes of sulfur oxidation and denitrification are distributed among organisms and microhabitats in SDN reactors. Characterizing the diversity and proportional representation of such functional genes, for example, through meta-omic sequencing, would help clarify the relationship between taxonomic composition and metabolic outcomes in marine SDN systems as well as suggest potential interactions among taxa.
Here, we investigated the taxonomic and metabolic diversity in the SDN vessels supporting Georgia Aquarium's Ocean Voyager exhibit. We used a combination of 16S rRNA gene amplicon and shotgun DNA and RNA sequencing (metagenomics and metatranscriptomics) along with an analysis of metagenome-assembled genomes (MAGs) from dominant SDN taxa. The analysis targeted multiple habitats within the SDN vessels, including communities attached to sulfur and aragonite pellets and in the SDN (interstitial) water. The results are described in light of efficiency differences between the pads and confirm a high level of variation and community complexity in marine SDN reactors.
RESULTS AND DISCUSSION
Physical and chemical variation in SDN pads.
Figure 1A provides a schematic of the Ocean Voyager SDN system, which has been operating since April 2015. Approximately 0.15% of the exhibit's circulating flow is diverted to two sets of equipment pads, each supporting four SDN vessels (8 total), with each vessel measuring 1.2 m in diameter by 3.3 m in height. Two circulation loops provide the optimal hydraulic retention time of water in the system, with the first loop pumped and recirculated continuously through the vessels while the second loop titrates a small amount of water from the system into the first loop and back out to the exhibit. Within a pad, water is able to circulate throughout all four towers (Fig. 1B), with water pumped from the bottom to the top of each vessel at a rate of 2.7 liters/m2/s. At the time of initiation (2015), each vessel was filled with 2,000 kg of elemental (S0) sulfur prills (to within 5 cm of the upper drain), 900 kg of pea gravel to act as a supporting substrate for the sulfur, and a 0.3-m layer of reactor grade aragonite (800 kg) for pH buffering (Fig. 1B). Maintenance was performed on both pads at irregular intervals primarily by adding aragonite or increasing the flow rate to prevent clogging (see Table S1 in the supplemental material).
FIG 1.

Schematic representations of the Ocean Voyager denitrification system consisting of eight denitrification towers split between two pads (A) and of an individual denitrification tower (B). (C) Change in percent dissolved oxygen (sO2), nitrate removal (mg/liter), pH, and flow rate (liters/min) for each pad over a 4-month time bracketing the two sampling events. Dashed vertical lines represent maintenance events performed on the pads and solid vertical lines represent sampling dates. Horizontal dashed lines represent mean values over the 4-month period.
We took samples of sulfur prills, water from within the SDN towers (interstitial), and aragonite (if present; see Materials and Methods) on two dates: 18 May and 12 July 2016 (Fig. 1C). The physical and chemical parameters varied between the two SDN pads over the 4-month period bracketing these sampling events. Oxygen saturation (sO2) in the SDN water remained fairly consistent in both pads, averaging near 0% sO2, but spiked intermittently due to maintenance or sampling events that introduced oxygen (vertical bars, Fig. 1C; Table S1). In contrast, the flow rates differed notably between the pads, averaging ∼120 liters/min for pad 1 but only ∼69 liters/min for pad 2. The longer water residence time in pad 2 corresponded with consistently lower water pH in pad 2 (∼6.0) than in pad 1 (∼6.5). As diverse metabolic processes, notably, the oxidation of sulfur and sulfide, release protons or produce acid end products, the consistently lower pH observed in pad 2 may reflect an overall increased microbial activity.
Additionally, pad 2 was more efficient at removing nitrate. In the 4 months surrounding the sampling period, the passage of water through pad 2 removed an average of 40 mg nitrate/liter compared to approximately 20 mg/liter in pad 1.
Microbial community structure.
16S rRNA gene sequencing revealed a high diversity of microbial taxa in the SDN system, with community composition structured on the basis of niche type (interstitial water, sulfur, and aragonite) and also between pads. Overall, 7,214 unique operational taxonomic units (OTUs) were identified across all samples, with some samples having up to 1,400 OTUs (after rarefaction to 14,661 sequences) (see Table S3). OTU richness was highest in the input water, SDN interstitial water, and aragonite samples (averages of 1,064, 1,017, and 939, respectively), whereas sulfur prills contained significantly fewer taxa (average, 629 OTUs). Aragonite samples exhibited the highest taxon evenness, with an average Simpson's E score of 0.094, whereas sulfur (0.014), interstitial water (0.012), and input water (0.027) were substantially less even and more likely to be dominated by a subset of taxa (Table S3). On the basis of the weighted UniFrac metric, taxonomic compositions clearly differed among interstitial water, sulfur, and aragonite communities, with the input water community most similar to that of the aragonite (Fig. 2). These patterns indicate distinct microhabitats with the SDN system, with those associated with the interstitial water and sulfur prills having the strongest effect on restructuring the incoming microbial community.
FIG 2.
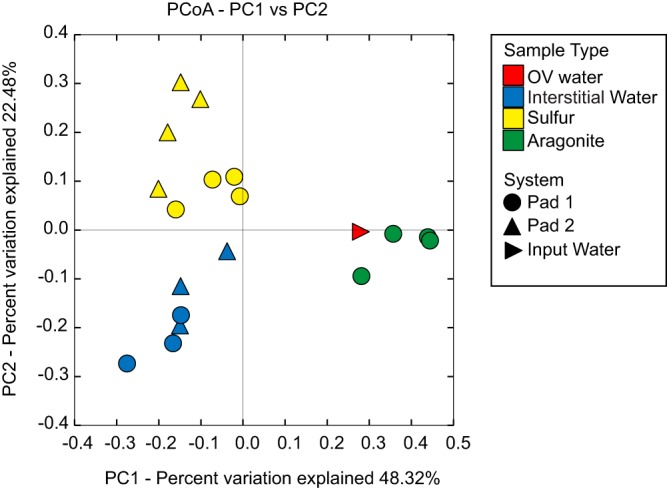
Microbial community relatedness based on 16S rRNA gene amplicon sequencing as quantified by the weighted UniFrac distance metric. Niche type is denoted by color and sample origin by shape. OV water, exhibit water prior to entry into the denitrification system; interstitial water, water from within the system.
Pad-specific patterning was also observed, being most evident among the sulfur samples (Fig. 2 and 3). Notably, sequences affiliated with the sulfur-oxidizing betaproteobacterial genus Thiobacillus (family Hydrogenophilaceae) were significantly enriched (up to 50% of sequences) in the sulfur prills of pad 2, and to a lesser extent in the pad 2 water. In contrast, Thiobacillus was a minor component of the pad 1 sulfur and water communities (Fig. 3). Sequences matching the sulfur-oxidizing epsilonproteobacterial genus Sulfurimonas were widely distributed and abundant in both pads, ranging from 15% to 40% of the total communities in pad 1 and 2 sulfur prills, and 30% to 70% of the community in interstitial water. In a study applying microbial colonization traps to marine sediments, Epsilonproteobacteria, including members of Sulfurimonas, preferentially colonized traps containing S0 (56% of sequences) compared to those containing either pyrite or glass beads (<5%). In the same study, pure cultures of S. denitrificans were able to oxidize cyclooctasulfur (S8), a major component of S0 (26), suggesting that Sulfurimonas species may be uniquely suited to oxidize S0. Even in pad 2, where Thiobacillus species were enriched in the sulfur prills, Sulfurimonas was still the dominant microbe in the surrounding water (Fig. 3). Interestingly, Sulfurimonas flagellum genes were the most highly transcribed genes in the SDN system (data not shown), raising the possibility that the high Sulfurimonas abundance is linked to a capacity for motility and attachment, both of which may be mediated by flagella (27). Pad communities also contained a number of other groups traditionally associated with dissimilatory sulfur oxidation, including the Chromatiales and Thiotrichales, both of which were proportionally enriched in the pad 1 sulfur prills where Thiobacillus was at a low abundance. In contrast, Thiotrichales was effectively absent from pad 2, where Thiobacillus was dominant. Similar to both Thiobacillus and Sulfurimonas, members of the Chromatiales and Thiotrichales have been shown to couple sulfur oxidation with reductive nitrogen metabolism (28). Certain members of the Chromatiales genera Thioalkalispira and Thioalkalivibrio can reduce nitrate but have not been shown to reduce other nitrogen oxides (29, 30), whereas others, such as Thioalkalivibrio thiocyanodenitrificans, can further reduce nitrite but appear unable to perform full denitrification (31). Similarly, Thiothrix species, members of the order Thiotrichales, have diverse metabolisms, with some members performing full denitrification and others only coupling sulfur oxidation to nitrate reduction (32). The presence of these groups in the SDN system suggests potentially diverse routes of sulfur-driven denitrification, independent of the presumed contributions by Thiobacillus and Sulfurimonas.
FIG 3.

Proportional abundances of microbial taxa based on 16S rRNA gene amplicon sequencing. The figure shows only those bacterial families averaging greater than 0.01 proportional representation. Aragonite samples have been omitted for clarity. *, family containing Sulfurimonas species; **, family containing Thiobacillus species.
We further investigated the diversity of Thiobacillus-like and Sulfurimonas-like sequences, given their dominance in the samples and the prior association of these taxa with SDN metabolism. Across the rarefied data set, a total of 169 and 632 OTUs were assigned to Thiobacillus and Sulfurimonas, respectively (Table S3). The evenness of OTU representation differed notably between the genera. A single OTU (547362) accounted for nearly 100% of all sequences assigned to Thiobacillus, regardless of the sample type (Fig. 4). This OTU shared 100% identity (over the 253-bp amplicon) to uncultured clones from diverse marine and freshwater environments. The OTU shares slightly less identity (98%) to the cultured SDN species T. denitrificans; rather, its closest cultured relative (99% identity) is Thiobacillus thioparus DSM 505T. Intriguingly, despite containing all of the genes for denitrification, T thioparus DSM 505T appears incapable of growing by denitrification under fully anoxic conditions, raising the (untested) hypothesis that denitrification by this taxon may be linked to microoxic conditions (33). Here, OTU 547362 was also detected in water from the Ocean Voyager system (OV water), suggesting that SDN-associated microbes are dispersed throughout the exhibit.
FIG 4.

Proportional abundances of the top 10 most abundant Thiobacillus or Sulfurimonas OTUs based on 16S rRNA gene amplicon sequencing. For each sample, the left bar corresponds to the abundance of Thiobacillus or Sulfurimonas reads as a proportion of the total microbial 16S rRNA pool in either water (blue), sulfur (yellow), or aragonite (green). The right bar, either in shades of purple (Sulfurimonas) or orange (Thiobacillus), shows the proportional representation of the top 10 most abundant OTUs within each genus.
The Sulfurimonas community, in contrast to that of Thiobacillus, was markedly diverse, with multiple OTUs present in all samples at significant proportions (Fig. 4 and 5). The 20 most abundant OTUs spanned the Sulfurimonas phylogeny, clustering in diverse clades with sequences recovered from environments ranging from groundwater to deep-sea hydrothermal vents (Fig. 5). The most abundant OTU (4406648) clustered in a clade with other SDN OTUs and few reference sequences, being most closely related to an uncultured bacterium from the East Pacific Rise vent field. The reference isolates most closely related to the OTUs in the SDN system have been associated with diverse metabolic functions. For example, the cultured bacterium most closely related to OTU 440664 is Sulfurimonas gotlandica GD1 (Fig. 5), isolated from the pelagic sulfidic zone of the Baltic Sea (19). S. gotlandica can grow chemolithoautotrophically via sulfide or thiosulfate oxidation linked to diverse steps of denitrification as well as heterotrophically on various organic compounds. Furthermore, S. gotlandica also can utilize amino acids as electron donors and appears to be tolerant of relatively high oxygen levels (10% saturation) (19, 34). In contrast, other OTUs, such as OTUs 65, 170, and 240, with the latter being particularly abundant in interstitial water, clustered loosely (bootstrap, >50%) with species such as S. denitrificans and Sulfurimonas autotrophica that are more typically associated with a strictly autotrophic lifestyle and higher oxygen sensitivity (35).
FIG 5.

Maximum likelihood phylogeny tree showing the relatedness of the top 20 most abundant Sulfurimonas OTUs (red) based on partial 16S rRNA gene sequences and their proportional abundance in the total 16S rRNA amplicon pool. The tree includes reference sequences identified in NCBI nt database as top matches in BLAST queries of each OTU, as well as RDP sequences representing the Sulfurimonas genus. Campylobacter sequences (n = 7) from RDP were used as an outgroup. The tree was constructed on the basis of an alignment (256 nt) of a total of 73 sequences using the Kimura 2 model iterated for 1,000 bootstraps.
Such results suggest a potentially high diversity of functional roles in the SDN Sulfurimonas community. Indeed, Sulfurimonas community structures varied among water, sulfur, and aragonite microhabitats, further suggesting that the observed diversity may be associated with ecological niche variation (see Fig. S1A). It remains unclear to what extent this diversity reflects the metabolic heterogeneity among lineages versus genetic partitioning due to barriers in gene flow. At the least, the taxonomic richness observed in the Sulfurimonas data set suggests that community structuring in this system may not be constrained by the limited connectivity to inocula from environments outside the exhibit. The tractability of the SDN, coupled with its high genetic diversity, suggests this system is a model for exploring the processes of functional diversification in Sulfurimonas. Collectively, the microbial community data suggest that wide inter- and intraspecies diversity contributes to nitrogen loss through sulfur oxidation in the SDN system.
Metabolic gene content and transcription in the SDN system.
We used metagenomic analysis of sulfur prill samples to test the extent to which diverse steps of SDN metabolism are distributed across taxonomic groups. Sixty MAGs were recovered from the metagenomic reads: of these, 24 had completeness ≥40% and contamination ≤10%, with 15 of these being more than 90% complete (see Table S4 and Data S1). Taxonomic assignment using MiGA confirmed that these MAGs represent a broad range of phylogenetic groups, including taxa such as Thiobacillus and Sulfurimonas sp. identified by amplicon sequencing (Table S4 and Data S1). As is common in metagenome binning, we recovered few 16S rRNA sequences in the MAG set and were therefore unable to definitively link MAGs to OTUs. However, consistent with the amplicon results (Fig. 5), a phylogenetic analysis based on concatenated single-copy genes confirmed that the MAGs include a nearly complete (>98%) genome (MAG pad1.007) related to Sulfurimonas gotlandica, as well as two genomes (98 and 100% complete; MAGs pad1.009 and pad2.002) highly similar to one another and both clustering within the Thiobacillus clade containing T. thioparus and T. denitrificans (see Fig. S2). In addition, the nearly complete (>90) MAG set contains diverse members of the Alphaproteobacteria, Gammaproteobacteria, FCB (Fibrobacteres, Chlorobi, and Bacteroidetes), Terrabacteria, and PVC (Planctomycetes, Verrucomicrobia, and Chlamydiae) groups (Fig. 6 and Fig. S2).
FIG 6.

Abundances of transcripts and presence/absence of nitrogen and sulfur metabolism genes associated with metagenome-assembled genomes (MAGs). (A) Heatmap showing the percentages of total mRNA reads mapping to individual MAGs by BLAST (bit, >50; ID, >90%), with warmer colors indicating increased transcript representation. MAG IDs are listed at the far right. Thiobacillus MAGs (Betaproteobacteria) are pad1.009 and pad2.002; Sulfurimonas MAGs (Epsilonproteobacteria) are pad1.007, pad2.009, and pad2.010. (B) Heatmap showing the presence (dark gray) or absence (light gray) of key genes involved in sulfur oxidation and denitrification within individual MAGs. Marker genes were identified by the bidirectional best BLAST hit in KAAS using the “for prokaryotes” database. MAGs were organized by phylogenetic analysis (left) using 30 shared single-copy marker genes identified by HMMER and aligned with MAFFT. Scale bar represents AA substitutions per site. dsrA, dissimilatory sulfite reductase; aprA, adenylylsulfate reductase; sat, sulfate adenylyltransferase; sqr, sulfide:quinone reductase; soxABXYZ, SOX (sulfur oxidation) system; narG, cytoplasmic nitrate reductase; napA, periplasmic nitrate reductase; nirK, nirS, nitrite reductase; norB, nitric oxide reductase; nosZ, nitrous oxide reductase.
Key genes of SDN metabolism were present in phylogenetically diverse MAGs (Fig. 6). As in SDN systems elsewhere, the Ocean Voyager's towers are packed with elemental sulfur (S0) in the form of prills. This elemental sulfur, as well as thiosulfate (S2O32−), if present, is presumably oxidized to sulfate (SO42−) via the sox multienzyme system (36). In reactors with low oxygen conditions, the resulting sulfate may be reduced by sulfate-reducing bacteria to sulfide (S2−), which can be reoxidized either using the sulfide:quinone reductase enzyme (Sqr) or through a reverse form of the dissimilatory sulfite reductase enzyme (oDsrAB), forming sulfite (SO3−). The sulfite can be further oxidized to sulfate by the adenylylsulfate reductase (AprAB) and sulfate adenylyltransferase (Sat) enzymes. A marker gene for at least one of these steps was detected in all but one of the 24 MAGs (those with completeness >40%), with sqr being the most broadly distributed gene, identified in 19 of the 24 genomes (Fig. 6B). Genes coding for all three pathways (Sox, Sqr, and oDsr) were detected in the two Thiobacillus MAGs (pad1.009 and pad2.002), as well as in diverse gammaproteobacterial MAGs (Fig. 6B). The latter include a nearly complete (99.9% estimated) MAG (pad1.005) closely related to Sedimenticola thiotaurini (Fig. S2), a metabolically plastic species known to both oxidize sulfur autotrophically as well as supplement energy production by oxidizing organic matter and to exhibit various levels of oxygen sensitivity depending on reductant availability (37). Other MAGs may have the potential for a more limited set of sulfur transformations. The sqr gene was the only sulfur metabolism gene found in all three Sulfurimonas MAGs (pad1.007, pad2.009, and pad2.010). The sat gene was detected in only Sulfurimonas MAG pad1.007, and no genes of the Sox pathway were detected in any Sulfurimonas MAG (Fig. 6B), despite a high proportional recovery of epsilonproteobacterial sox transcripts (likely from Sulfurimonas; see below). However, these MAGs are not complete and we caution that conclusions based on the absence of genes require further validation.
In SDN metabolism, electrons generated from the above reactions are used to reduce inorganic nitrogen compounds. Initially, nitrate (NO3−) is reduced to nitrite (NO2−) either by the periplasmic nitrate reductase (Nap) or the cytoplasmic nitrate reductase (Nar). Nitrite is reduced to nitric oxide (NO) by nitrite reductases (NirS or NirK), and nitric oxide to nitrous oxide (N2O) by nitric oxide reductase (NorBC). Ultimately, complete denitrification involves nitrous oxide reduction to dinitrogen (N2) by nitrous oxide reductase (NosZ). Genes for one or more steps of this pathway were detected in 22 of the 24 MAGs (with completeness of >40%). Genes encoding nitrate reductases (Nar or Nap) were most frequently detected, appearing in 16 of the 24 MAGs. However, genes for all pathway steps were common, and four of the MAGs (pad1.008, pad1.009, pad2.002, and pad2.019), including Thiobacillus MAGs pad1.009 and pad2.002, contained all genes necessary for the reduction of nitrate to dinitrogen (Fig. 6B). The dominance of Thiobacillus, particularly in pad 2, may be partially explained by the presence of the full denitrification pathway, which should provide more energy than partial denitrification (38). In contrast, we did not detect nitrite reduction potential (nirK or nirS) in any of the Sulfurimonas MAGs (pad1.007, pad2.009, and pad2.010), despite one of these (pad1.007) estimated at >98% completeness and containing the rest of the denitrification pathway (Fig. 6B; Table S4). This absence of nir from SDN Sulfurimonas requires confirmation, notably as nirS homologs, albeit divergent from those of other bacteria (39), have been found in diverse Sulfurimonas species, including S. gotlandica (19), a close relative of MAG pad1.007 (Fig. S2). In other MAGs, such as Flavobacteriia MAG pad1.010 (99% completeness), we detected multiple denitrification genes but no genes for dissimilatory sulfur metabolism, suggesting that reductants other than inorganic sulfur species may play a role in nitrogen loss in these towers.
High numbers of transcripts from the interstitial water and sulfur prill metatranscriptomes were recruited to the MAGs (Fig. 6A), suggesting MAG taxa as key contributors to the metabolically active community. However, few transcripts from the aragonite data sets were recruited; this was not unexpected given that the MAGs were assembled from sulfur-associated metagenomes. Notably, sequences recruited to Thiobacillus MAGs (pad1.009 and pad2.002) represented ∼5% to 6.5% of sulfur prill transcripts from pad 2 (Fig. 6A), consistent with the Thiobacillus enrichment in pad 2 amplicon data sets (Fig. 3). In contrast, transcripts recruited to the Sulfurimonas MAGs (pad1.007, pad2.009, and pad2.010) were most abundant (up to 4% of total) in the interstitial water (Fig. 6A), again, in agreement with the amplicon data (Fig. 3). Transcripts of the Sedimenticola MAG (pad1.005) were detected at relatively high abundances (0.7 to 4%) in all sulfur and water samples. Other MAGs with denitrifying potential, notably members of the Flavobacteriia and Gammaproteobacteria, were also broadly represented across samples, suggesting these diverse taxa as potentially important contributors to bulk SDN metabolism.
Transcripts affiliated with all major steps of SDN metabolism were detected in roughly equivalent proportions in interstitial water and sulfur habitats (although not in aragonite samples) and were associated with diverse microbial lineages (Fig. 7). The taxonomic classification was based on least common ancestry (LCA), as assessed for all transcripts rather than only those affiliated with the MAGs. Many transcript fragments were not classifiable at a resolution lower than the phylum or domain level, suggesting that these genes are conserved and challenging to distinguish across multiple taxa (Fig. 7B). Different taxa nonetheless appear to dominate the transcript pool at different pathway steps. For example, transcripts classified as Epsilonproteobacteria, likely Sulfurimonas, were major contributors to steps of nitrate reduction (napAB), nitric oxide reduction (norBC), and sulfur/thiosulfate oxidation via the Sox pathway. This result is in contrast with the lack of identifiable sox genes in the Sulfurimonas MAGs (Fig. 6B). This may be due in part to the incompleteness of the MAGs. Alternatively, as the sox genes are commonly found in an operon and our LCA analysis indicates that they are conserved and identifiable only to the phylum level, it is possible that Sulfurimonas sox-containing metagenomic fragments were difficult to bin to specific MAGs. In contrast, a relative lack of epsilonproteobacterial nirKS transcripts is consistent with the absence of nirKS genes in Sulfurimonas MAGs, although these MAGs contained the rest of the denitrification pathway (Fig. 6B, Fig. 7). Transcripts suggesting sulfate production via sulfate adenylyltransferase (Sat) were classified almost exclusively as Betaproteobacteria, likely Thiobacillus. Transcripts encoding the oxidative form of the dissimilatory sulfite reductase (odsrAB) were abundant in all samples, although the majority of these were not well classified via LCA, suggesting that these sequences were highly conserved (Fig. 7B). Transcripts for the reductive form of this enzyme were also detected, but only in the sulfur prills of one sample, suggesting a relatively limited potential for dissimilatory sulfate reduction to sulfide in this system (Fig. 7A). In some samples, notably the sulfur prills of pad 1, Gammaproteobacteria and Bacteroidetes/Chlorobi sequences together dominated the nos transcripts associated with nitrous oxide reduction to dinitrogen (Fig. 7B). Intriguingly, nos transcripts were at a much lower abundance relative to those encoding Nor nitric oxide reductases, which were enriched severalfold compared to those of all other denitrification steps (Fig. 7A). This pattern suggests that nitrous oxide, rather than dinitrogen, might be the dominant gaseous end product in this SDN system. This hypothesis cannot be verified with gene expression data, however, and suggests a need for further studies to evaluate energy and element partitioning among SDN steps and community members.
FIG 7.

Taxonomic affiliation of transcripts matching key genes involved in sulfur oxidation and denitrification. Data sets were normalized to a uniform number of mRNA reads. (A) Abundances of transcripts (read counts/gene length in kb) matching genes (or gene groups) from aragonite (green), interstitial water (blue), and sulfur (yellow) metatranscriptome data sets. (B) Proportions of reads assigned to taxonomic groups via the LCA algorithm in MEGAN5. napAB, periplasmic nitrate reductase; narGHI, cytoplasmic nitrate reductase; nirKS, nitrite reductase; norBC, nitric oxide reductase; nosZ, nitrous oxide reductase; odsrAB, oxidative form of dissimilatory sulfite reductase; rdsrAB, dissimilatory sulfite reductase; aprAB, adenylylsulfate reductase; sat, sulfate adenylyltransferase; soxABCDXYZ, SOX (sulfur oxidation) system; sqr, sulfide:quinone reductase.
Conclusions.
Ocean Voyager's SDN system sustains a highly diverse microbial community capable of coupling sulfur oxidation to denitrification. The observed diversity of bacteria and their metabolic genes suggests a modular approach to sulfur-based denitrification, with SDN steps partitioned among diverse community members and certain steps potentially strongly influenced by taxa not canonically associated with SDN reactors (e.g., Gammaproteobacteria such as Sedimenticola). The fragmented distribution of microbial denitrification and sulfur oxidation pathways has been documented in diverse taxa from other systems (3, 36). Indeed, it is not uncommon that isolates from the same source contain only a partial pathway even in the presence of isolates containing the full pathway (40, 41). Such modularity suggests partial denitrification or sulfur oxidation as establishing distinct microbial niches in the same system and therefore the potential for chemical dependencies among SDN members.
Furthermore, the presence of multiple substrates in the SDN system, in the form of aragonite and sulfur prills as well as the surrounding interstitial water, enables the ecological separation of microbes across these niches. The diversity of specific taxa, for example, as seen in the high number of OTUs within the genus Sulfurimonas, suggests the potential for ecological separation within a presumptive niche and therefore a need for further genomic or culture-based analyses to discriminate the functional properties at the strain or substrain level.
Although set up simultaneously with identical inputs, the two pads of the SDN system developed distinct physical and chemical characteristics and significantly different microbial communities. Nonetheless, both pads and community types remove nitrate, albeit at differing efficiencies. It is unknown to what extent these microbial differences are a cause or effect (or both) of differences in the environmental conditions between the pads. Presumably, the pronounced difference in water retention time between pads 1 and 2 affects dissolved substrate (including oxygen) availability and potentially the rates of diffusion and physical mixing within the towers; changes in these parameters are likely to affect microbial activity and potentially also spatial organization. It is unknown how microbial community compositions and functions changed in the SDN system from the time of system initiation to our first sampling roughly 1 year later. The microbial differences between pads suggest that the system is dynamic, and further investigation is needed to determine if these differences are static or if individual pads are at different stages in a succession event.
Importantly, these results indicate the potential for the manipulation of SDN setups to maximize nitrate removal efficiency, either by changing physical factors, such as lowering the flow rate or pH, or potentially by microbial seeding. We suggest, however, that the latter strategy is unlikely to be successful. Indeed, the observed high diversity of OTUs, spanning a wide range of phylogenetic groups and genetic divergence levels, suggests that the Ocean Voyager exhibit is not inoculum limited, despite being a closed system with incoming marine microbes limited primarily to those attached to food or animals. Such inocula are sufficient for fostering highly complex SDN communities that, given their relative tractability, may be valuable models for exploring the mechanisms that maintain metabolic and taxonomic diversity in the environment.
MATERIALS AND METHODS
Denitrification system parameters and sampling.
The SDN system was sampled on two occasions, 18 May and 12 July 2016. Samples of water from within each pad (interstitial water) were obtained through a spigot connected to the recirculating flow of the pad. For each water sample, 2 liters were filtered through a 0.2-μm pore size Sterivex filter (EMD Millipore) using a peristaltic pump. The filter was filled with RNA/DNA stabilizing buffer (25 mM sodium citrate, 10 mM EDTA, 5.3 M ammonium sulfate, pH 5.2) and immediately frozen on dry ice. In addition, a sample of the influent water from the Ocean Voyager system (OV water) prior to entry into the SDN system was sampled, filtered, and preserved using the same methods. To obtain samples of SDN substrate (sulfur and aragonite), the lids of the towers were unscrewed and the systems were sampled from above. Sulfur prills and aragonite pellets (when present) were sampled from each tower and placed in sterile conical tubes; the prills/pellets were immersed in RNA preservation solution and immediately frozen on dry ice. At the time of sampling, aragonite was absent from all towers from pad 2; thus, all aragonite samples are from pad 1. All samples were stored at −80°C until further processing. Samples for 16S rRNA amplicon sequencing were taken from both time points, and samples for metagenomic and metatranscriptomic analyses were taken only on the 18 May sampling event. Table S2 in the supplemental material provides an overview of which samples were used in different analyses.
Physical and chemical parameters.
The flow rate, pH, and dissolved oxygen were recorded daily for each pad using in-line probes. The flow rate was measured using a Signet Magmeter flow meter (Georg Fischer). The pH of the outflowing SDN water was measured using a Signet 2750 DryLoc ph/ORP sensor (Georg Fischer), and the dissolved oxygen concentration was measured using an Orbisphere model 410 analyzer (Hach). The concentrations of nitrate in the inflowing and outflowing water were measured using a Dionex ICS-5000+ ion chromatograph with UV detection (Thermo Fisher Scientific).
Nucleic acid extraction.
DNA and RNA were extracted from the sulfur and aragonite samples using the PowerSoil DNA isolation kit (Mo Bio Laboratories) and the PowerMicrobiome RNA isolation kit (Mo Bio Laboratories) as instructed by the manufacturer. DNA was extracted from the interstitial and OV water samples (Sterivex filters) by adding lysozyme (2 mg in 40 μl of lysis buffer per filter) directly to the filter cartridges, sealing the ends, and incubating for 45 min at 37°C. Proteinase K (1 mg in 100 μl lysis buffer, with 100 μl 20% SDS) was added, and the cartridges were resealed and incubated for 2 h at 55°C. The lysate was removed, and DNA was extracted once with phenol-chloroform-isoamyl alcohol (25:24:1) and once with chloroform-isoamyl alcohol (24:1) and then concentrated by spin dialysis using Ultra-4 (100 kDa; Amicon) centrifugal filters. RNA from Sterivex filters was isolated using a modification of the mirVana miRNA isolation kit (Ambion). The filters were thawed on ice, and RNA stabilizing buffer was expelled from Sterivex cartridges via a syringe and discarded. The cells were lysed by adding lysis buffer and miRNA homogenate additive (Ambion) directly to the cartridge. Following vortexing and incubation on ice, the lysates were transferred to RNase-free tubes and processed via acid-phenol-chloroform extraction according to the kit protocol. The Turbo DNA-free kit (Ambion) was used to remove DNA from RNA samples, and the extracts were purified using the RNeasy MinElute cleanup kit (Qiagen). Purified DNA and RNA were quantified using the Qubit 2.0 fluorometer (Thermo Fisher Scientific).
16S rRNA gene amplicon sequencing and analysis.
The V3 to V4 hypervariable region of the 16S rRNA gene was amplified from 1.5 μl (total reaction volume of 25 μl) of extracted DNA for each sample using Platinum PCR SuperMix (Life Technologies) and universal 16S rRNA gene primers F515 (5′-GTGCCAGCMGCCGCGGTAA-3′) and R806 (5′-GGACTACHVGGGTWTCTAAT-3′). Both primers were modified with sample-specific barcode sequences and Illumina sequencing adapters according to Kozich et al. 2013 (42). Primers were added to the reaction mix at a final concentration of 0.2 μM. Ten micrograms of bovine serum albumin (BSA; New England BioLabs Inc.) was added as a PCR adjuvant. The PCR cycling conditions were an initial denaturation at 94°C for 3 min, followed by 30 cycles of a 45-s denaturation step (94°C), 45-s primer annealing step (55°C), and 90-s extension step (72°C), with a final extension step of 10 min at 72°C. The PCR products were run on a 1% agarose-Tris-acetate-EDTA (TAE) gel to verify amplicon size and the absence of contamination. The products were purified using Diffinity RapidTips (Sigma-Aldrich) and quantified using the Qubit 2.0 fluorometer. Equimolar concentrations of each amplicon were pooled, mixed with 10% PhiX DNA to increase template diversity, and sequenced using a 500-cycle paired-end MiSeq reagent V2 kit on an Illumina MiSeq sequencer.
After demultiplexing, barcoded sequences were trimmed and filtered using Trim Galore! (http://www.bioinformatics.babraham.ac.uk/projects/trim_galore/; Phred score, >25; minimum sequence length, >100 nucleotides [nt]). Paired-end sequences were merged using FLASH with the criteria of an average read length of >250 nt, fragment length of >300 nt, and fragment standard deviation of <30 nt (43). Chimeric sequences were detected and removed from the data set using USEARCH v6.1 (44). Operational taxonomic units (OTUs) were identified by clustering at 97% sequence similarity using the UCLUST algorithm in QIIME (44, 45). Taxonomy was assigned to OTUs by comparisons to the SILVA database v7.2. Singletons and OTUs affiliated with chloroplast or mitochondrial sequences were removed from further analysis. OTU counts were standardized to a uniform sequence depth (n = 14,661), and alpha diversity was calculated using Shannon's diversity index. To compare community compositions between samples, sequences were aligned in QIIME using PyNAST, and beta diversity was calculated using the weighted UniFrac metric (46).
Sulfurimonas phylogenetics.
A phylogenetic analysis was used to characterize the diversity of 16S rRNA gene OTUs classified to the genus Sulfurimonas, which was highly abundant in our samples. The analysis included the top 20 most abundant Sulfurimonas OTUs recovered in the data set, as well as (i) reference Sulfurimonas sequences retrieved from the Ribosomal Database Project (RDP), (ii) sequences identified as the top two matches when querying (BLASTN) each of the top 20 OTUs against the NCBI nonredundant (nr) database, and (iii) RDP sequences classified as Campylobacter species for use as an outgroup. The sequences (n = 73) were aligned in MEGA7 (alignment length, 256 nt) using MUSCLE (47), followed by manual curation. A phylogenetic tree was constructed in MEGA7 using maximum likelihood with the Kimura 2 model and complete gap deletion (48, 49). The tree was bootstrapped over 1,000 iterations and visualized in FigTree.
Metagenome sequencing and analysis.
Four sulfur prill DNA samples—two from each pad from the 18 May 2016 sampling—were used for the metagenomic analysis. DNA libraries were constructed using the Nextera XT DNA sample prep kit (Illumina) per the manufacturer's instructions. The resultant libraries were verified on the 2100 Bioanalyzer (Agilent) and sequenced on an Illumina MiSeq instrument using a 600-cycle paired-end MiSeq reagent V3 kit. Barcoded sequences were demultiplexed, trimmed, and quality controlled as described above. The quality trimmed forward and reverse sequences were assembled into contigs using SPAdes 3.7.0 with the “meta” tag enforced (50). Contigs of ≥1,000 bp were organized into metagenome-assembled genomes (MAGs) on the basis of tetranucleotide frequency and sequence coverage using MaxBin2.0 (51). MAG completeness and contamination were determined on the basis of the representation of lineage-specific marker gene sets using the program CheckM (52).
The taxonomic affiliation of MAGs was estimated using MiGA (http://enve-omics.ce.gatech.edu:3000/) by comparing MAG contigs against the NCBI Prok database. For those MAGs with an estimated completeness of ≥80% (n = 18), a phylogenetic tree was constructed on the basis of 107 concatenated single-copy marker genes. The tree also contained (i) the top two database genomes most similar to each MAG, as identified in MiGA on the basis of amino acid identity, (ii) other genomes spanning diverse bacterial phyla, and (iii) two Thaumarchaeota genomes for use as the outgroups. For all selected genomes (n = 101), marker genes were identified using hidden Markov models (HMMs) via HMMER3 with default settings (53). The identified marker genes were aligned using clustalW and then concatenated using the alignment tool “Aln.cat.rb” in the enve-omics package (54, 55). The resulting alignment was manually verified and used to generate a maximum likelihood phylogeny inferred with the Dayhoff substitution model and iterated 100 times for bootstrapping (49, 56).
For each MAG with ≥40% completeness and ≤10% contamination (n = 23), open reading frames (ORFs) were identified using Prodigal (57). ORFs were annotated by BLASTP queries against the NCBI nr database (June 2017) using the program DIAMOND, with match thresholds of amino acid identity of ≥40% for ≥70% of the alignment and a bit score of ≥50 (58). A secondary level of annotation was performed with the KEGG automatic annotation server (KAAS) using a bidirectional BLAST best hit against the “for prokaryotes” database (59).
Metatranscriptome sequencing and analysis.
Eight RNA samples from the 18 May 2016 sampling—two sulfur prill samples from each pad, one water sample from each pad, and two aragonite prill samples from pad 1 (aragonite was not present in pad 2)—were selected for metatranscriptomic analysis. Metatranscriptome samples were collected from the same towers sampled for the metagenomic analysis. rRNA was removed from RNA extracts (described above) using the Ribo-Zero rRNA removal kit (Illumina) and purified using the RNA MinElute cleanup kit according to the manufacturer's instructions. Sequencing libraries were constructed using the ScriptSeq v2 RNA-Seq library preparation kit (Illumina). The resultant libraries were verified on the 2100 Bioanalyzer (Agilent) and sequenced on an Illumina MiSeq instrument using a 600-cycle paired-end MiSeq reagent V3 kit. Barcoded sequences were demultiplexed, trimmed, and quality controlled as described above. Any remaining rRNA sequences were removed from the metatranscriptomic data sets using the riboPicker program (60).
The metatranscriptomic data sets were then rarefied to a uniform number of non-rRNA reads (185, 000) on the basis of the sample with the lowest read count. The normalized data sets were queried against the NCBI nr database (June 2017) using the BLASTX command in DIAMOND with the following parameters: f, 6 (output format, m8); k, 10 (retain top 10 hits); minimum score, 50 (bit score, ≥50); id, 70 (percent identification [ID] ≥ 70); sensitive. The resulting accession numbers were converted into GeneInfo identifier (GI) numbers using the biological DataBase network (bioDBnet) server (https://biodbnet-abcc.ncifcrf.gov/db/db2dbRes.php). The GI-containing DIAMOND output was imported in MEGAN5 in order to use a KEGG analysis to identify marker genes of sulfur oxidation and denitrification. The taxonomic breadth of the marker gene transcript pools was evaluated using the LCA algorithm in MEGAN5 to assign taxonomy to each marker gene fragment.
The rarefied metatranscriptome data sets were mapped to MAGs with ≥40 completeness and ≤10% contamination. Each MAG was converted to a DIAMOND database, and metatranscriptomes were queried with each database using the BLASTX command in DIAMOND with the following parameters: f, 6 (output format, m8); k, 1 (retain top hit); minimum score, 50 (bit score, ≥50); id, 95 (percent ID ≥ 95); sensitive.
Accession number(s).
All sequence data generated in this study have been deposited in the NCBI Sequence Read Archive under BioProject PRJNA397990.
Supplementary Material
ACKNOWLEDGMENTS
The authors of the manuscript would like to thank the following life support systems (LSS), water quality (WQ), and animal care facility (ACF) team members at Georgia Aquarium Inc. for efforts and contributions above and beyond on the Ocean Voyager sulfur autotrophic denitrification system project. Specific thanks are extended to John Masson (LSS), Steve Rusk (ACF), Robert Frazier (LSS), Alan Grosse (LSS), Barrett Rhoades (ACF), Calvin Phillips (LSS), Christopher Kyle Sears (LSS), Steven Keuper (LSS), Matthew O'Grady (LSS), Joseph Bayliss (LSS), Heather Crish (LSS), Angel Jungwirth (LSS), Shannon Wood (LSS), Parker Ivey (LSS), Rivers Ludden (LSS), Corey Groom (LSS), Brian Chesnut (LSS), and Myles Stevens (LSS), as well as other members of the LSS technician group at Georgia Aquarium past and present for their efforts and contributions in assisting with the design, construction, operation, and maintenance of the system. We also thank Susan Walsh (WQ), Lea Merriwether (WQ), Harry Webb (WQ), Cynthia Hartter (WQ), Rocky Ienna (WQ), Morgan Kendall (WQ), and Audrey McQuagge, as well as other members of the water quality laboratory technician and analyst group at Georgia Aquarium past and present for their efforts and contributions in performing daily water quality testing and water manipulation, as well as data interpretation and analysis that lead to critical operational adjustments of the system. Without the insight, expertise, effort, and knowledge of these individuals and groups, the manuscript would not be possible.
This work was supported by the Simons Foundation (award 346253 to F.J.S.), the NSF Advances in Bioinformatics Program (award 1564559 to F.J.S.), the NSF Biological oceanography program (award 1151698 to F.J.S.), and the Teasley Endowment to the Georgia Institute of Technology.
The authors declare no financial, commercial, or personal conflict of interest involving publication of this work.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AEM.01250-18.
REFERENCES
- 1.Philippot L. 2002. Denitrifying genes in bacterial and Archaeal genomes. Biochim Biophys Acta 1577:355–376. doi: 10.1016/S0167-4781(02)00420-7. [DOI] [PubMed] [Google Scholar]
- 2.Simon J, Klotz MG. 2013. Diversity and evolution of bioenergetic systems involved in microbial nitrogen compound transformations. Biochim Biophys Acta 1827:114–135. doi: 10.1016/j.bbabio.2012.07.005. [DOI] [PubMed] [Google Scholar]
- 3.Graf DRH, Jones CM, Hallin S. 2014. Intergenomic comparisons highlight modularity of the denitrification pathway and underpin the importance of community structure for N2O Emissions. PLoS One 9:e114118. doi: 10.1371/journal.pone.0114118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Davidson J, Good C, Welsh C, Summerfelt ST. 2014. Comparing the effects of high vs. low nitrate on the health, performance, and welfare of juvenile rainbow trout Oncorhynchus mykiss within water recirculating aquaculture systems. Aquac Eng 59:30–40. doi: 10.1016/j.aquaeng.2014.01.003. [DOI] [Google Scholar]
- 5.Hamlin HJ. 2006. Nitrate toxicity in Siberian sturgeon (Acipenser baeri). Aquaculture 253:688–693. doi: 10.1016/j.aquaculture.2005.08.025. [DOI] [Google Scholar]
- 6.van Bussel CGJ, Schroder JP, Wuertz S, Schulz C. 2012. The chronic effect of nitrate on production performance and health status of juvenile turbot (Psetta maxima). Aquaculture 326–329:163–167. doi: 10.1016/j.aquaculture.2011.11.019. [DOI] [Google Scholar]
- 7.Camargo JA, Alonso A, Salamanca A. 2005. Nitrate toxicity to aquatic animals: a review with new data for freshwater invertebrates. Chemosphere 58:1255–1267. doi: 10.1016/j.chemosphere.2004.10.044. [DOI] [PubMed] [Google Scholar]
- 8.Shao M-F, Zhang T, Fang HH-P. 2010. Sulfur-driven autotrophic denitrification: diversity, biochemistry, and engineering applications. Appl Microbiol Biotechnol 88:1027–1042. doi: 10.1007/s00253-010-2847-1. [DOI] [PubMed] [Google Scholar]
- 9.Christianson L, Lepine C, Tsukuda S, Saito K, Summerfelt S. 2015. Nitrate removal effectiveness of fluidized sulfur-based autotrophic denitrification biofilters for recirculating aquaculture systems. Aquac Eng 68:10–18. doi: 10.1016/j.aquaeng.2015.07.002. [DOI] [Google Scholar]
- 10.Sher Y, Schneider K, Schwermer CU, van Rijn J. 2008. Sulfide-induced nitrate reduction in the sludge of an anaerobic digester of a zero-discharge recirculating mariculture system. Water Res 42:4386–4392. doi: 10.1016/j.watres.2008.07.031. [DOI] [PubMed] [Google Scholar]
- 11.Lampe DG, Zhange T. 1996. Evaluation of sulfur-based autotrophic denitrification. Proceedings of the HSRC/WERC Joint Conference on the Environment, Great Plains/Rocky Mountain Hazardous Substance Research Center, Albuquerque, NM. [Google Scholar]
- 12.Sahinkaya E, Kilic A, Duygulu B. 2014. Pilot and full scale applications of sulfur-based autotrophic denitrification process for nitrate removal from activated sludge process effluent. Water Res 60:210–217. doi: 10.1016/j.watres.2014.04.052. [DOI] [PubMed] [Google Scholar]
- 13.An S, Tang K, Nemati M. 2010. Simultaneous biodesulphurization and denitrification using an oil reservoir microbial culture: effects of sulphide loading rate and sulphide to nitrate loading ratio. Water Res 44:1531–1541. doi: 10.1016/j.watres.2009.10.037. [DOI] [PubMed] [Google Scholar]
- 14.Sun Y, Nemati M. 2012. Evaluation of sulfur-based autotrophic denitrification and denitritation for biological removal of nitrate and nitrite from contaminated waters. Bioresour Technol 114:207–216. doi: 10.1016/j.biortech.2012.03.061. [DOI] [PubMed] [Google Scholar]
- 15.Tang K, An S, Nemati M. 2010. Evaluation of autotrophic and heterotrophic processes in biofilm reactors used for removal of sulphide, nitrate and COD. Bioresour Technol 101:8109–8118. doi: 10.1016/j.biortech.2010.06.037. [DOI] [PubMed] [Google Scholar]
- 16.Claus G, Kutzner HJ. 1985. Physiology and kinetics of autotrophic denitrification by Thiobacillus denitrificans. Appl Microbiol Biotechnol 22:283–288. doi: 10.1007/BF00252031. [DOI] [Google Scholar]
- 17.Frey C, Hietanen S, Jürgens K, Labrenz M, Voss M. 2014. N and O isotope fractionation in nitrate during chemolithoautotrophic denitrification by Sulfurimonas gotlandica. Environ Sci Technol 48:13229–13237. doi: 10.1021/es503456g. [DOI] [PubMed] [Google Scholar]
- 18.Gadekar S, Nemati M, Hill GA. 2006. Batch and continuous biooxidation of sulphide by Thiomicrospira sp. CVO: reaction kinetics and stoichiometry. Water Res 40:2436–2446. doi: 10.1016/j.watres.2006.04.007. [DOI] [PubMed] [Google Scholar]
- 19.Grote J, Schott T, Bruckner CG, Glöckner FO, Jost G, Teeling H, Labrenz M, Jürgens K. 2012. Genome and physiology of a model epsilonproteobacterium responsible for sulfide detoxification in marine oxygen depletion zones. Proc Natl Acad Sci U S A 109:506–510. doi: 10.1073/pnas.1111262109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Han Y, Perner M. 2015. The globally widespread genus Sulfurimonas: versatile energy metabolisms and adaptation to redox clines. Front Microbiol 6:989. doi: 10.3389/fmicb.2015.00989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Villahermosa D, Corzo A, González JM, Portillo MC, García-Robledo E, Papaspyrou S. 2013. Reduction of net sulfide production rate of nitrate in wastewater bioreactors. Kinetics and changes in the microbial community. Water Air Soil Pollut 224:1738. doi: 10.1007/s11270-013-1738-3. [DOI] [Google Scholar]
- 22.Lee HW, Park YK, Choi E, Lee JW. 2008. Bacterial community and biological nitrate removal: comparisons of autotrophic and heterotrophic reactors for denitrifications with raw sewage. J Microbiol Biotechnol 18:1826–1835. [DOI] [PubMed] [Google Scholar]
- 23.Zhou W, Li Y, Liu X, He S, Huang JC. 2017. Comparison of microbial communities in different sulfur-based autotrophic denitrification reactors. Appl Microbiol Biotechnol 101:447–453. doi: 10.1007/s00253-016-7912-y. [DOI] [PubMed] [Google Scholar]
- 24.Delbeek JC, Sprung J. 2005. The reef aquarium, vol 3. Science, art, and technology. Two Little Fishies, Inc., d.b.a. Ricordea Publishing, Miami Gardens, FL. [Google Scholar]
- 25.Tremblay-Gratton A, Boussin J-C, Bennachi A, Tamigneaux É, Vandenberg G, Le François NR. 2017. Implementation of sulphur dentrification in a large-scale fully recirculated cold-SW aquarium: a sustainability practice. J Zoo Aquar Res 5:104–108. doi: 10.19227/jzar.v5i2.288. [DOI] [Google Scholar]
- 26.Pjevac P, Kamyshny A Jr, Dyksma S, Mußmann M. 2014. Microbial consumption of zero-valence sulfur in marine benthic habitats. Environ Microbiol 16:3416–3430. doi: 10.1111/1462-2920.12410. [DOI] [PubMed] [Google Scholar]
- 27.Haiko J, Westerlund-Wikström B. 2013. The role of the bacterial flagellum in adhesion and virulence. Biology 2:1242–1267. doi: 10.3390/biology2041242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sorokin DY, Tourova TP, Lysenko AM, Muyzer G. 2006. Diversity of culturable halophilic sulfur-oxidizing bacteria in hypersaline habitats. Microbiology 152:3013–3023. doi: 10.1099/mic.0.29106-0. [DOI] [PubMed] [Google Scholar]
- 29.Sorokin DY, Tourova TP, Kolganova TV, Sjollema KA, Kuenen JG. 2002. Thioalkalispira microaerophila gen. nov., sp. nov., a novel lithoautotrophic, sulfur-oxidizng bacterium from a soda lake. Int J Syst Evol Microbiol 52:2175–2182. doi: 10.1099/00207713-52-6-2175. [DOI] [PubMed] [Google Scholar]
- 30.Sorokin DY, Tourova TP, Sjollema KA, Kuenen JG. 2003. Thialkalivibrio nitratireducens sp. nov., a nitrate-reducing member of an autotrophic denitrifying consortium from a soda lake. Int J Syst Evol Microbiol 53:1779–1783. doi: 10.1099/ijs.0.02615-0. [DOI] [PubMed] [Google Scholar]
- 31.Sorokin DY, Tourova TP, Antipov AN, Muyzer G, Kuenen JG. 2004. Anaerobic growth of the haloalkaliphilic denitrifying sulfur-oxidizing bacterium Thialkalivibrio thiocyanodenitrificans sp. nov. with thiocyanate. Microbiology 150:2435–2442. doi: 10.1099/mic.0.27015-0. [DOI] [PubMed] [Google Scholar]
- 32.Trubitsyn IV, Belousova EV, Tutukina MN, Merkel AY, Dubinina GA, Grabovich MY. 2014. Expansion of ability of denitrification within the filamentous colorless sulfur bacteria of the genus Thiothrix. FEMS Microbiol Lett 358:72–80. doi: 10.1111/1574-6968.12548. [DOI] [PubMed] [Google Scholar]
- 33.Hutt LP, Huntemann M, Clum A, Pillay M, Palaniappan K, Varghese N, Mikhailova N, Stamatis D, Reddy T, Daum C, Shapiro N, Ivanova N, Kyrpides N, Woyke T, Boden R. 2017. Permanent draft genome of Thiobacillus thioparus DSM 505T, an obligately chemolithoautotrophic member of the Betaproteobacteria. Stand Genomic Sci 12:10. doi: 10.1186/s40793-017-0229-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Labrenz M, Grote J, Mammitzsch K, Boschker HTS, Laue M, Jost G, Glaubitz S, Jürgens K. 2013. Sulfurimonas gotlandica sp. nov., a chemoautotrophic and psychrotolerant epsilonproteobacterium isolated from a pelagic redoxcline, and an emended description of the genus Sulfurimonas. Int J Syst Evol Microbiol 63:4141–4148. doi: 10.1099/ijs.0.048827-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sievert SM, Scott KM, Klotz MG, Chain PSG, Hauser LJ, Hemp J, Hugler M, Land M, Lapidus A, Larimer FW, Lucas S, Malfatti SA, Meyer F, Paulsen IT, Ren Q, Simon J, USF Genomics Class. 2008. Genome of the epsilonproteobacterial chemolithoautotroph Sulfurimonas denitrificans. Appl Environ Microbiol 74:1145–1156. doi: 10.1128/AEM.01844-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ghosh W, Dam B. 2009. Biochemistry and molecular biology of lithotrophic sulfur oxidation by taxonomically and ecologically diverse bacteria and archaea. FEMS Microbiol Rev 33:999–1043. doi: 10.1111/j.1574-6976.2009.00187.x. [DOI] [PubMed] [Google Scholar]
- 37.Flood BE, Jones DS, Bailey JV. 2015. Sedimenticola thiotaurini sp. nov., a sulfur-oxidizing bacterium isolated from salt marsh sediments, and emended descriptions of the genus Sedimenticola and Sedimenticola selenatireducens. Int J Syst Evol Microbiol 65:2522–2530. doi: 10.1099/ijs.0.000295. [DOI] [PubMed] [Google Scholar]
- 38.Koike I, Hattori A. 1975. Energy yield of denitrification: an estimate from growth yield in continuous cultures of Pseudomonas denitrificans under nitrate-, nitrite-, and nitrous oxide-limited conditions. J Gen Microbiol 88:11–19. doi: 10.1099/00221287-88-1-11. [DOI] [PubMed] [Google Scholar]
- 39.Murdock SA, Juniper SK. 2017. Capturing compositional variation in denitrifying communities: a multiple-primer approach that includes Epsilonproteobacteria. Appl Environ Microbiol 83:e02753-16. doi: 10.1128/AEM.02753-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lycus P, Bøthun KL, Berguast L, Shapleigh JP, Bakken LR, Frostegård Å. 2017. Phenotypic and genotypic richness of denitrifiers revealed by a novel isolation strategy. ISME J 11:2219–2232. doi: 10.1038/ismej.2017.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Roco CA, Bergaust LL, Bakken LR, Yavitt JB, Shapleigh JP. 2017. Modularity of nitrogen-oxide reducing soil bacteria: linking phenotype to genotype. Environ Microbiol 19:2507–2519. doi: 10.1111/1462-2920.13250. [DOI] [PubMed] [Google Scholar]
- 42.Kozich JJ, Westcott SL, Baxter NT, Highlander SK, Schloss PD. 2013. Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform. Appl Environ Microbiol 79:5112–5120. doi: 10.1128/AEM.01043-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Magoč T, Salzberg SL. 2011. FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics 27:2957–2963. doi: 10.1093/bioinformatics/btr507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Edgar RC. 2010. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26:2460–2461. doi: 10.1093/bioinformatics/btq461. [DOI] [PubMed] [Google Scholar]
- 45.Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Peña AG, Goodrich JK, Gordon JI, Huttley GA, Kelley ST, Knights D, Koenig JE, Ley RE, Lozupone CA, McDonald D, Muegge BD, Pirrung M, Reeder J, Sevinsky JR, Turnbaugh PJ, Walters WA, Widmann J, Yatsunenko T, Zaneveld J, Knight R. 2010. QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7:335–336. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lozupone C, Knight R. 2005. UniFrac: a new phylogenetic method for comparing microbial communities. Appl Environ Microbiol 71:8228–8235. doi: 10.1128/AEM.71.12.8228-8235.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Edgar RC. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kimura M. 1980. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J Mol Evol 16:111–120. doi: 10.1007/BF01731581. [DOI] [PubMed] [Google Scholar]
- 49.Kumar S, Stecher G, Tamura K. 2016. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol 33:1870–1874. doi: 10.1093/molbev/msw054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nurk S, Bankevich A, Antipov D, Gurevich AA, Korobeynikov A, Lapidus A, Prjibelski AD, Pyshkin A, Sirotkin A, Sirotkin Y, Stepanauskas R, Clingenpeel SR, Woyke T, McIean JS, Lasken R, Tesler G, Alekseyev MA, Pevzner PA. 2013. Assembling single-cell genomes and mini-metagenomes from chimeric MDA products. J Comput Biol 20:714–737. doi: 10.1089/cmb.2013.0084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wu Y-W, Simmons BA, Singer SW. 2016. MaxBin 2.0: an automated binning algorithm to recover genomes from multiple metagenomic datasets. Bioinformatics 32:605–607. doi: 10.1093/bioinformatics/btv638. [DOI] [PubMed] [Google Scholar]
- 52.Parks DH, Imelfort M, Skennerton CT, Hugenholtz P, Tyson GW. 2015. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res 25:1043–1055. doi: 10.1101/gr.186072.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Finn RD, Clements J, Eddy SR. 2011. HMMER web server: interactive sequence similarity searching. Nucleic Acids Res 39:W29–W37. doi: 10.1093/nar/gkr367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rodriguez-R LM, Konstantinidis KT. 2016. The enveomics collection: a toolbox for specialized analyses of microbial genomes and metagenomes. PeerJ Prepr 4:e1900v1. [Google Scholar]
- 55.Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Söding J, Thompson JD, Higgings DD. 2011. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol 7:539. doi: 10.1038/msb.2011.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dayhoff MO, Schwartz RM, Orcutt BC. 1978. A model for evolutionary change in proteins, p 345–352. In Dayhoff MO. (ed), Atlas of protein sequences and structure, vol 5 National Biomedical Research Foundation, Washington, DC. [Google Scholar]
- 57.Hyatt D, Chen G-L, LoCascio PF, Land ML, Larimer FW, Hauser LJ. 2010. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 11:119. doi: 10.1186/1471-2105-11-119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Buchfink B, Xie C, Huson DH. 2015. Fast and sensitive protein alignment using DIAMOND. Nat Methods 12:59–60. doi: 10.1038/nmeth.3176. [DOI] [PubMed] [Google Scholar]
- 59.Moriya Y, Itoh M, Okuda S, Yoshizawa AC, Kanehisa M. 2007. KAAS: an automatic genome annotation and pathway reconstruction server. Nucleic Acids Res 35:W182–W185. doi: 10.1093/nar/gkm321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Schmieder R, Lim YW, Edwards R. 2012. Identification and removal of ribosomal RNA sequences from metatranscriptomes. Bioinformatics 28:433–435. doi: 10.1093/bioinformatics/btr669. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.