

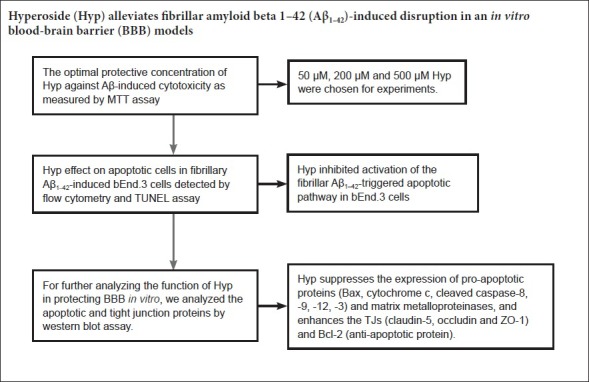
Keywords: nerve regeneration, Alzheimer's disease, amyloid beta 1–42, blood-brain barrier, bEnd.3 cells1, tight junction proteins, hyperoside, anti-apoptosis, neural regeneration
Abstract
Mounting evidence indicates that amyloid β protein (Aβ) exerts neurotoxicity by disrupting the blood-brain barrier (BBB) in Alzheimer's disease. Hyperoside has neuroprotective effects both in vitro and in vivo against Aβ. Our previous study found that hyperoside suppressed Aβ1–42-induced leakage of the BBB, however, the mechanism remains unclear. In this study, bEnd.3 cells were pretreated with 50, 200, or 500 µM hyperoside for 2 hours, and then exposed to Aβ1–42 for 24 hours. Cell viability was determined using 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2-H-tetrazolium bromide assay. Flow cytometry and terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling assay were used to analyze cell apoptosis. Western blot assay was carried out to analyze expression levels of Bax, Bcl-2, cytochrome c, caspase-3, caspse-8, caspase-9, caspase-12, occludin, claudin-5, zonula occludens-1, matrix metalloproteinase-2 (MMP-2), and MMP-9. Exposure to Aβ1–42 alone remarkably induced bEnd.3 cell apoptosis; increased ratios of cleaved caspase-9/caspase-9, Bax/Bcl-2, cleaved caspase-8/caspase-8, and cleaved caspase-12/caspase-12; increased expression of cytochrome c and activity of caspase-3; diminished levels of zonula occludens-1, claudin-5, and occludin; and increased levels of MMP-2 and MMP-9. However, hyperoside pretreatment reversed these changes in a dose-dependent manner. Our findings confirm that hyperoside alleviates fibrillar Aβ1–42-induced BBB disruption, thus offering a feasible therapeutic application in Alzheimer's disease.
Introduction
Current evidence suggests that the progressive deposition of amyloid β-protein (Aβ) in cerebral microvasculature induces blood-brain barrier (BBB) disruption in Alzheimer's disease (AD) patients (Erickson and Banks, 2013; Rosenberg, 2014; Yamazaki and Kanekiyo, 2017). The BBB is a complex barrier that plays a crucial role in protecting the cerebral parenchyma from harmful substances in peripheral circulation. Specialized endothelial cells expressing tight junction proteins (TJs), which connected endothelial cells, form the main structure of the BBB. TJs primarily secreted by endothelial cells, including occludin, claudin-5, and cytoplasmic zonula occludens-1 (ZO-1), are used as sensitive indices for structural variations in the BBB during disease pathogenesis. Normal structure of the BBB is easily damaged by endothelial cell apoptosis and malformation of TJs in many central nervous system disturbances (Bednarczyk and Lukasiuk, 2011; Chow and Gu, 2015). Several studies have shown that Aβ-induced disruption of claudin-5, occludin, and ZO-1 increased vascular permeability both in vivo and in vitro (Carrano et al., 2011; Kook et al., 2012; Wan et al., 2015).
Matrix metalloproteinases (MMPs), which are mainly secreted by endothelial cells, are a family of zinc and calcium enzymes that digest components of the extracellular matrix such as collagen, proteoglycan, and laminin. Several studies have shown that Aβ can increase MMP-2 and MMP-9 expression, and decrease claudin-5, occludin, and ZO-1, eventually leading to BBB disruption (Gonzalez-Velasquez et al., 2008; Hartz et al., 2012).
Hyperoside (Hyp), a flavone glycoside isolated from Rhododendron brachycarpum G., has demonstrated anti-oxidant, anti-inflammatory, anti-cancer, anti-depressant, and anti-apoptosis properties (Zhang et al., 2010; Zheng et al., 2012; Ku et al., 2014; Park et al., 2016). Several studies have shown that Hyp elicits a protective effect against Aβ-induced neurotoxicity (Zeng et al., 2011). Moreover, Hyp can inhibit fibrillar Aβ1–42-induced BBB damage and hyperpermeability in an in vitro co-culture model (Liu et al., 2017). However, the protective mechanism by which Hyp alleviates Aβ-induced BBB leakiness remains unclear. Notably, as the yield of primary endothelial cell isolation and culture is very low, immortalized brain endothelial cell lines, such as bEnd.3, RBE4, or hCMEC/D3 cells, are frequently used as in vitro models of the BBB. Hence, this study explored the potential mechanism of Hyp on mitigation of BBB hyperpermeability under fibrillar Aβ1–42-induced condition in bEnd.3 endothelial cells.
Materials and Methods
Preparation of Hyp and Aβ1–42 fibrils
Aβ1–42 fibrils were prepared as previously described (Jekabsone et al., 2006). Briefly, 1 mg of Aβ1–42 (China Peptides, Shanghai, China) was dissolved in 100 µL of phosphate-buffered saline (PBS) and then cultivated at 37°C for 7 days to accelerate aggregation into fibrous peptides. Hyp powder (98% purity; Chengdu ManSiTe Biotechnology, Sichuan, China) was dissolved in PBS and diluted to 8 mM.
Immortalized brain endothelial cell cultures
bEnd.3 cells were obtained from ATCC (Manassas, VA, USA) and cultured in Dulbecco's Modified Eagle's Medium (Gibco, Grand Island, NY, USA) containing 10% fetal bovine serum (Gibco), 100 U/mL penicillin, 100 mg/mL streptomycin, and 100 mg/mL amphotericin (Gibco) at 37°C in a humidified atmosphere containing 5% CO2; cells were subcultured every 2 days.
Cells were allowed to reach 90% confluence and then divided into five groups: control, 20 µM Aβ1–42, 20 µM Aβ1–42 and 50 µM Hyp, 20 µM Aβ1–42 and 200 µM Hyp, and 20 µM Aβ1–42 and 500 µM Hyp. After pretreatment with or without Hyp for 2 hours, bEnd.3 cells were cultivated with or without Aβ 20 µM for 24 hours, and then examined by different experimental approaches.
MTT assay of cell viability
3-(4,5-Dimethyl-2-thiazolyl)-2,5-diphenyl-2-H-tetrazolium bromide (MTT) assay, which is a sensitive measurement of cell metabolic status, was used to determine the optimum concentration of Hyp. Twenty microliters of MTT (Sigma-Aldrich, St. Louis, MO, USA; 5 mg/mL) was added and incubated for 4 hours under 5% CO2 at 37°C. After discarding the culture medium, formazan products were dissolved in dimethyl sulfoxide (Sigma-Aldrich). Absorbance at 490 nm was measured for each well by a Scientific Microplate Reader (Bio-Tek, Winooski, VT, USA).
Flow cytometry and TUNEL assay for apoptosis
For flow cytometry, cells were harvested and washed twice with ice-cold PBS (137 mM NaCl, 2.7 M KCl, 10 mM Na2HPO4 and 1.8 mM KH2PO4, pH 7.4), and resuspended in binding buffer. Next, cells were stained with Annexin V-fluorescein isothiocyanate (V-FITC) and propidium iodide according to the manufacturer's instructions (Miltenyi Biotec, Bergisch Gladbach, Germany). Briefly, each tube of resuspended cells was incubated with V-FITC (100:1, v:v) for 10 minutes in the dark at 4°C, and then incubated with propidium iodide (100:1, v:v) for 10 minutes in the dark at room temperature. Finally, cells were immediately examined on a FACS Calibur (BD Biosciences, Franklin Lakes, NJ, USA) flow cytometer. Data were analyzed with CellQuest software (BD Biosciences).
bEnd.3 cell apoptosis was evaluated using a terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL) assay. In addition, 4′,6-diamidino-2-phenylindole (DAPI) staining was performed for total nuclei quantification. The labeling protocol was performed according to the manufacturer's instructions (Beyotime, Shanghai, China). Briefly, bEnd.3 cells were washed in PBS and fixed with 4% neutral buffered formalin solution for 60 minutes. Cells were incubated in 0.1% Triton X-100 plus 0.1% sodium citrate in PBS at 4°C for 2 minutes and then washed three times in PBS at room temperature. Subsequently, cells were incubated for 60 minutes in 50 µL of a mixture containing 10 µL of TdT enzyme and 240 µL of equilibration buffer (Beyotime) at 37°C in a humidified chamber. After three washes in PBS at room temperature, bEnd.3 cells were counterstained with 50 µL of DAPI (Beyotime) for 5 minutes, followed by exhaustive washes in PBS. Finally, apoptosis and total nuclei were observed under an Olympus BX 50 microscope equipped with a reflected light fluorescent attachment (Olympus, Tatsuno, Japan), and 200× objective.
Western blot assay
After drug treatment, bEnd.3 cells were washed three times with ice-cold PBS and lysed in sodium dodecyl sulphate (SDS). Cell lysates containing equal amounts of protein were separated by SDS polyacrylamide gel electrophoresis and transferred to polyvinylidene difluoride membranes. After blocking in 5% nonfat milk in Tris-buffered saline with 0.1% Tween 20 (pH 7.6), membranes were incubated with appropriate primary antibodies against MMP-2 and occludin (rabbit) (Abcam, Cambridge, UK), MMP-9 (goat) (R&D, Minneapolis, MN), ZO-1 (goat) (Arigo, Taiwan, China), claudin-5 (rabbit) (Millipore, Bedford, MA), caspase-9 (mouse), Bax, Bcl-2, cytochrome c, caspase-8, caspase-12, caspase-3, and β-actin (rabbit) (Cell Signaling Technology, Shanghai, China) at 4°C overnight at a concentration of 1/1000. Afterwards, membranes were exposed to secondary antibodies (goat, rabbit, or mouse; 1:5000; Cell Signaling Technology) for 2 hours at room temperature. Immunoreactive proteins were visualized using an enhanced chemiluminescence system (Keygen, Nanjing, China). Images were analyzed with Image Lab software (Bio-Rad, Hercules, CA, USA).
Statistical analysis
All results are expressed as mean ± SEM. Statistical analysis was performed using SPSS 22.0 software (IBM Corp., Armonk, NY, USA). Comparisons were performed by one-way analysis of variance followed by Tukey's multiple comparison post hoc test. P values of less than 0.05 were considered statistically significant.
Results
Optimal protective concentration of Hyp against Aβ-induced cytotoxicity
To detect the optimal protective concentration of Hyp against Aβ-induced cytotoxicity, bEnd.3 cells were pretreated with various concentrations of Hyp (50, 100, 200, 300, 400, 500 µM) for 2 hours, and exposed to 20 µM fibrillar Aβ1–42. The results of MTT assay indicated that the protective effect of Hyp against Aβ-induced cytotoxicity occurred in a concentration-dependent manner. The difference between concentrations of 200 µM and 500 µM was statistically significant (P < 0.05; Figure 1). To explore the strength of the protective effect of different concentrations of Hyp, 50 µM, 200 µM and 500 µM Hyp were chosen for experiments.
Figure 1.
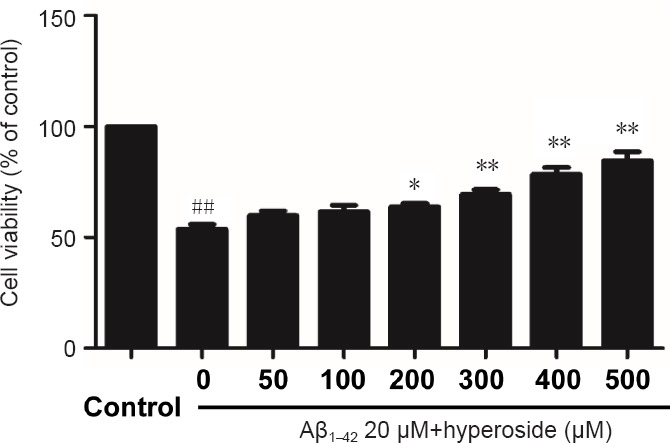
Effect of various concentrations of hyperoside on inhibiting Aβ1–42-induced damage in bEnd.3 cells.
3-(4,5-Dimethyl-2-thiazolyl)-2,5-diphenyl-2-H-tetrazolium bromide assay was used to detect the viability of bEnd.3 cells. Results were measured as optical density at 490 nm and presented as the mean ± SEM, and analyzed by one-way analysis of variance followed by Tukey's multiple comparison post hoc test. The experiment was conducted in triplicate. ##P < 0.01, vs. control group; *P < 0.05, **P < 0.01, vs. 20 µM Aβ1–42 group. Aβ1–42: Amyloid beta 1–42.
Hyp inhibited activation of the fibrillar Aβ1–42-triggered apoptotic pathway in bEnd.3 cells
Cerebral microvascular endothelial cells are vital components of the BBB. The survivability of bEnd.3 cells was crucial for the integrity of our BBB model in vitro (Abbott et al., 2010). To evaluate the effect of Hyp on Aβ1–42-induced bEnd.3 cell apoptosis, cells were pretreated with or without Hyp for 2 hours, then exposed to fibrillar Aβ1–42 for an additional 24 hours. In flow cytometry, the early apoptosis rates of fibrillar Aβ1–42-treated bEnd.3 cells, which were cultured with concentrations of Hyp from 50–500 µM, decreased from 28.5% to 9.5%, and the difference between 200 µM and 500 µM was statistically significant (P < 0.05; Figure 2). For the TUNEL assay, TUNEL-positive nuclei (representing apoptotic cells) were stained with bright green fluorescence, while total nuclei stained with DAPI and exhibited blue fluorescence (Figure 3). Compared with the control, increased intensity of green fluorescence and nuclei fragmentation were obvious after Aβ treatment. However, pretreatment with Hyp markedly reversed this phenomenon in a dose-dependent manner. Thus, the anti-apoptotic influences of Hyp on fibrillar Aβ1–42-treated bEnd.3 cells occurred in a dose-dependent manner.
Figure 2.

Protection of Hyp against Aβ1–42-induced apoptosis in bEnd.3 cells as detected by flow cytometry (FITC-V/PI) analysis.
The apoptosis rate of fibrillar Aβ1–42-treated bEnd.3 cells obviously increased, and dramatically decreased after pretreatment with concentrations of Hyp from 50 to 500 µM. The results are presented as the mean ± SEM, and analyzed by one-way analysis of variance followed by Tukey's multiple comparison post hoc test. The experiment was conducted in triplicate. ##P < 0.01, vs. control group; *P < 0.05, **P < 0.01, vs. 20 µM Aβ1–42 group. Aβ1–42: Amyloid beta 1–42; Hyp: hyperoside.
Figure 3.

Effect of Hyp on apoptotic cells in fibrillar Aβ1–42-induced bEnd.3 cells (fluorescence microscopy, 200× magnification).
Apoptotic nuclei stained by TUNEL present green fluorescence and even nuclear fragmentation (red arrows). All nuclei are stained by DAPI with dark blue fluorescence. The experiment was conducted in triplicate. Aβ1–42: Amyloid beta 1–42; Hyp: hyperoside; TUNEL: terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling; DAPI: 4′,6-diamidino-2-phenylindole.
To further analyze the anti-apoptotic mechanism of Hyp, we analyzed protein expression of Bcl-2, Bax, caspase-9, and cleaved caspase-9 by western blot assay after pretreatment with various concentrations of Hyp and 24 hours of cultivation with Aβ1–42. Ratios of cleaved caspase-9/caspase-9 and Bax/Bcl-2 rose significantly upon cultivation with Aβ1–42 (P < 0.01). Increases of these two ratios was reversed by pretreatment with various concentrations of Hyp (P < 0.05). Cytochrome c levels were significantly increased in cytosol after treatment with Aβ1–42 (P < 0.01), but this was also reversed by pretreatment with Hyp (P < 0.05). Similarly, the caspase-like activity of caspase-3 was significantly increased after 24 hours of Aβ1–42 exposure (P < 0.01). Pretreatment with Hyp also inhibited the activity of caspase-3 induced by Aβ1–42 (P < 0.05; Figure 4).
Figure 4.

Hyp prevents Aβ1–42-induced apoptosis in bEnd.3 cells via inhibiting mitochondrial, endoplasmic reticulum, and death receptor signaling pathways.
Results of western blot assay showed that ratios of Bax/Bcl-2, cleaved caspase-9/caspase-9, cleaved caspase-12/caspase-12, cleaved caspase-8/caspase-8, and cleaved caspase-3/caspase-3 were increased, while cytochrome c protein was reduced in Aβ1–42-induced apoptosis in bEnd.3 cells. Equal amount of protein loading was confirmed by actin levels. All results are represented as the mean ± SEM (one-way analysis of variance followed by Tukey's multiple comparison post hoc test). The experiment was conducted in triplicate. ##P < 0.01, vs. control group; **P < 0.01, vs. 20 µM Aβ1–42 group. Aβ1–42: Amyloid beta 1–42; Hyp: hyperoside.
Activation of caspase-8 and caspase-12 was analyzed by western blot assay. After exposing bEnd.3 cells to Aβ1–42 for 24 hours, ratios of cleaved caspase-8/caspase-8 and cleaved caspase-12/caspase-12 were significantly increased and reversed by pretreatment with Hyp (P < 0.05; Figure 4).
Hyp increased protein levels of ZO-1, claudin-5, and occludin, but decreases MMP-2 and MMP-9 in fibrillar Aβ1–42-treated bEnd.3 cells
Tight junctions, which ensure the integrity of the BBB, are the most prominent feature of endothelial cells in the central nervous system,. Occludin, claudin-5 and ZO-1 are the main TJs involved in BBB permeability (Yamazaki and Kanekiyo, 2017). MMP-2 and MMP-9, as principle members of the MMP family, are primarily responsibility for degrading TJs (Gonzalez-Velasquez et al., 2008; Hartz et al., 2012). Based on our results, we gauged the effect of Hyp on altered expression of ZO-1, claudin-5, and occludin, as well as the degrading proteins MMP-2 and MMP-9 in fibrillar Aβ1–42-treated bEnd.3 cells. Cells pretreated with or without 50–500 µM Hyp for 2 hours were exposed to 20 µM Aβ1–42 for 24 hours. Western blot assay results showed that treatment with 20 µM Aβ1–42 alone significantly decreased levels of ZO-1, claudin-5, and occludin and increased levels of MMP-2 and MMP-9 in bEnd.3 cells (P < 0.01). Levels of ZO-1, claudin-5, and occludin increased, while levels of MMP-2 and MMP-9 decreased after pretreatment with Hyp (P < 0.05). Hyp elicits a protective effect by ZO-1, claudin-5, and occludin in a concentration-dependent manner. Pretreatment with Hyp remarkably increased levels of ZO-1, claudin-5, and occludin, and decreased levels of MMP-2 and MMP-9 (Figure 5). The protective effect of Hyp on ZO-1, claudin-5, and occludin occurred in a concentration-dependent manner from 50–500 µM. In addition, the inhibitory effect of Hyp on MMP-2 and MMP-9 also occurred in a concentration-dependent manner from 50–500 µM. Taken together, Hyp mitigated BBB paracellular leakiness by decreasing MMP-2 and MMP-9, thereby reducing the degradation of TJ proteins. The protective effect of Hyp is concentration-dependent in the range of 50–500 µM.
Figure 5.

Effect of Hypeon expression of TJs and MMPs in fibrillar Aβ1–42-treated bEnd.3 cells.
Results of western blot assay showed expression of MMP-2, MMP-9 and TJs (ZO-1, occludin and claudin-5). Equal amount of protein loading was confirmed by β-actin levels. Data are represented as mean ± SEM (one-way analysis of variance followed by Tukey's multiple comparison post hoc test). The experiment was conducted in triplicate. ##P < 0.01, vs. control group; *P < 0.05, **P < 0.01, vs. 20 µM Aβ1–42 group. Aβ1–42: Amyloid beta 1–42; Hyp: hyperoside; TJs: tight junction proteins; MMP: matrix metalloproteinase; ZO-1: zonula occludens-1.
Discussion
Current evidence suggests that Aβ deposition inside the brain can result in BBB dysfunction, which will in turn increase the passage of ions, hydrophilic compounds, and macromolecules through the paracellular pathway in AD patients (Marco and Skaper, 2006; Biron et al., 2011; Erickson and Banks, 2013; Rosenberg, 2014). Our previous study found that Hyp alleviated Aβ-induced high permeability of the BBB model in vitro (Liu et al., 2017), however the molecular mechanism remains unclear. This study investigated the protective mechanism of Hyp on fibrillar Aβ1–42-induced disruption in an in vitro BBB model by analyzing cell toxicity, TJs, MMPs expression, and apoptotic signaling pathways.
Cerebral endothelial cells are the main component supporting the essential functions of the BBB. Therefore, a key point for protecting BBB integrity is protection of endothelial cells. in vitro studies demonstrated that Aβ exerts a marked toxic effect on cerebral endothelial and neuronal cells through endoplasmic reticulum (ER), death receptor, and mitochondria-dependent apoptosis pathways (Folin et al., 2006; Zhao et al., 2008; Fossati et al., 2012; Feng et al., 2015). The caspase family, such as cysteine proteases, are critical mediators of programmed cell death. First, in the mitochondria-dependent apoptosis pathway, the pro-apoptotic protein Bax facilitates release of cytochrome c inside mitochondria by increasing mitochondrial membrane permeability. Cytochrome c is also a pro-apoptotic protein that can induce cell apoptosis via activation of the caspase family. Bcl-2 acts as an anti-apoptosis protein by binding to Bax and inhibiting excessive Bax, leading to high permeability of the mitochondrial membrane. In the caspase cascade, caspase-9 and the apoptosis effector caspase-3 can be activated by cytochrome c. Moreover, activation of caspase-3, a downstream effector involved in the execution of apoptotic cascade, ultimately leads to cell apoptosis (Wang et al., 2016). Second, in the ER apoptosis pathway, Aβ induces release of Ca2+ and misfolded protein aggregation in the ER which leads to activation of the ER. On the one hand, excessive misfolded protein in the ER activates caspase-12 into cleaved caspase-12, which is a key effector in the ER apoptosis pathway. Next, caspase-9 and caspase-3 are activated sequentially via the caspase cascade reaction. On the other hand, Ca2+ overload in mitochondria resulting from excessive uptake of Ca2+ released from ER, increases the ratio of Bax/Bcl-2, activates the mitochondria-dependent apoptotic pathway, eventually inducing cell apoptosis (Kim et al., 2009; Fonseca et al., 2013). Third, caspase-8, a key effector in the death receptor apoptosis pathway, binds to specific receptors on the cell surface and is activated as cleaved caspase-8, which ultimately activates the extrinsic apoptosis pathway (Folin et al., 2006). In this study, Aβ specifically activated the three apoptosis pathways described above, as indicated by activated caspase-8, -12, -9, and -3, and induced apoptosis of bEnd.3 cells. Furthermore, Hyp reduced ratios of Bax/Bcl-2, cleaved caspase-8/caspase-8, cleaved caspase-12/caspase-12, cleaved caspase-9/caspase-9, and cleaved caspase-3/caspase-3, as well as the level of cytochrome c in Aβ1–42 fibril-induced bEnd.3 cells. These data indicated that Hyp may play an anti-apoptotic role by suppressing death receptor-, ER-, and mitochondrial-dependent apoptosis pathways. Moreover, these results demonstrate that Aβ1–42 induced apoptosis in bEnd.3 cells by mitochondria-independent, death receptor, and ER apoptosis pathways. Caspase-8 and caspase-12 served as indicators of death receptor and ER apoptosis pathways, respectively (Folin et al., 2006). In addition, mitochondria-dependent pathways may rely on cytochrome c release from mitochondria into the cytosol, an increased Bax/Bcl-2 ratio, and subsequent activation of caspase-9 and caspase-3. Hyp prevented Aβ1–42-induced bEnd.3 cell apoptosis by simultaneously restraining mitochondria-dependent, death receptor, and ER apoptosis pathways.
Intercellular TJs, the most prominent junctional complex between endothelial cells, are responsible for BBB integrity (Chen et al., 2012). TJs, including occludin, cytoplasmic ZO proteins, and claudins, separate the apical and basolateral domains, causing cell polarization and regulation of BBB permeability (Förster, 2008; Zlokovic, 2008). Among TJs, occludin, claudin-5, and ZO-1 are sensitive indicators of structural changes in the BBB during disease pathogenesis (Yang and Rosenberg, 2011; Naik et al., 2014). Losing one of these proteins may severely compromise BBB integrity and functionality, and deteriorate AD progression (Yamazaki and Kanekiyo, 2017). Some research has indicated that MMPs may be the main factor for damaging TJs (Seo et al., 2012). MMPs, a family of zinc and calcium enzymes, secreted by endothelial cells and neurons, can digest the extracellular matrix and evoke BBB damage in physiological states (Mott and Werb, 2004). Among the MMP family, MMP-2 and MMP-9 have very important effects on the pathogenesis of AD and degradation of TJs, such as claudin-5 and ZO-1, by disturbing cerebral microvessels and BBB integrity (Gonzalez-Velasquez et al., 2008; Hartz et al., 2012). Our previous studies showed that Aβ1–42 caused significant BBB leakage, but a certain concentration of Hyp could reverse this phenotype. This study explored the underlying molecular mechanism of Hyp and found that Hyp treatment upregulated ZO-1, occludin, and claudin-5, while downregulating MMP-2 and MMP-9, to protect against Aβ1–42-induced toxicity in bEnd.3 cells in vitro.
In summary, we provide fresh evidence to show that Hyp can prevent fibrillar Aβ1–42-induced brain endothelial cell damage by effectively suppressing both mitochondria-dependent death receptor and ER apoptosis pathways. Furthermore, Hyp reversed fibrillar Aβ1–42-induced downregulation of TJs and prevented fibrillar Aβ1–42-induced upregulation of MMP-2 and MMP-9. These results indicate that Hyp may serve as a potential therapeutic agent for prevention and/or treatment of BBB disruption in AD. Notably, as the pathogenesis of AD is varied, damaged or weakened barrier protection of the BBB is just one factor. Indeed, neuronal cell damage induced by Aβ and Tau proteins, and increased production and abnormal clearance of Aβ are all important pathogenic factors. As a result of limitations associated with in vitro study conditions, further studies are needed to verify specific effects for treating AD in vivo.
Footnotes
Conflicts of interest: None declared.
Financial support: This study was financially supported by the National Natural Science Foundation of China, No. 81573771; the Natural Science Foundation of Jiangsu Province of China, No. BK20151599. The funding bodies played no role in the study design, collection, analysis and interpretation of data, in the writing of the paper, or in the decision to submit the paper for publication.
Copyright license agreement: The Copyright License Agreement has been signed by all authors before publication.
Data sharing statement: Datasets analyzed during the current study are available from the corresponding author on reasonable request.
Plagiarism check: Checked twice by iThenticate.
Peer review: Externally peer reviewed.
Funding: This study was financially supported by the National Natural Science Foundation of China, No. 81573771; the Natural Science Foundation of Jiangsu Province of China, No. BK20151599.
(Copyedited by Amy Van Deusen, Raye W, Yu J, Li CH, Qiu Y, Song LP, Zhao M)
References
- 1.Abbott NJ, Patabendige AA, Dolman DE, Yusof SR, Begley DJ. Structure and function of the blood-brain barrier. Neurobiol Dis. 2010;37:13–25. doi: 10.1016/j.nbd.2009.07.030. [DOI] [PubMed] [Google Scholar]
- 2.Bednarczyk J, Lukasiuk K. Tight junctions in neurological diseases. Acta Neurobiol Exp (Wars) 2011;71:393–408. doi: 10.55782/ane-2011-1861. [DOI] [PubMed] [Google Scholar]
- 3.Biron KE, Dickstein DL, Gopaul R, Jefferies WA. Amyloid triggers extensive cerebral angiogenesis causing blood brain barrier permeability and hypervascularity in Alzheimer's disease. PLoS One. 2011;6:e23789. doi: 10.1371/journal.pone.0023789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Carrano A, Hoozemans JJ, van der Vies SM, Rozemuller AJ, van Horssen J, de Vries HE. Amyloid Beta induces oxidative stress-mediated blood-brain barrier changes in capillary amyloid angiopathy. Antioxid Redox Signal. 2011;15:1167–1178. doi: 10.1089/ars.2011.3895. [DOI] [PubMed] [Google Scholar]
- 5.Chen X, Threlkeld SW, Cummings EE, Juan I, Makeyev O, Besio WG, Gaitanis J, Banks WA, Sadowska GB, Stonestreet BS. Ischemia-reperfusion impairs blood-brain barrier function and alters tight junction protein expression in the ovine fetus. Neuroscience. 2012;226:89–100. doi: 10.1016/j.neuroscience.2012.08.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chow BW, Gu C. The molecular constituents of the blood-brain barrier. Trends Neurosci. 2015;38:598–608. doi: 10.1016/j.tins.2015.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Erickson MA, Banks WA. Blood-brain barrier dysfunction as a cause and consequence of Alzheimer's disease. J Cereb Blood Flow Metab. 2013;33:1500–1513. doi: 10.1038/jcbfm.2013.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Förster C. Tight junctions and the modulation of barrier function in disease. Histochem Cell Biol. 2008;130:55–70. doi: 10.1007/s00418-008-0424-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Feng J, Meng C, Xing D. Abeta induces PUMA activation: a new mechanism for Abeta-mediated neuronal apoptosis. Neurobiol Aging. 2015;36:789–800. doi: 10.1016/j.neurobiolaging.2014.10.007. [DOI] [PubMed] [Google Scholar]
- 10.Folin M, Baiguera S, Fioravanzo L, Conconi MT, Grandi C, Nussdorfer GG, Parnigotto PP. Caspase-8 activation and oxidative stress are involved in the cytotoxic effect of beta-amyloid on rat brain microvascular endothelial cells. Int J Mol Med. 2006;17:431–435. [PubMed] [Google Scholar]
- 11.Fonseca AC, Ferreiro E, Oliveira CR, Cardoso SM, Pereira CF. Activation of the endoplasmic reticulum stress response by the amyloid-beta 1-40 peptide in brain endothelial cells. Biochim Biophys Acta. 2013;1832:2191–2203. doi: 10.1016/j.bbadis.2013.08.007. [DOI] [PubMed] [Google Scholar]
- 12.Fossati S, Ghiso J, Rostagno A. Insights into caspase-mediated apoptotic pathways induced by amyloid-beta in cerebral microvascular endothelial cells. Neurodegener Dis. 2012;10:324–328. doi: 10.1159/000332821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gonzalez-Velasquez FJ, Kotarek JA, Moss MA. Soluble aggregates of the amyloid-beta protein selectively stimulate permeability in human brain microvascular endothelial monolayers. J Neurochem. 2008;107:466–477. doi: 10.1111/j.1471-4159.2008.05618.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hartz AM, Bauer B, Soldner EL, Wolf A, Boy S, Backhaus R, Mihaljevic I, Bogdahn U, Klunemann HH, Schuierer G, Schlachetzki F. Amyloid-beta contributes to blood-brain barrier leakage in transgenic human amyloid precursor protein mice and in humans with cerebral amyloid angiopathy. Stroke. 2012;43:514–523. doi: 10.1161/STROKEAHA.111.627562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jekabsone A, Mander PK, Tickler A, Sharpe M, Brown GC. Fibrillar beta-amyloid peptide Abeta1-40 activates microglial proliferation via stimulating TNF-alpha release and H2O2 derived from NADPH oxidase: a cell culture study. J Neuroinflammation. 2006;3:24. doi: 10.1186/1742-2094-3-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim SM, Park HS, Jun DY, Woo HJ, Woo MH, Yang CH, Kim YH. Mollugin induces apoptosis in human Jurkat T cells through endoplasmic reticulum stress-mediated activation of JNK and caspase-12 and subsequent activation of mitochondria-dependent caspase cascade regulated by Bcl-xL. Toxicol Appl Pharmacol. 2009;241:210–220. doi: 10.1016/j.taap.2009.08.024. [DOI] [PubMed] [Google Scholar]
- 17.Kook SY, Hong HS, Moon M, Ha CM, Chang S, Mook-Jung I. Abeta(1)(-)(4)(2)-RAGE interaction disrupts tight junctions of the blood-brain barrier via Ca(2)(+)-calcineurin signaling. J Neurosci. 2012;32:8845–8854. doi: 10.1523/JNEUROSCI.6102-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ku SK, Kwak S, Kwon OJ, Bae JS. Hyperoside inhibits high-glucose-induced vascular inflammation in vitro and in vivo. Inflammation. 2014;37:1389–1400. doi: 10.1007/s10753-014-9863-8. [DOI] [PubMed] [Google Scholar]
- 19.Liu CY, Bai K, Yu GR. Effect of hyperoside on damaged blood-brain barrier induced by Aβ1-42 in vitro model. Zhongguo Shiyan Fangji Xue Zazhi. 2017;23:138–143. [Google Scholar]
- 20.Marco S, Skaper SD. Amyloid beta-peptide1-42 alters tight junction protein distribution and expression in brain microvessel endothelial cells. Neurosci Lett. 2006;401:219–224. doi: 10.1016/j.neulet.2006.03.047. [DOI] [PubMed] [Google Scholar]
- 21.Mott JD, Werb Z. Regulation of matrix biology by matrix metalloproteinases. Curr Opin Cell Biol. 2004;16:558–564. doi: 10.1016/j.ceb.2004.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Naik P, Fofaria N, Prasad S, Sajja RK, Weksler B, Couraud PO, Romero IA, Cucullo L. Oxidative and pro-inflammatory impact of regular and denicotinized cigarettes on blood brain barrier endothelial cells: is smoking reduced or nicotine-free products really safe? BMC Neurosci. 2014;15:51. doi: 10.1186/1471-2202-15-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Park JY, Han X, Piao MJ, Oh MC, Fernando PM, Kang KA, Ryu YS, Jung U, Kim IG, Hyun JW. Hyperoside induces endogenous antioxidant system to alleviate oxidative stress. J Cancer Prev. 2016;21:41–47. doi: 10.15430/JCP.2016.21.1.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rosenberg GA. Blood-brain barrier permeability in aging and Alzheimer's disease. J Prev Alzheimers Dis. 2014;1:138–139. doi: 10.14283/jpad.2014.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Seo JH, Guo S, Lok J, Navaratna D, Whalen MJ, Kim KW, Lo EH. Neurovascular matrix metalloproteinases and the blood-brain barrier. Curr Pharm Des. 2012;18:3645–3648. doi: 10.2174/138161212802002742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wan W, Cao L, Liu L, Zhang C, Kalionis B, Tai X, Li Y, Xia S. Abeta(1-42) oligomer-induced leakage in an in vitro blood-brain barrier model is associated with up-regulation of RAGE and metalloproteinases, and down-regulation of tight junction scaffold proteins. J Neurochem. 2015;134:382–393. doi: 10.1111/jnc.13122. [DOI] [PubMed] [Google Scholar]
- 27.Wang Y, Miao Y, Mir AZ, Cheng L, Wang L, Zhao L, Cui Q, Zhao W, Wang H. Inhibition of beta-amyloid-induced neurotoxicity by pinocembrin through Nrf2/HO-1 pathway in SH-SY5Y cells. J Neurol Sci. 2016;368:223–230. doi: 10.1016/j.jns.2016.07.010. [DOI] [PubMed] [Google Scholar]
- 28.Yamazaki Y, Kanekiyo T. Blood-brain barrier dysfunction and the pathogenesis of Alzheimer's disease. Int J Mol Sci. 2017;18:E1965. doi: 10.3390/ijms18091965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yang Y, Rosenberg GA. MMP-mediated disruption of claudin-5 in the blood-brain barrier of rat brain after cerebral ischemia. Methods Mol Biol. 2011;762:333–345. doi: 10.1007/978-1-61779-185-7_24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zeng KW, Wang XM, Ko H, Kwon HC, Cha JW, Yang HO. Hyperoside protects primary rat cortical neurons from neurotoxicity induced by amyloid beta-protein via the PI3K/Akt/Bad/Bcl(XL)-regulated mitochondrial apoptotic pathway. Eur J Pharmacol. 2011;672:45–55. doi: 10.1016/j.ejphar.2011.09.177. [DOI] [PubMed] [Google Scholar]
- 31.Zhang XN, Li JM, Yang Q, Feng B, Liu SB, Xu ZH, Guo YY, Zhao MG. Anti-apoptotic effects of hyperoside via inhibition of NR2B-containing NMDA receptors. Pharmacol Rep. 2010;62:949–955. doi: 10.1016/s1734-1140(10)70356-x. [DOI] [PubMed] [Google Scholar]
- 32.Zhao L, Qian ZM, Zhang C, Wing HY, Du F, Ya K. Amyloid beta-peptide 31-35-induced neuronal apoptosis is mediated by caspase-dependent pathways via cAMP-dependent protein kinase A activation. Aging Cell. 2008;7:47–57. doi: 10.1111/j.1474-9726.2007.00352.x. [DOI] [PubMed] [Google Scholar]
- 33.Zheng M, Liu C, Pan F, Shi D, Zhang Y. Antidepressant-like effect of hyperoside isolated from Apocynum venetum leaves: possible cellular mechanisms. Phytomedicine. 2012;19:145–149. doi: 10.1016/j.phymed.2011.06.029. [DOI] [PubMed] [Google Scholar]
- 34.Zlokovic BV. The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron. 2008;57:178–201. doi: 10.1016/j.neuron.2008.01.003. [DOI] [PubMed] [Google Scholar]