

Abstract
The infectious template-mediated protein conversion is a unique mechanism for the onset of rare and fatal neurodegenerative disorders known as transmissible spongiform encephalopathies, or prion diseases, which affect humans and other animal species. However, emerging studies are now demonstrating prion-like mechanisms of self-propagation of protein misfolding in a number of common, non-infectious neurodegenerative diseases such as Alzheimer's disease and Parkinson's disease. It has been proposed that distinct and unrelated proteins (beta-amyloid, tau, α-synuclein, TAR DNA-binding protein 43 and huntingtin, etc.) associated with common neurodegenerative disorders can seed conversion and spread via cell-to-cell transfer, sustaining the transmission of neurotoxic agents along a stereotypic route, sharing features at the heart of the intrinsic nature of prions. Here we review the most recent development on both the molecular mechanisms underlying the pathogenesis of prion-like neurodegenerative diseases as well as innovative methods and strategies for potential therapeutic applications.
Keywords: prion-like, synuclein, tau, TAR DNA-binding protein 43, beta-amyloid, Parkinson's disease, frontotemporal dementia, amyotrophic lateral sclerosis, Alzheimer's disease, neurodegeneration
Propagation of Tau, α-Synuclein (α-syn) and TAR DNA-Binding Protein 43 (TDP-43) in Neurodegenerative Disorders
Major neurodegenerative diseases, such as Alzheimer's disease (AD), Parkinson's disease (PD) and dementia with Lewy bodies (DLB), frontotemporal dementia (FTD) and amyotrophic lateral sclerosis (ALS) share the defining features of neuronal and/or glial proteinaceous aggregations. Another common feature is that the pathological proteins assemble at initiation sites that direct the spreading to other locations of the brain following a relatively predictable sequence unique to the principle protein. AD-related neurofibrillary tau tangles is initially detectable in the transentorhinal and entorhinal regions (Braak et al., 2006). Lesions extend into the limbic allocortex and adjoining neocortex at more advanced stages. In Lewy body disease including PD or DLB, α-syn pathology is detected in dorsal motor nucleus in medulla and anterior olfactory nucleus at an early stage (Braak et al., 2003), thereafter in amygdala, cholinergic nuclei of the basal forebrain and substantia nigra at later stages, or further spread from the temporal lobe to the frontal and parietal lobes in DLB. Similarly, TDP‐43 pathology in ALS likely disseminates in a sequential pattern that permits recognition of four neuropathological stages (Brettschneider et al., 2013). In the last ten years, a growing body of evidence strongly supports the idea that the progression of these major neurodegenerative diseases is the consequence of prion-like propagation of amyloid-like protein pathologies (Herrera et al., 2011; Kurowska et al., 2011; Jaunmuktane et al., 2015; Smethurst et al., 2016; Duyckaerts et al., 2018). Experiments performed in vitro, in cultured cells and most convincingly in animal models have collectively shown that proteins including tau, α-syn and TDP-43 that aggregate and deposit in disparate neurodegenerative disorders share with prions the molecular characters of nucleation, templating, misfolding, multiplicating and spreading. Here we will not discuss the supporting evidence in detail again as they have been extensively reviewed recently (Scheckel and Aguzzi, 2018), but rather focus on the outstanding questions regarding how prion-like transfer is achieved and whether it could be targeted for therapeutic purpose. As the spreading and distributions of the proteins aggregates are closely associated with disease deterioration, understanding the mechanisms of interaction and propagation of these proteins will be vital to elucidate the fundamental pathogenesis and more importantly, to develop effective therapeutic targets to thwart the progression.
Possible Mechanisms for Prion-Like Spreading
By now, several possible mechanisms have been postulated for intercellular transmission. The traditional view is that misfolded proteins may be transmitted via binding to synaptic vesicles and released into the synaptic cleft, thereafter be taken up by the postsynaptic neuron. Tunneling nanotubes (TNTs) has emerged as a fast and direct route for prion-like aggregate transfer for long-range communication between otherwise unconnected cells (Rustom et al., 2004). TNTs are membranous tubes supported by actin polymerization that can extend up to 100 μm in length, creating a long-range transient communication between otherwise unconnected cells. Interestingly, TNT formation can be stimulated by intracellular buildup of prion-like aggregates, suggesting TNTs likely represent a self-protective function adopted by the neuron to rid itself of toxic protein aggregates and damaged organelles such as mitochondria and lysosomes. Unfortunately, it may simply provide a free highway for prion-like proteins to spread from one cell to another. Perhaps the most exciting development came from studies on the exocytosis/endocytosis pathways and the recent identification of receptors on cell surface for tau and α-syn to bind and enter cells (Rustom et al., 2004; Dujardin et al., 2014). Although there remains a fundamental question regarding how tau, α-syn, and TDP-43 are translocated into the endoplasmic reticulum (ER) vesicles as they do not contain apparent transmembrane domains or lipid anchors, their secretion into the extracellular space may be mediated by exosomes, intraluminal aggregate-containing vesicles within multivesicular bodies (MVBs) that can be released through fusion of MVBs with the plasma membrane (Asai et al., 2015). However, exosome-mediated secretion is likely only responsible for a small fraction of the released protein as two studies have demonstrated that the majority of the extracellular aggregates is membrane-free and binds directly to transmembrane receptor (Holmes et al., 2013; Mao et al., 2016). Holmes et al. (2013) found that tau fibril uptake occurs via heparan sulfate proteoglycan (HSPG) binding. HSPGs are transmembrane and lipid-anchored cell surface receptors that are extensively sulfated, allowing electrostatic interactions between the sugar polymers and short positively charged lysine or arginine stretches in heparin-binding domains of ligands. Prion-like proteins such as β-amyloid, tau, and α-syn all have putative heparin-binding domains. Importantly, tau aggregate binding, uptake, and seeding of intracellular aggregation could be potently blocked in cultured cells and primary neurons with multiple compounds specific to HSPG pathway: heparinase III, an enzyme that degrades cell surface HSPGs; heparin, a glycosaminoglycan that competitively inhibits tau binding to HSPGs; and sodium chlorate, a metabolic inhibitor of sulfation. Furthermore, genetic knockdown of a key HSPG synthetic enzyme, Ext1, effectively inhibited the internalization of tau aggregates. In a more recent study, Mao et al. (2016) screened a library encoding transmembrane proteins for α-syn pre-formed fibrils (PFF) binding candidates and identified lymphocyte-activation gene 3 (LAG3) with the highest ratio of selectivity for α-syn PFF over monomer. LAG3 colocalizes with the early endosomal marker Rab5 GTPase, confirming the endocytosis of α-syn PFF into endosomes. Importantly, neuron-to-neuron transmission of α-syn PFF, phosphorylation of α-syn at serine 129, synaptosomal-associated protein 25 (SNAP25) and other synaptic protein loss and accompanying neurotoxicity are substantially attenuated by deletion of LAG3. Remarkably, LAG3−/− mice had substantially delayed α-syn PFF induced loss of dopamine neurons, as well as significantly reduced motor deficits compared to the wildtype mice. Several import insights can be gained from these seminal studies. First, it is the cellular internalization of tau or α-syn fibrils, but not monomer, which is mediated by the cell surface receptors. Indeed, both HSPGs and LAG3 have a very low binding affinity to monomeric protein. Secondly, these receptors have relatively specific ligands. Tau and β-amyloid fibrils do not bind LAG3, indicating that LAG3 is specific for α-syn PFF. Likewise, HSPGs mediate internalization of tau and α-syn, but not Huntingtin which does not contain a heparin-binding domain. We thus exploit the genetic advantages of Drosophila model to investigate TDP-43 aggregate spreading. We used GMR-Gal4 to drive human TDP-43 A315T mutant protein expression in the fruit fly's optic lobe (Figure 1A). By day 15 after eclosion, TDP-43 A315T aggregates had spread beyond the photoreceptor neurons expressing red fluorescent protein (RFP) (unpublished data). By day 30 the spreading was even more diffuse, with aggregates present throughout the entire brain (Figure 1B). The absence of a clear heparin-binding domain in TDP-43 and the lack of a LAG3 homologous gene in Drosophila melanogaster strongly suggest that TDP-43 spreading is mediated by a different surface receptor or mechanism. Thirdly, the fact that both HSPGs and LAG3 can mediate α-syn aggregate uptake and the existence of other possible binding candidates, such as APLP1 and neurexins (Mao et al., 2016), suggesting one pathologic protein aggregate may be actively translocated into cells via alternative partners. Of course, whether these alternative binding partners might interact with each other or how important the receptors with suboptimal binding associations contribute to prion-like transmission and pathogenesis require further investigation.
Figure 1.
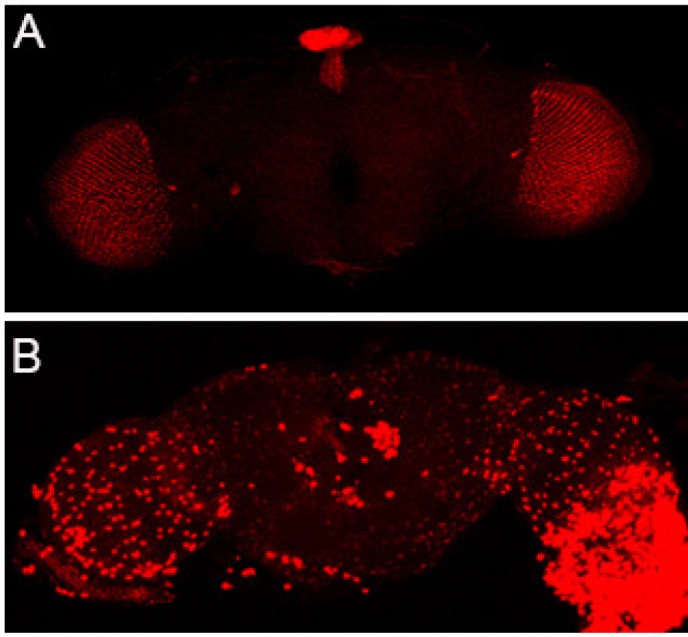
Mutant TDP-43 A315T aggregates spread throughout the Drosophila brain.
(A) The RFP marker alone remains in the optic lobe by day 30 after eclosion. (B) TDP-43-A315T-RFP fusion protein spreads throughout the brain as the fly ages. TDP-43: TAR DNA-binding protein 43; RFP: red fluorescent protein.
Therapeutically Targeting the Transmission of Pathological Proteins
Studies of prion-like transcellular spreading bear important implication on the fervently sought embryonic stem cell or genetically engineered cell transplantation trials. As PD patients who had survived long-term transplantation of fetal nigral neurons developed Lewy body pathology in grafted neurons (Kordower et al., 2008), cell transplantation therapy, if used alone, may not provide a long-term benefit for neurodegenerative proteinopathies. It may have to be coupled with agents that could block either release or uptake of misfolded proteins in the active transmission process to halt the disease progression (Figure 2). There appears to be a unifying, chaperone-dependent mechanism for the release of pathogenic proteins in human neurodegenerative diseases (Fontaine et al., 2016). Several proteins, including α-syn, tau, and TDP-43 can be released out of the cell by DnaJC5, a heat shock protein 70 (Hsc70) cochaperone. Small molecule inhibitor of Hsc70 such as YM-01 which binds to the DnaJ-binding region of Hsc70 and inhibits the allosteric inter-omain communication may hold therapeutic potential. On the uptaking side, synthetic heparin mimetics that have potent activity in blocking aggregate uptake via HSPGs without detrimental anticoagulation activity would conceivably to stop the spreading of pathology (Holmes et al., 2013). LAG3-blocking antibodies were used by Mao et al. (2016), and were able to reduce α-syn PFF toxicity and cell-to-cell transmission. Although LAG3 is enriched in brain and expressed predominantly in neurons including dopaminergic neurons, it is strongly expressed on activated T, natural killer, and B cells, where it dampens autoimmune diseases and coordinates humoral immune responses (Jucker and Heikenwalder, 2016). Because LAG3 is likely to play important parts in autoimmune or hyperimmune activation in the brain as well, drugs or antibodies specifically suppressing LAG3 call for in-depth research to minimize the risk of substantial side effects.
Figure 2.

Membrane receptor facilitates pathological protein fibrils endocytosis into neurons (left), triggering the seeding and spreading of the toxic protein. Antibodies generated against the pathologic proteins (right) may block cells from taking up the fibrils (Adapted from Mao et al., 2016).
Conclusions
Significant progress has been made over the last decade and emerging evidence supports the view that prion-like infectivity of aggregates in the nervous system may be a common theme of major neurodegenerative disorders such as AD, PD, ALS-FTD, and Huntington's disease. Understanding the mechanisms of pathologic protein spreading would be essential to develop novel diagnostic and therapeutic strategies. For future direction, a search of possible binding receptors for TDP-43 may bring significant benefits to the field. TDP-43 proteinopathy, a pathology first linked to ALS and FTD, is one of the most common non-canonical pathologies observed in AD cases and is closely associated with most chronic traumatic encephalopathy brains, suggesting a convergent mechanism of neurodegeneration (Chen, 2018). Therefore, developing antibodies or reagents to block TDP-43 spreading is likely the most cost-effective avenue to treat all the neurodegenerative disorders that shares TDP-43 proteinopathy.
Additional file: Open peer review report 1 (107.9KB, pdf) .
Acknowledgments
We thank Dr. Xiaobo Mao (Johns Hopkins University School of Medicine) for constructive comments.
Footnotes
Conflicts of interest: None declared.
Financial support: None.
Copyright license agreement: The Copyright License Agreement has been signed by all authors before publication.
Plagiarism check: Checked twice by iThenticate.
Peer review: Externally peer reviewed.
Open peer reviewer: Panlong Li, Institute of High Energy Physics, Chinese Academy of Sciences, China.
References
- 1.Asai H, Ikezu S, Tsunoda S, Medalla M, Luebke J, Haydar T, Wolozin B, Butovsky O, Kügler S, Ikezu T. Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat Neurosci. 2015;18:1584–1593. doi: 10.1038/nn.4132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Braak H, Alafuzoff I, Arzberger T, Kretzschmar H, Del Tredici K. Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol. 2006;112:389–404. doi: 10.1007/s00401-006-0127-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Braak H, Del Tredici K, Rub U, de Vos RA, Jansen Steur EN, Braak E. Staging of brain pathology related to sporadic Parkinson's disease. Neurobiol Aging. 2003;24:197–211. doi: 10.1016/s0197-4580(02)00065-9. [DOI] [PubMed] [Google Scholar]
- 4.Brettschneider J, Del Tredici K, Toledo JB, Robinson JL, Irwin DJ, Grossman M, Suh E, Van Deerlin VM, Wood EM, Baek Y, Kwong L, Lee EB, Elman L, McCluskey L, Fang L, Feldengut S, Ludolph AC, Lee VM, Braak H, Trojanowski JQ. Stages of pTDP-43 pathology in amyotrophic lateral sclerosis. Ann Neurol. 2013;74:20–38. doi: 10.1002/ana.23937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen L. What triggers tauopathy in chronic traumatic encephalopathy? Neural Regen Res. 2018;13:985–986. doi: 10.4103/1673-5374.233439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dujardin S, Bégard S, Caillierez R, Lachaud C, Delattre L, Carrier S, Loyens A, Galas MC, Bousset L, Melki R, Aurégan G, Hantraye P, Brouillet E, Buee L, Colin M. Ectosomes: a new mechanism for non-exosomal secretion of tau protein. PLoS One. 2014;9:e100760. doi: 10.1371/journal.pone.0100760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Duyckaerts C, Sazdovitch V, Ando K, Seilhean D, Privat N, Yilmaz Z, Peckeu L, Amar E, Comoy E, Maceski A, Lehmann S, Brion JP, Brandel JP, Haïk S. Neuropathology of iatrogenic Creutzfeldt-Jakob disease and immunoassay of French cadaver-sourced growth hormone batches suggest possible transmission of tauopathy and long incubation periods for the transmission of Abeta pathology. Acta Neuropathol. 2018;135:201–212. doi: 10.1007/s00401-017-1791-x. [DOI] [PubMed] [Google Scholar]
- 8.Fontaine SN, Zheng D, Sabbagh JJ, Martin MD, Chaput D, Darling A, Trotter JH, Stothert AR, Nordhues BA, Lussier A, Baker J, Shelton L, Kahn M, Blair LJ, Stevens SM, Jr, Dickey CA. DnaJ/Hsc70 chaperone complexes control the extracellular release of neurodegenerative-associated proteins. EMBO J. 2016;35:1537–1549. doi: 10.15252/embj.201593489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Herrera F, Tenreiro S, Miller-Fleming L, Outeiro TF. Visualization of cell-to-cell transmission of mutant huntingtin oligomers. PLoS Curr. 2011;3:RRN1210. doi: 10.1371/currents.RRN1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Holmes BB, DeVos SL, Kfoury N, Li M, Jacks R, Yanamandra K, Ouidja MO, Brodsky FM, Marasa J, Bagchi DP, Kotzbauer PT, Miller TM, Papy-Garcia D, Diamond MI. Heparan sulfate proteoglycans mediate internalization and propagation of specific proteopathic seeds. Proc Natl Acad Sci U S A. 2013;110:E3138–3147. doi: 10.1073/pnas.1301440110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jaunmuktane Z, Mead S, Ellis M, Wadsworth JD, Nicoll AJ, Kenny J, Launchbury F, Linehan J, Richard-Loendt A, Walker AS, Rudge P, Collinge J, Brandner S. Evidence for human transmission of amyloid-β pathology and cerebral amyloid angiopathy. Nature. 2015;525:247–250. doi: 10.1038/nature15369. [DOI] [PubMed] [Google Scholar]
- 12.Jucker M, Heikenwalder M. Immune receptor for pathogenic α-synuclein. Science. 2016;353:1498–1499. doi: 10.1126/science.aai9377. [DOI] [PubMed] [Google Scholar]
- 13.Kordower JH, Chu Y, Hauser RA, Freeman TB, Olanow CW. Lewy body-like pathology in long-term embryonic nigral transplants in Parkinson's disease. Nat Med. 2008;14:504–506. doi: 10.1038/nm1747. [DOI] [PubMed] [Google Scholar]
- 14.Kurowska Z, Englund E, Widner H, Lindvall O, Li JY, Brundin P. Signs of degeneration in 12-22-year old grafts of mesencephalic dopamine neurons in patients with Parkinson's disease. J Parkinsons Dis. 2011;1:83–92. doi: 10.3233/JPD-2011-11004. [DOI] [PubMed] [Google Scholar]
- 15.Mao X, Ou MT, Karuppagounder SS, Kam TI, Yin X, Xiong Y, Ge P, Umanah GE, Brahmachari S, Shin JH, Kang HC, Zhang J, Xu J, Chen R, Park H, Andrabi SA, Kang SU, Gonçalves RA, Liang Y, Zhang S, et al. Pathological α-synuclein transmission initiated by binding lymphocyte-activation gene 3. Science. 2016;353:aah3374. doi: 10.1126/science.aah3374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rustom A, Saffrich R, Markovic I, Walther P, Gerdes HH. Nanotubular highways for intercellular organelle transport. Science. 2004;303:1007–1010. doi: 10.1126/science.1093133. [DOI] [PubMed] [Google Scholar]
- 17.Scheckel C, Aguzzi A. Prions, prionoids and protein misfolding disorders. Nat Rev Genet. 2018;19:405–418. doi: 10.1038/s41576-018-0011-4. [DOI] [PubMed] [Google Scholar]
- 18.Smethurst P, Newcombe J, Troakes C, Simone R, Chen YR, Patani R, Sidle K. In vitro prion-like behaviour of TDP-43 in ALS. Neurobiol Dis. 2016;96:236–247. doi: 10.1016/j.nbd.2016.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.