

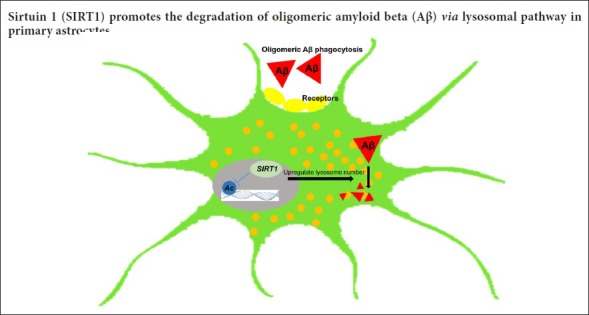
Keywords: nerve regeneration, amyloid beta peptide, Alzheimer's disease, neurodegeneration, astrocytes, gliocytes, sirtuin1, quantitative proteomics, lysosome, time point model, peptide degradation, neural regeneration
Abstract
Previous studies have shown that sirtuin 1 (SIRT1) reduces the production of neuronal amyloid beta (Aβ) and inhibits the inflammatory response of glial cells, thereby generating a neuroprotective effect against Aβ neurotoxicity in animal models of Alzheimer's disease. However, the protective effect of SIRT1 on astrocytes is still under investigation. This study established a time point model for the clearance of Aβ in primary astrocytes. Results showed that 12 hours of culture was sufficient for endocytosis of oligomeric Aβ, and 36 hours sufficient for effective degradation. Immunofluorescence demonstrated that Aβ degradation in primary astrocytes relies on lysosome function. Enzymatic agonists or SIRT1 inhibitors were used to stimulate cells over a concentration gradient. Aβ was co-cultured for 36 hours in medium. Western blot assay results under different conditions revealed that SIRT1 relies on its deacetylase activity to promote intracellular Aβ degradation. The experiment further screened SIRT1 using quantitative proteomics to investigate downstream, differentially expressed proteins in the Aβ degradation pathway and selected the ones related to enzyme activity of SIRT1. Most of the differentially expressed proteins detected are close to the primary astrocyte lysosomal pathway. Immunofluorescence staining demonstrated that SIRT1 relies on its deacetylase activity to upregulate lysosome number in primary astrocytes. Taken together, these findings confirm that SIRT1 relies on its deacetylase activity to upregulate lysosome number, thereby facilitating oligomeric Aβ degradation in primary astrocytes.
Introduction
The most common form of Alzheimer's disease (AD), which appears sporadically during aging, is characterized by the presence of extracellular senile plaques, intraneuronal aggregates of hyperphosphorylated tau, and synaptic and neuronal loss (Iqbal et al., 2005; Henry et al., 2010; Serrano-Pozo et al., 2011). Studies have revealed that there is no increase in amyloid beta (Aβ) production in sporadic AD, but that a deficiency in Aβ clearance is usually accompanied by age-related lysosomal dysfunction (LaFerla et al., 2007; Nixon, 2007; Nixon et al., 2008; Karran et al., 2011), which is considered to be the initiating factor of the disease. Therefore, it is important to investigate how Aβ clearance can be enhanced within the brain.
Astrocytes are fundamental for the homoeostasis, defense, and regeneration of the central nervous system, and they are the most abundant cell type in the brain, occupying most of the cerebral cortex (Chung et al., 2013; Verkhratsky et al., 2013). Attenuating astrocyte activation can accelerate plaque pathogenesis in amyloid precursor protein/presenilin 1 (APP/PS1) mice (Kraft et al., 2013). Activated astrocytes can surround Aβ in AD brains and subsequently take up Aβ and traffic it to lysosomes for degradation (Wisniewski et al., 1991; Verkhratsky et al., 2010; Basak et al., 2012; Lööv et al., 2012). Astrocytes are found in high numbers throughout all brain regions, so they can effectively clear Aβ from the brain, and they are susceptible to oligomeric Aβ (Nielsen et al., 2010; Jones et al., 2013), which highly correlates with markers of disease severity and is toxic to the central nervous system (McLean et al., 1999; Sokolowski et al., 2011). Thus, astrocytes may prevent excessive Aβ accumulation in the early stages of AD. Unlike microglia, astrocytes do not need to be stimulated by cytokines to take up Aβ (Chang et al., 2000; Magnus et al., 2002; Guénette, 2003). Wyss-Coray and his team have proposed that astroglial dysfunction aggravates progressive amyloid deposition (Wyss-Coray et al., 2003). Furthermore, impaired lysosomal function during aging has been implicated in AD (Cuervo et al., 2000; Mueller-Steiner et al., 2006; Bahr, 2009; Wolfe et al., 2013), which could be the underlying mechanism for the accumulation of Aβ within astrocytes, thus promoting AD pathology. Therefore, the role of astrocytes in Aβ degradation in AD needs further investigation, and additional characterization of the mechanisms regulating this process is required.
Sirtuin1 (SIRT1) can regulate a vast number of cellular processes, and it is associated with age-related diseases including AD. SIRT1 is the most conserved member of the sirtuin family of nicotinamide adenine dinucleotide (NAD+)-dependent protein deacetylases (Cohen et al., 2004; Sauve et al., 2006; Chang et al., 2014). Previous studies have indicated that SIRT1 protects against Aβ toxicity and cognitive deficits in animal models, mainly by decreasing Aβ production in neurons and inhibiting inflammatory responses in neuroglia (Han et al., 2004; Qin et al., 2006; Albani et al., 2009; Donmez et al., 2010). Apart from the observations mentioned previously, the molecular mechanisms underlying the protective effects of SIRT1 in astrocytes are still being investigated. The current study evaluates whether SIRT1 affects Aβ degradation in primary astrocytes via its deacetylase activity, and investigates the correlation with lysosome number using a quantitative proteomics approach.
Materials and Methods
Experimental animals
All animal experiments were approved by the Laboratory Animal Research Center, Peking University (approval number: LS-JiJG-3), certified by the Association for Assessment and Accreditation of Laboratory Animal Care International. All parts of the experimental animals were used for the isolation of primary cells. Wild-type specific-pathogen-free Sprague Dawley rats at the age of 1 to 2 days, irrespective of sex, were used to obtain primary astrocytes. Usually 10 rats weighing 5–6 g each were used for one cell isolation experiment. The brain cells of suckling mice of this age are well differentiated and vigorous. Cortical cells from 3–5 rats were isolated and plated in one T75 flask (Corning, Steuben County, NY, USA), and the whole isolation process was sterile and performed on ice.
Cell culture
Primary astrocytes were prepared from whole brains of Sprague-Dawley rats. Briefly, the pups were decapitated and the meninges and blood vessels were removed from the brains. The cerebellum, interbrain, midbrain, brainstem, and hippocampus were also removed. Only the cerebral cortex was used for the culture of astrocytes and microglia. The cerebral cortex was minced and digested with DNase I (0.01%) and trypsin (0.25%) for 30 minutes at 37°C. Digestion was stopped by suspending the cells in Dulbecco's Modified Eagle's Medium (DMEM) containing 10% fetal bovine serum and 1% penicillin-streptomycin. The cell suspension was then triturated and plated in a T75 flask, then maintained at 37°C in a 5% CO2 incubator. Media were replaced with fresh DMEM containing 10% fetal bovine serum 24 hours later, and the cells were cultured for an additional 14–21 days at 37°C with 5% CO2. Primary astrocytes were isolated from microglia by shaking the flask for 16 hours at 260 r/min. The suspended cells in the culture media were microglia, while the cells that remained adhered to the flask were astrocytes. Astrocytes were then digested and used for subsequent experiments, which were repeated three times. Primary astrocytes were principally divided into five groups, for the following experiments: 1) establishing a time point model for the clearance of Aβ in primary astrocytes, 2) investigating whether Aβ degradation in primary astrocytes relies on lysosomal function, 3) examining whether SIRT1 relies on its deacetylase activity to facilitate Aβ degradation in primary astrocytes, 4) exploring the potential downstream proteins of SIRT1 in facilitating lysosome-mediated Aβ degradation in primary astrocytes, and 5) investigating whether SIRT1 relies on its deacetylase activity to upregulate lysosome number in primary astrocytes.
Preparation of Aβ
Human Aβ1–42 (Cat. No. 20276; AnaSpec, Fremont, CA, USA) or Hilyte-Fluor-488 Aβ1–42 (Cat. No. 60479; AnaSpec, Fremont, CA, USA) was dissolved to 1 mM in 100% hexafluoroisopropanol. The specific methods of operation were performed as previously described by Stine et al. (2011).
Plasmids and transfection
Human SIRT1 was subcloned into pcDNA3.1 (+) and pEGFP-N3, separately. SIRT1-H363Y was mutated from SIRT1-pcDNA3.1 (+) and SIRT1-pEGFP-N3, separately. Transient transfection was conducted using Viafect according to the manufacturer's instructions (Cat. No. E4981; Promega, Madison, WI, USA). The cells were harvested at 48 hours for further use.
RNA interference
Primary astrocytes were transfected with a specific siRNA against the target gene (SIRT1 siRNA: Cat. No. sc-108043; Santa Cruz Biotechnology, Santa Cruz, CA, USA) or the scrambled siRNA (control) with Lipofectamine RNAiMAX (Invitrogen, Carlsbad, CA, USA). The cells were harvested at 72 hours for further use.
Aβ uptake and degradation
Primary astrocytes were principally divided into seven groups for different interventions and subsequent Aβ treatment. To analyze astroglial degradation, primary astrocytes were incubated with 1 µM Aβ1–42 for different time periods. The cells were transfected with scrambled (Group 1) or SIRT1 (Group 2) siRNA or SIRT1 (Group 4) or SIRT1-H363Y (Group 5) plasmids before incubation with 1 µM Aβ1–42 for an additional 12 or 36 hours, to track astroglial phagocytosis or degradation of Aβ, respectively. Subsequently, the cells were thoroughly washed with DMEM three times and lysed for western blot assay. The intensities of the protein bands, which indicated relative intracellular Aβ levels, were measured in three independent experiments.
To pinpoint the localization of the internalized Aβ, 1 µM Hilyte-Fluor-488 oligomeric Aβ1–42 was added to cells for 30 minutes. Cells were then stained with Lyso-Tracker Red (Cat. No. C1046; Beyotime, Nantong, China), and observed under a confocal microscope.
A lysosome inhibitor or neprilysin inhibitor was added to the cells 24 hours before adding Aβ and maintained in the media until cells were harvested. The lysosome inhibitors used were leupeptin (Cat. No. L2884; Sigma, St. Louis, MO, USA) and chloroquine (Cat. No. C6628; Sigma), while phosphoramidon (Cat. No. R7385; Sigma) was used as the neprilysin inhibitor.
Western blot assay
Cells were harvested and lysed with 1% sodium dodecyl sulphate and then sonicated. The protein concentration was determined with a 2-D Quantitative Kit (GE Healthcare, Little Chalfont, Buckinghamshire, UK). Equal amounts of lysates were resolved using 12.5% sodium dodecyl sulfate polyacrylamide gel electrophoresis and transferred onto polyvinylidene fluoride membranes using a semi-dry blotting apparatus (Bio-Rad, Hercules, CA, USA). After blocking with 5% non-fat milk in phosphate buffered saline (PBS) containing 0.05% Tween 20, the membranes were incubated overnight at 4°C with primary antibodies, including mouse monoclonal anti-β-actin (Cat. No. ab3280; Abcam, Cambridge, MA, USA), rabbit monoclonal anti-SIRT1 (Cat. No. 9475; Cell Signaling Technology, Danvers, MA, USA), mouse monoclonal anti-Aβ1–16 (6E10) (Cat. No. SIG39320, Covance, Princeton, NJ, USA), and rabbit polyclonal anti-Lamp1 (Cat. No. ab24170; Abcam). Afterwards, the membranes were incubated with horseradish-peroxidase-conjugated secondary antibodies for 2 hours at room temperature, including goat anti-mouse IgG (H+L) (Cat. No. 1031-05; Southern Biotech, Birmingham, AL, USA) and goat anti-rabbit IgG (H+L) (Cat. No. 4050-05; Southern Biotech). Protein bands were detected by chemiluminescence (Millipore, Billerica, MA, USA). The intensities of the protein bands were quantified using ImageJ software (Media Cybernetics, Silver Springs, MD, USA). The intensities of the protein bands were measured in three independent experiments and results were presented as normalized to actin.
Immunofluorescent staining
Cells were harvested for immunofluorescent staining after 24 hours of inhibitor treatment, 48 hours of plasmid transfection, or 72 hours of siRNA knockdown. The cells were fixed in 4% paraformaldehyde for 20 minutes, permeabilized in 1% Triton X-100/PBS for 20 minutes, and then blocked in 10% bovine serum albumin/0.05% Tris-buffered saline with Tween 20 (TBST) for 1 hour at room temperature. The cells were then incubated with primary antibodies in 1% bovine serum albumin/TBST overnight at 4°C, followed by an additional incubation in Alexa Fluor-conjugated secondary antibodies at room temperature for 1 hour. The images were acquired with an LSM 710 NLO & DuoScan System confocal laser-scanning microscope (Zeiss, Jena, Germany).
RNA isolation and quantitative real-time polymerase chain reaction (PCR)
Total RNA was isolated from cells that underwent different treatments using TRIzol Reagent (Invitrogen, Carlsbad, CA, USA), and 1 μg of total RNA was reverse transcribed to cDNA with the HiFi-MMLV cDNA First Strand Synthesis Kit (CW Bio, Shanghai, China). Quantitative PCR was performed by combining cDNA with GoTaq qPCR Master Mix (Promega, Madison, WI, USA). The reaction was carried out with the CFX96 Real Time PCR Detection System (Bio-Rad) using the following conditions: 95°C for 2 minutes, 95°C for 15 seconds, 60°C for 30 seconds, and 72°C for 25 seconds, followed by 40 cycles of 95°C for 15 seconds and 60°C for 2 minutes. Quantification of mRNA expression was calculated by the Livak method as described by the manufacturer instructions, shown in a 2-ΔΔCt method, and was finally presented in a column graph using Graphpad Prism 7 (La Jolla, San Diego, CA, USA).
Mass spectrometry analysis
Cells were harvested for mass spectrometry pretreatment after 48 hours of plasmid transfection and 36 hours of Aβ co-culture. Primary astrocytes were transfected with empty VECTOR, SIRT1, or SIRT1-H363Y for 48 hours. Subsequently, the cells were treated with 1 µM Aβ1–42 for an additional 36 hours. The cells were thoroughly washed and lysed with 1% sodium dodecyl sulphate. After sonication, concentrations of the lysates were measured with a Bicinchoninic Acid Kit (Thermo Fisher Scientific, Waltham, MA, USA). Filter-aided sample preparation was used to enzymatically digest the protein samples. The eluted peptide mixtures were then desalinated, tandem mass tag (TMT) labeled, and classified, followed by mass spectrometry analysis. The peptide identifications were processed with Proteome Discoverer 2.1 Software (Thermo Fisher Scientific). After identifying protein expression ratios greater than 1.3 or lower than 0.75, a list of differentially expressed proteins was obtained. The proteins induced by SIRT1-H363Y overexpression were excluded from the ones induced by SIRT1 overexpression. The proteins involved in SIRT1 deacetylase activity are shown in Table 1. Gene ontology (GO) clustering analysis was performed using GENECODIS 3, and this online analytical tool automatically generated pie charts and column graphs to show clusters of proteins.
Table 1.
Differentially expressed lysosome-related proteins

Statistical analysis
Data were analyzed using GraphPad Prism 7.0 Software (La Jolla) and presented as the mean ± SEM. Multiple sets of data were analyzed by one-way analysis of variance with Tukey's post hoc test, while the unpaired Student's t-test was used to analyze two sets of data. The significance level was set at P < 0.05.
Results
Primary astrocytes cleared oligomeric Aβ via lysosomes
The purity of primary astrocytes isolated from the cerebral cortex of rats was validated to be greater than 98% and confirmed by the immunofluorescent intensity and area of glial fibrillary acidic protein (GFAP), a marker of astrocytes (Additional Figure 1 (1.7MB, tif) ). To ascertain a preference for different states of Aβ, astrocytes were treated with fibrillar and oligomeric Aβ, harvested at different time points, and lysed. Our results revealed that primary astrocytes rapidly cleared oligomeric Aβ, whereas fibrillar Aβ clearance was much slower. With increasing time, the internalized fibrillar Aβ level slowly increased to its peak level at 36 hours, and then gradually decreased by 96 hours (Figure 1A, B). However, the internalized oligomeric Aβ level increased to its peak level at 12 hours, and then gradually disappeared by 48 hours (Figure 1C). This experiment allowed us to conclude that primary astrocytes have a preference for oligomeric Aβ when clearing extracellular Aβ aggregates. For this reason, oligomeric Aβ was used in the following experiments, and 12 hours represented the maximal astroglial capabilities of Aβ phagocytosis, while 36 hours represented maximal Aβ degradation. Oligomeric Aβ conjugated to a green fluorescent label was observed to be rapidly taken up and trafficked into lysosomes within 30 minutes, showing definite co-localization (Figure 1D). Aβ was mostly degraded within lysosomes, because lysosome inhibitors such as chloroquine or leupeptin remarkably weakened the astroglial degradation of Aβ; in contrast, phosphoramidon, an inhibitor of neprilysin, which is involved in Aβ degradation, exerted little impact on this process (Figure 1E).
Figure 1.

Primary astrocytes clear oligomeric Aβ [Aβ1–16 (6E10)] using lysosomes.
(A–C) Primary astrocytes clear oligomeric Aβ: Primary astrocytes were incubated with 1 µM fibrillar Aβ (A, B) or oligomeric Aβ (C) at 37°C, and the cells were harvested at different time points and lysed, followed by the detection of the intracellular Aβ level by western blot assay. The intensities of the protein bands, which indicated the relative intracellular Aβ level, were measured in three independent experiments, and results were presented as the mean ± SEM (lower panels in A–C). (D) Aβ is rapidly trafficked into lysosomes: Confocal imaging of live astrocytes 30 minutes after the addition of 1 µM Hilyte488-labeled oligomeric Aβ (green). The cells were then stained with Lyso-Tracker Red. Scale bar: 10 µm. (E) Internalized Aβ is degraded in lysosomes. Primary astrocytes were pretreated with DMSO, phosphoramidon (neprilysin inhibitor, 10 µM), or chloroquine or leupeptin (lysosome inhibitors, 10 µM) for 24 h. The cells were then incubated with Aβ (1 µM) in the presence of DMSO or inhibitors for an additional 36 h. The intensities of the protein bands indicated the relative intracellular Aβ level. Results were measured in three independent experiments, and were presented as the mean ± SEM (one-way analysis of variance with Tukey's post hoc test). *P < 0.05, ***P < 0.001, vs. DMSO control. Aβ: Amyloid beta; DMSO: dimethyl sulfoxide; h: hour.
Enriched astrocytes cultured on coverslips were immunofluorescently stained with different nerve cell markers.
The purity of enriched astrocytes was greater than 98%. Glial fibrillary acidic protein (GFAP) staining was specific for astrocytes (green). Iba1 staining was specific for microglia (red). Microtubule-associated protein-2 (MAP-2) staining was specific for neurons (red). Nuclei were stained by 4’,6-diamidino-2-phenylindole (DAPI) (blue). LSM 710 NLO & DuoScan System confocal laser-scanning microscope (Zeiss, Jena, Germany) was used. Scale bar: 50 μm.
SIRT1 relied on its deacetylase activity to facilitate Aβ degradation
To investigate the role of SIRT1 in Aβ clearance, the changes in endogenous SIRT1 levels were detected after prolonged Aβ treatment. Endogenous SIRT1 levels decreased and then increased, even exceeding the initial level at 48 hours (Figure 2A). Combined with the results on the degradation of oligomeric Aβ, intracellular Aβ levels gradually increased from 0 to 3 hours, which may have been due to the stress that induced the temporary reduction in SIRT1 levels. With a further increase in intracellular Aβ levels, the cells most likely began to respond accordingly and upregulated the expression of endogenous SIRT1, which appeared to be ready for Aβ degradation prior to the peak of intracellular Aβ levels. Endogenous SIRT1 was at its highest level at 48 hours, whereas intracellular Aβ was at its lowest level, suggesting that SIRT1 might directly promote Aβ clearance (Figure 2B). Supporting this finding, the siRNA-specific knockdown of SIRT1 in astrocytes greatly suppressed their ability to degrade Aβ (Figure 2C).
Figure 2.

SIRT1 relies on its deacetylase activity to facilitate Aβ [Aβ1–16 (6E10)] degradation.
(A, B) Changes in endogenous SIRT1 levels after Aβ stimulation: Primary astrocytes were incubated with oligomeric Aβ (1 µM) at 37°C. The cells were harvested at different time points and lysed, followed by the detection of endogenous SIRT1 levels by western blot assay (A). Combined with the results shown in Figure 1, there were differences in the levels of Aβ and SIRT1 with increasing time after treatment. The green line represents the relative SIRT1 level, whereas the red line represents the Aβ level. The y-axis indicates the relative intracellular protein content levels (B). The intensities of the protein bands, which indicate the relative endogenous SIRT1 level, were measured in three independent experiments, and the results are presented as the mean ± SEM (lower panels in A, B). (C–F) SIRT1 facilitates Aβ degradation by relying on its deacetylase activity. Scrambled or SIRT1 siRNA was transfected into primary astrocytes (C) and the cells were then incubated with Aβ (1 µM) for 36 hours. The cells were harvested to detect intracellular Aβ levels by western blot assay. Vector or SIRT1 (WT or H363Y) was overexpressed in astrocytes, and the cells were incubated with Aβ (1 µM) for 36 hours. The cells were harvested to detect intracellular Aβ levels by western blot assay (D). Primary astrocytes were pretreated with increasing concentrations of resveratrol (E) or NAM (f) for 24 hours. The cells were then incubated with Aβ (1 µM) in the presence of resveratrol or NAM for an additional 36 hours, followed by western blot assay. The intensities of the protein bands, which indicated the relative intracellular Aβ level, were measured in three independent experiments, and the results were presented as the mean ± SEM (lower panel in C–F). **P < 0.01 (unpaired Student's t-test; C). ***P < 0.001 (one-way analysis of variance with Tukey's post hoc test; D). **P < 0.01, ***P < 0.001, vs. control (one-way analysis of variance with Tukey's post hoc test; E, F). SIRT1: Sirtuin 1; SIRT1-WT: wild-type SIRT1; SIRT1-HY: mutant SIRT1 with the 363rd amino acid mutated from histidine to tyrosine; Aβ: amyloid beta; NAM: nicotinamide.
Based on the evidence that SIRT1 plays a positive role in the maintenance of homeostasis, resistance to aging, and alleviation of the AD pathological process as a deacetylase (Qin et al., 2006; Donmez et al., 2010), we hypothesized that SIRT1 might facilitate Aβ degradation via its enzymatic activity. To address this, primary astrocytes were transfected with SIRT1-WT as well as with its catalytically inactive mutant SIRT1-H363Y. Intracellular Aβ levels were significantly lower after 36 hours in astrocytes overexpressing SIRT1-WT than in those overexpressing VECTOR or SIRT1-H363Y (Figure 2D), indicating that only catalytically active SIRT1 can efficiently facilitate the astroglial degradation of Aβ. In addition, pretreating astrocytes with resveratrol, an SIRT1 agonist, enhanced the clearance of intracellular Aβ in a dose-dependent manner (Figure 2E), whereas the SIRT1 inhibitor nicominatide (NAM) abolished the ability of astrocytes to degrade Aβ (Figure 2F). However, SIRT1 did not affect Aβ endocytosis because the intracellular Aβ level was unchanged after treatment for 12 hours (Additional Figure 2 (661.8KB, tif) ).
SIRT1 does not affect Aβ endocytosis in primary astrocytes.
VECTOR or SIRT1 was overexpressed in primary astrocytes. The cells were incubated with Aβ (1 μM) for 12 hours and then harvested to detect the intracellular Aβ level by western blot analysis. The intensities of the protein bands, which indicated the relative intracellular Aβ level, were measured in three independent experiments. The results are presented as the mean ± SEM (unpaired Student's t-test). SIRT1: Sirtuin 1; Aβ: beta amyloid peptide.
Identification of potential downstream proteins of SIRT1 in facilitating Aβ degradation in primary astrocytes
Based on the important role of SIRT1 in facilitating Aβ degradation in primary astrocytes, we investigated the potential downstream proteins of SIRT1. After Aβ treatment, all proteins were screened by excluding differentially expressed proteins induced by SIRT1-H363Y overexpression from the ones induced by SIRT1 overexpression, thus identifying those involved in SIRT1 deacetylase activity. We identified several lysosome-related proteins (Table 1 and Additional Figure 3 (4.1MB, tif) ), and several of these differentially expressed proteins were then subjected to quantitative PCR with SIRT1 overexpression to further strengthen the relationship between SIRT1 and lysosomal function. The levels of several lysosome-related genes changed with SIRT1, which likely influences lysosomal function, thus affecting Aβ degradation in primary astrocytes (Figure 3).
Figure 3.
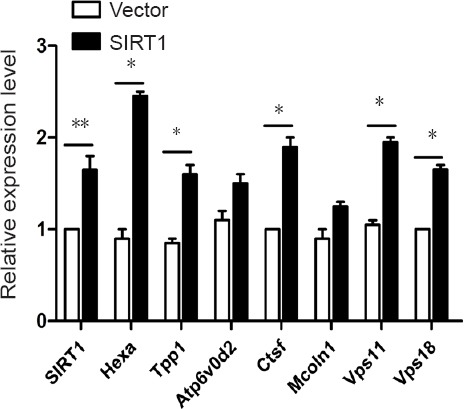
SIRT1 upregulates the levels of lysosome-related genes.
VECTOR or SIRT1 was overexpressed in primary astrocytes and the cells were then processed for quantitative polymerase chain reaction to detect the levels of lysosome-related genes. The expression levels were measured in three independent experiments, and the results are presented as the mean ± SEM (unpaired Student's t-test), *P < 0.05, **P < 0.01. SIRT1: Sirtuin 1.
Comprehensive GO clustering analysis of differentially expressed proteins.
The Biological Process, Molecular Function, Cellular Component and KEGG Pathway of differentially expressed proteins were analyzed comprehensively. The P value was set to < 0.001. KEGG: Kyoto encyclopedia of genes and genomes.
SIRT1 relied on its deacetylase activity to upregulate lysosome number in primary astrocytes
Lysosomal dysfunction is associated with aging in AD (Cuervo et al., 2000) and SIRT1 alters several lysosome-related genes. Similarly, we found that the number of lysosomes stained with Lyso-Tracker Red or the lysosome membrane marker LAMP1 decreased in primary astrocytes when endogenous SIRT1 levels were downregulated (Figure 4A). Conversely, an increase in lysosome number was observed when cells were exogenously transfected with GFP-tagged SIRT1-WT compared with VECTOR or GFP-tagged SIRT1-H363Y (Figure 4B). Furthermore, lysosome numbers were lower in primary astrocytes pretreated with NAM than in control cells, whereas there was no difference between trichostatin A (TSA)-treated astrocytes and control cells, indicating that SIRT1, but not other NAD-dependent histone deacetylases (HDACs), control lysosome number in primary astrocytes (Figure 4C). Taken collectively, these results suggest that SIRT1 facilitates Aβ degradation by upregulating lysosome numbers in primary astrocytes.
Figure 4.

SIRT1 relied on its deacetylase activity to upregulate lysosome numbers in primary astrocytes.
(A) SIRT1 knockdown decreased the number of lysosomes in primary astrocytes. Primary astrocytes were transfected with scrambled or SIRT1 siRNA for 72 hours, followed by staining with SIRT1 (green) and Lyso-Tracker Red or antibodies against LAMP1 (red). (B) SIRT1 stimulated an increase in lysosome number by relying on its deacetylase activity in primary astrocytes. The empty vector or GFP-tagged SIRT1 (WT or H363Y) was overexpressed in primary astrocytes, followed by staining with Lyso-Tracker Red or an antibody against LAMP1 (red). (C) NAM, but not TSA, downregulated lysosome numbers in primary astrocytes. Primary astrocytes were pretreated with NAM or TSA, followed by staining with Lyso-Tracker Red. The fluorescent signal in each cell was estimated by examining more than 50 cells, and the results are presented as the mean ± SEM (right panel in A–C; unpaired Student's t-test), **P < 0.01, ***P < 0.001 (A). One-way analysis of variance with Tukey's post hoc test, **P < 0.01, ***P < 0.001 (B, C). Scale bars: 15 µm. SIRT1: Sirtuin 1; SIRT1-WT: wild-type SIRT1; SIRT1-HY: mutant SIRT1 with the 363rd amino acid mutated from histidine to tyrosine; DMSO: dimethyl sulfoxide; NAM: nicotinamide; TSA: trichostatin A.
Discussion
Early in the onset of AD, oligomeric Aβ progressively aggregates into fibrils. To reduce the amount of Aβ that is deposited, it is therefore important to efficiently clear oligomeric Aβ. Astrocyte activation precedes extracellular Aβ deposition, indicating that astrocytes maintain a normal Aβ level under physiological conditions (Funato et al., 1998; Nagele et al., 2003). Astroglial atrophy in the brains of AD patients (Rodríguez et al., 2011) weakens the ability of astrocytes to clear Aβ to a certain extent, which may be the main cause of AD pathogenesis. In addition, Aβ removal is affected by lysosomal biogenesis (Xiao et al., 2014). Our results describe the timeline of fibrillar and oligomeric Aβ clearance, respectively. With a series of specific time points, we confirmed a preference of primary astrocytes to degrade oligomeric Aβ. We also observed that either an acidic environment or the hydrolase function of lysosomes can influence Aβ degradation, thus demonstrating the importance of lysosomal function in primary astrocytes.
Astrocytes play an important role in maintaining homeostasis, whether under normal physiological conditions or as part of a pathological process (Li et al., 2016). As the most abundant cell type in the brain, astrocytes cover the brain parenchyma comprehensively and maintain homeostasis by providing energy, eliminating waste, regulating blood-brain barrier transport, dumping redundant synaptic neurotransmitters, and regulating cell repair processes and ion flux (Parpura et al., 2012; Verkhratsky et al., 2013). Our results demonstrate that primary astrocytes can enhance the degradation of oligomeric Aβ with SIRT1 upregulation, which may provide important clues for the early prevention and treatment of AD. However, our primary astrocytes were obtained from the whole cerebral cortex of Sprague-Dawley rats, suggesting a coordinating function of astrocytes from different brain regions to degrade Aβ. It is well known that the cerebral cortex has many subregions that represent and control different body functions, which are precisely regulated. Future experiments should further explore the astroglial function of each part of the cortex, such as the precentral gyrus and olfactory cortex, to correlate cerebral Aβ degradation with overall dementia performance. Furthermore, aside from the cerebral cortex, the hippocampus also contains astrocytes, and hippocampal cells are more related to memory. A previous study has shown that astrocytes in the CA1 and dentate gyrus of 3xTg-AD animals showed atrophic signs at the age of 6 months, demonstrating a functional relationship between hippocampal astrocytes and the pathological process of dementia (LaFerla et al., 2007). Therefore, it would be valuable to study whether hippocampal astrocytes could affect Aβ degradation, and in which pathway this might occur.
SIRT1 alleviates AD pathology by decreasing Aβ production, promoting tau degradation, and protecting neurons from inflammation (Chen et al., 2005; Qin et al., 2006; Kim et al., 2007; Donmez et al., 2010). By contrast, little is known about the role of SIRT1 in Aβ clearance. It has been reported that lysosomal dysfunction gradually increases with aging, and data from AD patients have also shown that SIRT1 is lower in these individuals than in control subjects (Patel et al., 2005; Julien et al., 2009). Considering the important role of astrocytes in Aβ clearance, and the effectiveness of the lysosome pathway in Aβ clearance, our results indicate that SIRT1 could enhance the ability of astrocytes to clear Aβ during early stages, which would essentially delay the formation of amyloid deposits.
Protein acetylation has been recently recognized as a promising approach to control autophagic processes, such as the elimination of damaged organelles or toxic protein aggregates (Lee et al., 2008, 2009; Chakrabarti et al., 2011; Bánréti et al., 2013). Deacetylase SIRT1 can stimulate basal rates of autophagy under increased expression, and SIRT1 /- mouse embryonic fibroblasts do not fully activate autophagy under starved conditions (Lee et al., 2008; Hariharan et al., 2010; Chang et al., 2015). From this aspect, of autophagy, SIRT1's facilitating role on lysosomes is again confirmed. It is highly possible that SIRT1 acts through downstream factors, such as the ones identified in this study, which control deacetylase activity and facilitate Aβ degradation via a lysosome-mediated pathway. More studies are needed to further discover exact downstream factors of SIRT1.
In summary, this study has shown for the first time that SIRT1 relies on its enzymatic activity to deacetylate several lysosome-related proteins and upregulate lysosome number, thereby facilitating oligomeric Aβ degradation in primary astrocytes; however, possible mechanisms need additional investigation. Our results provide important insights for the early prevention and treatment of AD.
Additional files:
Additional Figure 1 (1.7MB, tif) : Enriched astrocytes cultured on coverslips were immunofluorescently stained with different nerve cell markers.
Additional Figure 2 (661.8KB, tif) : SIRT1 does not affect Aβ endocytosis in primary astrocytes.
Additional Figure 3 (4.1MB, tif) : Comprehensive GO clustering analysis of differentially expressed proteins.
Acknowledgments
We are very grateful to Jin-Tao Bao from College of Life Sciences, Peking University, China for assistance with the foundation of earlier animal model and cell model.
Footnotes
Conflicts of interest: The authors declare that there are no conflicts of interest.
Financial support: This study was supported by the National Natural Science Foundation of China, No. 31670832, 31470807, 31270872; a grant from the National Key Research and Development Program of China, No. 2016YFA0500301; a grant from the State Key Laboratory of Protein and Plant Gene Research, College of Life Sciences, Peking University, China. The funders had no role in study conception and design, data analysis or interpretation, paper writing or deciding to submit this paper for publication.
Institutional review board statement: All experimental procedures and protocols were approved by Laboratory Animal Research Center Peking University, certificated by Association for Assessment and Accreditation of Laboratory Animal Care international on February 15, 2008 (approval number LS-JiJG-3, approval date 2017-07-10). All experimental procedures described here were in accordance with the National Institutes of Health (NIH) guidelines for the Care and Use of Laboratory Animals.
Copyright license agreement: The Copyright License Agreement has been signed by all authors before publication.
Data sharing statement: Datasets analyzed during the current study are available from the corresponding author on reasonable request.
Plagiarism check: Checked twice by iThenticate.
Peer review: Externally peer reviewed.
Funding: This study was supported by the National Natural Science Foundation of China, No. 31670832, 31470807, 31270872; a grant from the National Key Research and Development Program of China, No. 2016YFA0500301; and a grant from the State Key Laboratory of Protein and Plant Gene Research, College of Life Sciences, Peking University, China.
(Copyedited by Gardner B, Pack M, Wang J, Li CH, Qiu Y, Song LP, Zhao M)
References
- 1.Albani D, Polito L, Batelli S, De Mauro S, Fracasso C, Martelli G, Colombo L, Manzoni C, Salmona M, Caccia S, Negro A, Forloni G. The SIRT1 activator resveratrol protects SK-N-BE cells from oxidative stress and against toxicity caused by α-synuclein or amyloid-β (1-42) peptide. J Neurochem. 2009;110:1445–1456. doi: 10.1111/j.1471-4159.2009.06228.x. [DOI] [PubMed] [Google Scholar]
- 2.Bahr BA. Lysosomal modulatory drugs for a broad strategy against protein accumulation disorders. Curr Alzheimer Res. 2009;6:438–445. doi: 10.2174/156720509789207903. [DOI] [PubMed] [Google Scholar]
- 3.Bánréti Á, Sass M, Graba Y. The emerging role of acetylation in the regulation of autophagy. Autophagy. 2013;9:819–829. doi: 10.4161/auto.23908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Basak JM, Verghese PB, Yoon H, Kim J, Holtzman DM. Low-density lipoprotein receptor represents an apolipoprotein E-independent pathway of Aβ uptake and degradation by astrocytes. J Biol Chem. 2012;287:13959–13971. doi: 10.1074/jbc.M111.288746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chakrabarti P, English T, Karki S, Qiang L, Tao R, Kim J, Luo Z, Farmer SR, Kandror KV. SIRT1 controls lipolysis in adipocytes via FOXO1-mediated expression of ATGL. J Lipid Res. 2011;52:1693–1701. doi: 10.1194/jlr.M014647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chang C, Su H, Zhang D, Wang Y, Shen Q, Liu B, Huang R, Zhou T, Peng C, Wong CC, Shen HM, Lippincott-Schwartz J, Liu W. AMPK-dependent phosphorylation of GAPDH triggers sirt1 activation and is necessary for autophagy upon glucose starvation. Mol Cell. 2015;60:930–940. doi: 10.1016/j.molcel.2015.10.037. [DOI] [PubMed] [Google Scholar]
- 7.Chang GH, Barbaro NM, Pieper RO. Phosphatidylserine-dependent phagocytosis of apoptotic glioma cells by normal human microglia, astrocytes, and glioma cells. Neuro Oncol. 2000;2:174–183. doi: 10.1093/neuonc/2.3.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chang HC, Guarente L. SIRT1 and other sirtuins in metabolism. Trends Endocrinol Metab. 2014;25:138–145. doi: 10.1016/j.tem.2013.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen J, Zhou Y, Mueller-Steiner S, Chen LF, Kwon H, Yi S, Mucke L, Gan L. SIRT1 protects against microglia-dependent amyloid-β toxicity through inhibiting NF-κB signaling. J Biol Chem. 2005;280:40364–40374. doi: 10.1074/jbc.M509329200. [DOI] [PubMed] [Google Scholar]
- 10.Chung WS, Clarke LE, Wang GX, Stafford BK, Sher A, Chakraborty C, Joung J, Foo LC, Thompson A, Chen C, Smith SJ, Barres BA. Astrocytes mediate synapse elimination through MEGF10 and MERTK pathways. Nature. 2013;504:394–400. doi: 10.1038/nature12776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cohen HY, Miller C, Bitterman KJ, Wall NR, Hekking B, Kessler B, Howitz KT, Gorospe M, de Cabo R, Sinclair DA. Calorie restriction promotes mammalian cell survival by inducing the SIRT1 deacetylase. Science. 2004;305:390–392. doi: 10.1126/science.1099196. [DOI] [PubMed] [Google Scholar]
- 12.Cuervo AM, Dice JF. When lysosomes get old. Exp Gerontol. 2000;35:119–131. doi: 10.1016/s0531-5565(00)00075-9. [DOI] [PubMed] [Google Scholar]
- 13.Donmez G, Wang D, Cohen DE, Guarente L. SIRT1 Suppresses β-amyloid production by activating the α-secretase gene ADAM10. Cell. 2010;142:320–332. doi: 10.1016/j.cell.2010.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 14.Funato H, Yoshimura M, Yamazaki T, Saido TC, Ito Y, Yokofujita J, Okeda R, Ihara Y. Astrocytes containing amyloid β-protein (Aβ)-positive granules are associated with Aβ40-positive diffuse plaques in the aged human brain. Am J Pathol. 1998;152:983–992. [PMC free article] [PubMed] [Google Scholar]
- 15.Guénette SY. Astrocytes: a cellular player in Aβ clearance and degradation. Trends Mol Med. 2003;9:279–280. doi: 10.1016/s1471-4914(03)00112-6. [DOI] [PubMed] [Google Scholar]
- 16.Han YS, Zheng WH, Bastianetto S, Chabot JG, Quirion R. Neuroprotective effects of resveratrol against betaamyloid-induced neurotoxicity in rat hippocampal neurons: involvement of protein kinase C. Br J Pharmacol. 2004;141:997–1005. doi: 10.1038/sj.bjp.0705688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hariharan N, Maejima Y, Nakae J, Paik J, Depinho RA, Sadoshima J. Deacetylation of FoxO by Sirt1 plays an essential role in mediating starvation-induced autophagy in cardiac myocytes. Circ Res. 2010;107:1470–1482. doi: 10.1161/CIRCRESAHA.110.227371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Henry W. Querfurth FML (2010) Alzheimer's Disease. N Engl J Med. 362:329–344. doi: 10.1056/NEJMra0909142. [DOI] [PubMed] [Google Scholar]
- 19.Iqbal K, Alonso Adel C, Chen S, Chohan MO, El-Akkad E, Gong CX, Khatoon S, Li B, Liu F, Rahman A, Tanimukai H, Grundke-Iqbal I. Tau pathology in Alzheimer disease and other tauopathies. Biochim Biophys Acta. 2005;1739:198–210. doi: 10.1016/j.bbadis.2004.09.008. [DOI] [PubMed] [Google Scholar]
- 20.Jones RS, Minogue AM, Connor TJ, Lynch MA. Amyloid-β-induced astrocytic phagocytosis is mediated by CD36, CD47 and RAGE. J Neuroimmune Pharmacol. 2013;8:301–311. doi: 10.1007/s11481-012-9427-3. [DOI] [PubMed] [Google Scholar]
- 21.Julien C, Tremblay C, Emond V, Lebbadi M, Salem N, Jr, Bennett DA, Calon F. Sirtuin 1 reduction parallels the accumulation of tau in Alzheimer disease. J Neuropathol Exp Neurol. 2009;68:48–58. doi: 10.1097/NEN.0b013e3181922348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Karran E, Mercken M, De Strooper B. The amyloid cascade hypothesis for Alzheimer's disease: an appraisal for the development of therapeutics. Nat Rev Drug Discov. 2011;10:698–712. doi: 10.1038/nrd3505. [DOI] [PubMed] [Google Scholar]
- 23.Kim D, Nguyen MD, Dobbin MM, Fischer A, Sananbenesi F, Rodgers JT, Delalle I, Baur JA, Sui G, Armour SM, Puigserver P, Sinclair DA, Tsai LH. SIRT1 deacetylase protects against neurodegeneration in models for Alzheimer's disease and amyotrophic lateral sclerosis. EMBO J. 2007;26:3169–3179. doi: 10.1038/sj.emboj.7601758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kraft AW, Hu X, Yoon H, Yan P, Xiao Q, Wang Y, Gil SC, Brown J, Wilhelmsson U, Restivo JL, Cirrito JR, Holtzman DM, Kim J, Pekny M, Lee JM. Attenuating astrocyte activation accelerates plaque pathogenesis in APP/PS1 mice. FASEB J. 2013;27:187–198. doi: 10.1096/fj.12-208660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.LaFerla FM, Green KN, Oddo S. Intracellular amyloid-β in Alzheimer's disease. Nat Rev Neurosci. 2007;8:499–509. doi: 10.1038/nrn2168. [DOI] [PubMed] [Google Scholar]
- 26.Lee IH, Cao L, Mostoslavsky R, Lombard DB, Liu J, Bruns NE, Tsokos M, Alt FW, Finkel T. A role for the NAD-dependent deacetylase Sirt1 in the regulation of autophagy. Proc Natl Acad Sci U S A. 2008;105:3374–3379. doi: 10.1073/pnas.0712145105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee IH, Finkel T. Regulation of autophagy by the p300 acetyltransferase. J Biol Chem. 2009;284:6322–6328. doi: 10.1074/jbc.M807135200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li JM, Zhang Y, Tang L, Chen YH, Gao Q, Bao MH, Xiang J, Lei DL. Effects of triptolide on hippocampal microglial cells and astrocytes in the APP/PS1 double transgenic mouse model of Alzheimer's disease. Neural Regen Res. 2016;11:1492–1498. doi: 10.4103/1673-5374.191224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lööv C, Hillered L, Ebendal T, Erlandsson A. Engulfing astrocytes protect neurons from contact-induced apoptosis following injury. PLoS One. 2012;7:e33090. doi: 10.1371/journal.pone.0033090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Magnus T, Chan A, Linker RA, Toyka KV, Gold R. Astrocytes are less efficient in the removal of apoptotic lymphocytes than microglia cells: implications for the role of glial cells in the inflamed central nervous system. J Neuropathol Exp Neurol. 2002;61:760–766. doi: 10.1093/jnen/61.9.760. [DOI] [PubMed] [Google Scholar]
- 31.McLean CA, Cherny RA, Fraser FW, Fuller SJ, Smith MJ, Beyreuther K, Bush AI, Masters CL. Soluble pool of Aβ amyloid as a determinant of severity of neurodegeneration in Alzheimer's disease. Ann Neurol. 1999;46:860–866. doi: 10.1002/1531-8249(199912)46:6<860::aid-ana8>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 32.Mueller-Steiner S, Zhou Y, Arai H, Roberson ED, Sun B, Chen J, Wang X, Yu G, Esposito L, Mucke L, Gan L. Antiamyloidogenic and neuroprotective functions of cathepsin B: implications for Alzheimer's disease. Neuron. 2006;51:703–714. doi: 10.1016/j.neuron.2006.07.027. [DOI] [PubMed] [Google Scholar]
- 33.Nagele R G, D’Andrea M R, Lee H, Venkataraman V, Wang HY. Astrocytes accumulate Aβ42 and give rise to astrocytic amyloid plaques in Alzheimer disease brains. Brain Res. 2003;971:197–209. doi: 10.1016/s0006-8993(03)02361-8. [DOI] [PubMed] [Google Scholar]
- 34.Nielsen HM, Mulder SD, Beliën JA, Musters RJ, Eikelenboom P, Veerhuis R. Astrocytic Aβ1-42 uptake is determined by Aβ-aggregation state and the presence of amyloid-associated proteins. Glia. 2010;58:1235–1246. doi: 10.1002/glia.21004. [DOI] [PubMed] [Google Scholar]
- 35.Nixon R. Autophagy, amyloidogenesis and Alzheimer disease. J Cell Sci. 2007;120:4081–4091. doi: 10.1242/jcs.019265. [DOI] [PubMed] [Google Scholar]
- 36.Nixon RA, Yang DS, Lee JH. Neurodegenerative lysosomal disorders: A continuum from development to late age. Autophagy. 2008;4:590–599. doi: 10.4161/auto.6259. [DOI] [PubMed] [Google Scholar]
- 37.Parpura V, Heneka MT, Montana V, Oliet SH, Schousboe A, Haydon PG, Stout RF, Jr, Spray DC, Reichenbach A, Pannicke T, Pekny M, Pekna M, Zorec R, Verkhratsky A. Glial cells in (patho)physiology. J Neurochem. 2012;121:4–27. doi: 10.1111/j.1471-4159.2012.07664.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Parpura V, Verkhratsky A. Homeostatic function of astrocytes: Ca2+ and Na+ signalling. Transl Neurosci. 2012;3:334–344. doi: 10.2478/s13380-012-0040-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Patel NV, Gordon MN, Connor KE, Good RA, Engelman RW, Mason J, Morgan DG, Morgan TE, Finch CE. Caloric restriction attenuates Aβ-deposition in Alzheimer transgenic models. Neurobiol Aging. 2005;26:995–1000. doi: 10.1016/j.neurobiolaging.2004.09.014. [DOI] [PubMed] [Google Scholar]
- 40.Qin W, Yang T, Ho L, Zhao Z, Wang J, Chen L, Zhao W, Thiyagarajan M, MacGrogan D, Rodgers JT, Puigserver P, Sadoshima J, Deng H, Pedrini S, Gandy S, Sauve AA, Pasinetti GM. Neuronal SIRT1 activation as a novel mechanism underlying the prevention of alzheimer disease amyloid neuropathology by calorie restriction. J Biol Chem. 2006;281:21745–21754. doi: 10.1074/jbc.M602909200. [DOI] [PubMed] [Google Scholar]
- 41.Rodríguez JJ, Verkhratsky A. Neuroglial roots of neurodegenerative diseases? Mol Neurobiol. 2011;43:87–96. doi: 10.1007/s12035-010-8157-x. [DOI] [PubMed] [Google Scholar]
- 42.Sauve AA, Wolberger C, Schramm VL, Boeke JD. The biochemistry of sirtuins. Annu Rev Biochem. 2006;75:435–465. doi: 10.1146/annurev.biochem.74.082803.133500. [DOI] [PubMed] [Google Scholar]
- 43.Serrano-Pozo A, Frosch MP, Masliah E, Hyman BT. Neuropathological alterations in Alzheimer disease. Cold Spring Harb Perspect Med. 2011;1:1–23. doi: 10.1101/cshperspect.a006189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sokolowski JD, Nobles SL, Heffron DS, Park D, Ravichandran KS, Mandell JW. Brain-specific angiogenesis inhibitor-1 expression in astrocytes and neurons: implications for its dual function as an apoptotic engulfment receptor. Brain Behav Immun. 2011;25:915–921. doi: 10.1016/j.bbi.2010.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stine WB, Jungbauer L, Yu C, LaDu MJ. Preparing synthetic Aβ in different aggregation states. Methods Mol Biol. 2011;670:13–32. doi: 10.1007/978-1-60761-744-0_2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Verkhratsky A, Butt A. Glial physiology and pathophysiology. Biomedical Sciences, UK. 2013 [Google Scholar]
- 47.Verkhratsky A, Olabarria M, Noristani HN, Yeh CY, Rodriguez JJ. Astrocytes in Alzheimer's disease. Neurotherapeutics. 2010;7:399–412. doi: 10.1016/j.nurt.2010.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wisniewski HM, Wegiel J. Spatial relationships between astrocytes and classical plaque components. Neurobiol Aging. 1991;12:593–600. doi: 10.1016/0197-4580(91)90091-w. [DOI] [PubMed] [Google Scholar]
- 49.Wolfe DM, Lee JH, Kumar A, Lee S, Orenstein SJ, Nixon RA. Autophagy failure in Alzheimer's disease and the role of defective lysosomal acidification. Eur J Neurosci. 2013;37:1949–1961. doi: 10.1111/ejn.12169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wyss-Coray T, Loike JD, Brionne TC, Lu E, Anankov R, Yan F, Silverstein SC, Husemann J. Adult mouse astrocytes degrade amyloid-β in vitro and in situ. Nat Med. 2003;9:453–457. doi: 10.1038/nm838. [DOI] [PubMed] [Google Scholar]
- 51.Xiao Q, Yan P, Ma X, Liu H, Perez R, Zhu A, Gonzales E, Burchett JM, Schuler DR, Cirrito JR, Diwan A, Lee JM. Enhancing astrocytic lysosome biogenesis facilitates Aβ clearance and attenuates amyloid plaque pathogenesis. J Neurosci. 2014;34:9607–9620. doi: 10.1523/JNEUROSCI.3788-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Enriched astrocytes cultured on coverslips were immunofluorescently stained with different nerve cell markers.
The purity of enriched astrocytes was greater than 98%. Glial fibrillary acidic protein (GFAP) staining was specific for astrocytes (green). Iba1 staining was specific for microglia (red). Microtubule-associated protein-2 (MAP-2) staining was specific for neurons (red). Nuclei were stained by 4’,6-diamidino-2-phenylindole (DAPI) (blue). LSM 710 NLO & DuoScan System confocal laser-scanning microscope (Zeiss, Jena, Germany) was used. Scale bar: 50 μm.
SIRT1 does not affect Aβ endocytosis in primary astrocytes.
VECTOR or SIRT1 was overexpressed in primary astrocytes. The cells were incubated with Aβ (1 μM) for 12 hours and then harvested to detect the intracellular Aβ level by western blot analysis. The intensities of the protein bands, which indicated the relative intracellular Aβ level, were measured in three independent experiments. The results are presented as the mean ± SEM (unpaired Student's t-test). SIRT1: Sirtuin 1; Aβ: beta amyloid peptide.
Comprehensive GO clustering analysis of differentially expressed proteins.
The Biological Process, Molecular Function, Cellular Component and KEGG Pathway of differentially expressed proteins were analyzed comprehensively. The P value was set to < 0.001. KEGG: Kyoto encyclopedia of genes and genomes.