

Abstract
Despite great therapeutic potential and development of a repertoire of delivery approaches addressing degradation and cellular uptake limitations, small interfering RNA (siRNA) exhibits poorly controlled tissue-specific localization. To overcome this hurdle, siRNA was complexed to nanoparticles (siRNA/NP) embedded within poly(ethylene glycol)-poly(lactic acid)- dimethacrylate (PEG-PLA-DM) hydrogels with the hypothesis that hydrolytic degradation of ester bonds within the PLA crosslinks would provide tunable, sustained siRNA/NP release. Hydrogels formed from macromers with increasing PLA repeats (e.g., 0 or non-degradable to 5 PLA repeats flanking PEG cores) and mixtures of nondegradable PEG-DM (0 PLA) and degradable PEG-PLA5-DM macromers were investigated. Hydrogels formed only with fully degradable crosslinks degraded rapidly over 6–14 days with limited control over siRNA/NP release. However, hydrogels formed with mixtures of nondegradable and 20%, 50%, and 100% degradable macromers resulted in siRNA/NP release over 3 to 28 days. Subsequently, gene silencing mediated by released siRNA/NP from 20% and 50% degradable hydrogels was sustained for ~28 days. Furthermore, in vivo imaging showed that hydrogel degradation controlled siRNA/NP localization, with sustained siRNA/NP release from 0%, 20% and 50% degradable hydrogels over 28, 21, and 15 days. A model, which accounts for hydrogel degradation rate and siRNA/NP diffusion, was developed to enable rational design of siRNA/NP delivery depots. Overall, this study shows that siRNA/NP release can be sustained via encapsulation in hydrogel depots with tunable degradation kinetics and modeled for a priori design of delivery depots.
Keywords: siRNA delivery, biomaterials, controlled delivery, degradable hydrogel
Introduction
Since the discovery of RNA interference (RNAi) as a mechanism for gene silencing through small interfering RNA (siRNA), there has been considerable advancement in the use of siRNA therapeutically [1, 2]. siRNA can be designed to knockdown nearly any gene and siRNA- mediated gene silencing in vivo has been used to treat diseases such as cancers and viral infections [3]. However, effective in vivo delivery of siRNA faces numerous challenges. siRNA is susceptible to ribonuclease (RNase) degradation and its relatively large molecular weight (~13 kDa) and negative charge prevent it from permeating negatively-charged cellular membranes [3] Another primary concern for siRNA delivery is tissue localization over timeframes consistent with therapeutic requirements. Systemic delivery has been used to deliver siRNA, but renal clearance, cell phagocytosis, aggregation with proteins in the bloodstream, and RNase degradation are some of the myriad barriers to target tissue delivery [3, 4]. Local injections allow for greater target tissue bioavailability of siRNA [5, 6], but has drawbacks including rapid diffusion limiting applicability of this delivery route [7]. Thus, localized and sustained delivery of siRNA remains a great challenge.
For siRNA mediated knockdown, cellular uptake and endosomal escape is required. Polyethyleneimine (PEI) is a commonly used cationic polymeric carrier for siRNA delivery [4]. However, PEI has molecular weight and dose-dependent toxicities, resulting in significant safety concerns [8]. To overcome these challenges, a biocompatible diblock copolymer nanoparticle (NP), poly(dimethylaminoethyl methacrylate)-b-poly-(dimethylaminoethyl methacrylate-co- propylacrylic acid-co-butyl methacrylate) (pDMAEMA-b-p(DMAEMA-co-PAA-co-BMA)) (Fig. 1A), has been developed to complex with siRNA, protecting it from RNase degradation and escape endo-lysosomal trafficking and degradation [9–14]. The copolymer is synthesized through reversible addition-fragmentation chain-transfer (RAFT) polymerization [9]. The cationic diblock copolymers self-assemble into ~23 nm spherical particles in aqueous environments and complexes with negatively charged siRNA via electrostatic interactions (Fig. 1B). DMAEMA and PAA protonate at endo-lysosomal pH, causing a conformational change that results in endosomal membrane disruption [9, 11, 12]. With this method, the siRNA-NP complex can be taken up by cells and translocated in the endosome, where the acidic environment triggers the release of siRNA from the nanoparticle, realizing potent gene silencing [9].
Figure. 1.
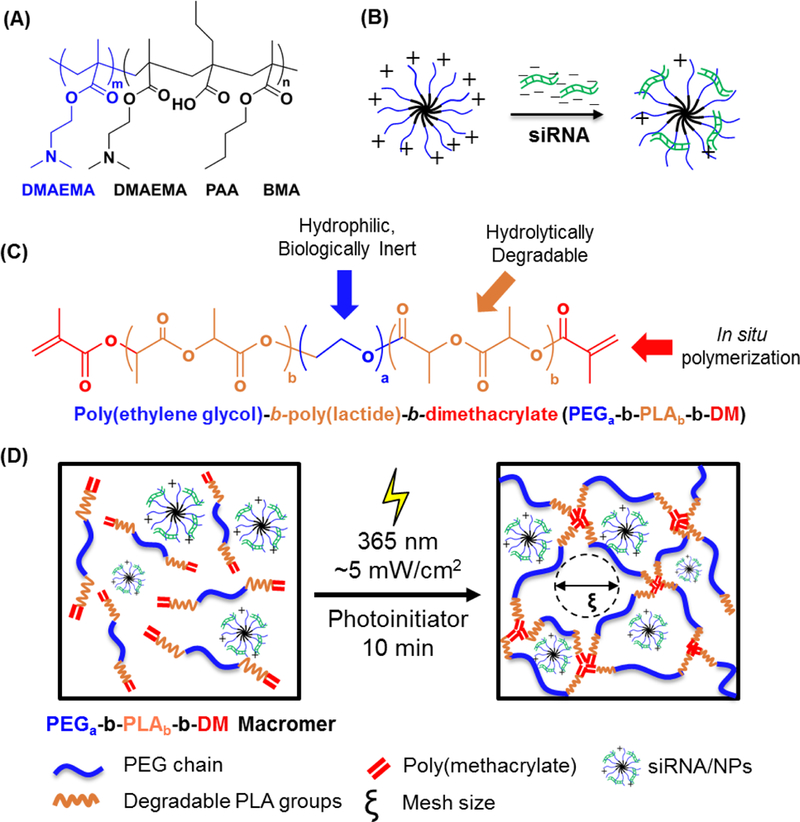
(A) Chemical structure of poly(dimethylaminoethyl methacrylate)-b-poly-(dimethylaminoethyl methacrylate-co-propylacrylic acid-co-butyl methacrylate) (pDMAEMA-b- p(DMAEMA-co-PAA-co-BMA)) (m ~140, n ~80) diblock polymer. (B) Complexation of negatively-charged siRNA to self-assembled nanoparticles with net positive charge. (C) Chemical structure of poly(ethylene glycol)-b-poly(lactide)-b-dimethacrylate (PEGa-b-PLAb- DM, a=91 for 4 kDa PEGa-b-PLAb-b-DM macromers; b=1, 3, or 5 for these studies). (D) The schematic for polymerzation of the hydrogel system with the incorporation of the siRNA-NP complex
While the siRNA-complexed NPs address the issue of biocompatibility, cellular uptake, and endosomal escape, tissue-localized and longitudinal delivery is still a hurdle. Delivery depots, most commonly hydrogels, have been investigated to address issues associated with localized, sustained delivery [10, 15-19]. Hydrogels are polymeric structures that exhibit high water content and whose chemical structure can be altered to change physical properties ranging from mesh size to degradation [20]. Hydrogel degradation can be used to control the release rate of drugs and cells and allow for hydrogel clearance after drug exhaustion [21-24]. In particular, poly(ethylene glycol) (PEG) hydrogels offer extensive flexibility as drug depots, as they are hydrophilic, biocompatible, bio-inert, and easy to functionalize [7, 25-27]. PEG is non- degradable, but by functionalizing PEG with poly(lactic acid) (PLA), a degradable network can be created, as esters within the PLA undergo hydrolysis. Hydrogel degradation can be tuned by varying the numbers of PLA repeats where greater numbers of repeats exhibit faster degradation rate [7]. Additionally, overall degradation can also be achieved by synthesizing hybrid hydrogels composed of macromers with variable degradation [28].
Previous efforts showed hydrogels exhibited degradation kinetics that provide siRNA/NP release with a half-life of ~ 4 days in vivo, resulting in enhanced fracture healing [29]. Here, we exploit two methods to further control hydrogel degradation to tune siRNA/NP release (Fig. 1C). One approach varies the number of PLA repeats to control degradation, and the other changes the ratio of non-degradable PEG-DM to PEG-PLA5-DM precursors to create hydrogels with 0%, 20%, 50% and 100% degradability. The two approaches provided tunable siRNA/NP release over time scales ranging from 3 to 28 days in vivo using a fracture model, as fracture healing occurs over 4–6 weeks and benefits from sustained therapeutic delivery. From these data, a model that predicts siRNA/NP release behavior from degradable hydrogels was established a priori design of delivery depots for future uses.
Materials and Methods
Diblock copolymer synthesis and characterization
The siRNA delivery copolymer is composed of two blocks. The first cationic block is composed of dimethylaminoethyl methacrylate (DMAEMA), which complexes and protects negatively charged siRNA molecules [9, 30, 31], Poly(DMAEMA) (pDMAEMA) was synthesized by reversible addition-fragmentation chain transfer (RAFT) polymerization at 60 °C for 6 hrs as previously described using the radical initiator 2,2’-Azobis(2-methylpropionitrile) (AIBN, Sigma) and chain transfer agent (CTA) 4-cyano-4-[(ethylsulfanylthiocarbonyl)sulfanyl]pentanoic acid) (ECT) under a nitrogen atmosphere in Ν,Ν-dimethylformamide (DMF) [30], The second block is a tercopolymer of DMAEMA, propylacrylic acid (PAA), and butyl methacrylate (BMA). This second block is pH-responsive and designed to enable endosomal escape of siRNA [9], AIBN and pDMAEMA macroCTA were added to DMAEMA, PAA, and BMA (DMAEMA:PAA:BMA=25:25:50) in DMF and the reaction was allowed to proceed at 60 °C for 24 hrs. The reaction was terminated via exposure to atmospheric oxygen and the diblock copolymers were then purified via precipitation in 80:20 pentane:diethyl ether and dried under vacuum. Polymer molecular weights (Mn) were determined using gel permeation chromatography (GPC) (Shimadzu Technologies) using a TSKgel Guard Super H-H guard column (Tosoh Biosciences) and a TSKgel Super HM-N for separation using a column oven at 60 °C. Polymer composition was determined using proton nuclear magnetic resonance (1H- NMR, Bruker Avance400), as previously described [9].
siRNA/NP complexation and characterization
Diblock polymers (4 mg/ml) were dissolved in ethanol and then diluted to 2 mg/ml in phosphate- buffered saline (PBS) [10]. The polymer solution was purified using dialysis with 3,500 Da molecular cutoff in distilled H2O for 24 hrs. The concentration of the resulting polymer solution was then determined gravimetrically after lyophilization. Dynamic light scattering analysis (DLS) via a Malvern Zetasizer was performed to determine NP sZe and zeta potential. The NP solution was sterilized using 0.2 μm filters then utilized to form complexes with Silencer® Select Negative Control No. 1 siRNA (Cat. # 4390843), Silencer® FAM-labeled Negative Control No. 1 siRNA (Cat. # AM4620, Ambion®), or Silencer® Select GAPDH mouse siRNA s98891 (Cat. # 4390771) from Ambion®, as previously described [9,30]. Briefly, siRNA, PBS, and diblock copolymer were added to Eppendorf tubes. The solution was then incubated for 20 min at room temperature for siRNA/NP complexation. The amount of polymers added was calculated to be 4-fold the critical charge ratio, which represents the ratio between positive charges within the pDMAEMA block (0.5 positive charge per mole as 50% of the DMAEMA tertiary amines are protonated at physiological pH) and negative charges of siRNA molecules (42 negative charges per mole) at which there is no uncomplexed siRNA, which was determined empiracally by gel electrophoresis [31].
Hydrogel formation and characterization of swelling and degradation behavior
Poly(ethylene glycol)-b-poly(lactide)-b-dimethacrylate (PEGa-PLAb-DM) macromers were synthesized as previously described by functionalizing linear PEG (Alfa Aesar, MW = 4 kDa, a=91) with d,l-lactide via microwave-assisted methacrylation using methacrylic anhydride [7,23]. Briefly, PEG and D,L-lactide at 1:2, 1:6, and 1:10 molar ratios were reacted via ring-opening polymerization with a catalyst, tin (II) 2-ethylhexanoate [7]. To form PEG-b-PLA-b-DM, a microwave-assisted methacrylation was performed as previously developed [7]. PEG polymers were then isolated via precipitation in ice-cold diethyl ether and dried under vacuum. 1H NMR analysis (Bruker Avance 400 MHz, CDCI3) was used to determine the number of lactide units and methacrylate groups per PEG macromer, as described previously [7]
siRNA/NP loaded hydrogels were formulated by mixing siRNA/NPs, PEG-b-PLA-b-DM (10 wt%), and the photoinitiator, lithium phenyl-2,4,6-trimethylbenzoylphosphinate (l-AP, synthesized as previously described [32]) (0.05 wt%) in PBS, pipetting 40 μl into each of a Teflon-based array of cylindrical molds (6×3 mm), and exposing to ~5 mW/cm2, 365 nm light for 10 min. To determine the swelling ratio of PEG hydrogels, wet weight (Ww) and dry (lyophilized) weights (Wd) were measured gravimetrically at various time points over 4 weeks. The swelling ratio (Q) was calculated using Q = wet weight (Ww) / initial dry weight (Wdi). The mesh size of hydrogels over the time course of degradation were calculated based on the swelling ratio using the Flory-Rehner equation [33], The degradation with was represented by percent mass loss was calculated by (Wdi -Wd)/Wdi χ 100.
Charaterizaiton of siRNA/NP release from hydrogels
NPs complexed with FAM-labeled siRNA, as previously described, were incorporated into 40 μΙ PEG hydrogels at 1 μΜ siRNA concentration. Hydrogels were washed briefly PBS to remove loosely associated siRNA/NP at the hydrogel surface. Hydrogels were placed in PBS buffer in well plates on a shaker at 37 °C with protection from light. siRNA/NP releasate were collected at various time points over hydrogel degradation. Release of siRNA was quantified using a plate reader (Tecan M200 Infinite, 480 nm/520 nm for FAM-labeled siRNA).
Analysis of sustained gene silencing efficiency of hydrogel-released siRNA/NPs
Mouse MSCs (mMSCs) (Cyagen) derived from C57BL/6 mice were cultured at 37 °C and 5% C02 in growth media consisting of low glucose Dulbecco’s Modified Eagle Medium (DMEM, Gibco) supplemented with 10% fetal bovine serum (FBS), and 100 units/ml Penicillin-Streptomycin (Gibco). mMSCs were seeded in 24-well plates at 8,000 cells/cm2 one day before treatment. For GAPDH siRNA/NP treated groups, nanoparticles complexed with GAPDH siRNA were used to treat cells at 40 nM. For hydrogel releasate treated groups, MSCs were treated with hydrogel releasate that was collected at day 1,4, 7, 16, 22, and 28, as described in the previous section. Two days after each treatment, cells were washed gently with 0.5 ml PBS twice and lysed using TRK Lysis Buffer (OMEGA Bio-Tek). RNA extraction, cDNA synthesis, and RT-PCR were performed as described previously [34], GAPDH expression was quantified based on the reaction efficiency and normalized to β-actin expression and comparing to untreated cell populations [34].
Characterizing in vivo sustained release of siRNA/NP from hydrogels
All animal experiments were approved by the Institutional Animal Care and University Committee of Animal Resources at the University of Rochester. In vivo studies were performed using a previously-developed mid-diaphyseal femur fracture model in female 6–8 week old Balb/c mice (Harlan Laboratories, Indianapolis, IN) after 1 week of acclimatization. Prior to surgeries, mice were anesthetized with ketamine (60 mg/kg) and xylazine (4 mg/kg) intraperitoneally. After treatment with iodine and alcohol for disinfection, an 8 mm long incision was made on the skin of the femur and blunt dissection of muscle was used to expose the midshaft of the femur. Femur fractures then were created using a rotary Dremel attached with a diamond blade. A 25-gauge needle was inserted into the medullary canal of the femur from the distal end to stabilize the fracture. Cy5-labeled non-targeted siRNA (Dharmacon®) complexed with NPs were loaded into PEG hydrogels as reported previously [35], Hydrogels (0.1 nmole siRNA in 20 μl hydrogels) were wrapped around femur fracture sites. After muscle reappositioning, skin was sutured and X-ray images (Faxitron X-ray, Wheeling, IL) were used to verify fracture and fixation directly after surgery. Buprenorphine (0.05 mg/kg IP) was administered prior to fractures and every 6–12 hours thereafter for 3 days post-fracture.
Localization and release of siRNA/NPs (649 nm/670 nm for Cy5) was monitored longitudinally using a XENOGEN/IVIS imaging system (PerkinElmer). IVIS was performed 0, 4, 7, 14, 18, 21 and 28 days after implantation using 2% isoflurane gas. Quantification of hydrogel localization from IVIS imaging was achieved by measuring the total radiant efficiency of regions of interest and normalizing to day 0.
Theoretical model for prediction of siRNA/NP release from hydrogels
The release of NPs from hydrogels prepared from varied ratios of PEG-DM and PEG-PLA5-DM was modeled. Generally, theoretical models for predicting molecule diffusion coefficients have the following general form [20]:
| (1) |
DA and D0 represent drug diffusion coeffients in the swollen hydrogel network and in pure solvent, respectively, while 𝑟𝑠is the size of the drug to be delivered and v2,S is the polymer volume fraction in the swollen state. This general expression takes into account factors affecting drug release such as the structure of gel, the polymer composition, the water content, and the size of the diffusing molecules. The theoretical estimate of the diffusion coefficients in water, D0, was made with the Stokes-Einstein equation:
| (2) |
where k is the Boltzmann constant (1.38×10–23 J-K−1), T the absolute temperature (310.6 K), η the dynamic viscosity of water (0.001 Pa-s), 𝑟sthe hydrodynamic radius of nanoparticles (23 nm).
For degradable hydrogels, DA changes as the network degrades due to an increase in mesh size and a decrease in polymer volume fraction over time. The diffusivity correlation shown in Eq. (3) was used to calculate DA during degradation:
| (3) |
Fick’s law of diffusion with either constant or variable diffusion coefficients was used in modeling diffusion-controlled release [36], The cumulative fractional drug release (Mt/Minf) can be described by the time-dependent diffusion coefficient and time-dependent thickness defined by diffusion time constant (β) and degradation time constant (k) [37], For the PEGa-PLAb-DM hydrogel system, k of hydrogels with vaired percentage of degradable PEG can be modeled by polynomial fitting based on release data from 0%, 20%, 50% and 100% degradable hydrogels (Figure S1) with R2 of 1. Assume hydrogels undergo bulk degradation, where thickness remains constant throughout the drug delivery period, NP release can be predicted using [37]:
| (4) |
Statistical Analysis
All studies had sample sizes of at least 3 per treatment, which are specifically denoted in figure legends. Differences between groups were compared using one-way analysis of variance (ANOVA) or two-way ANOVA with Tukey’s or Dunnett’s post-hoc testing, as indicated in figure legends. For in vivo studies, 6–12 mice per condition were used based on an a priori power analysis and previous in vivo live animal imaging. A p-value of less than 0.05 was considered significant. Statistics were assessed with GraphPad Prism 6 Software.
Results
Characterization of hydrogel degradation with varied macromer PLA
A series of methacrylate-functionalized PEG polymers were synthesized with 0, 1, 3, and 5 lactide repeats. These polymers were then used to form hydrogels at 10 wt% via photopolymerization as previously described [35] Hydrogels were incubated at 37 °C with buffered cell culture media to mimic standard physiological conditions. Hydrogel degradation (Fig. 2A) was monitored gravimetrically over ~1 month using wet and dry weights of hydrogels at equilibrium swelling ratio. From this data, the mesh size of hydrogels was calculated using the Flory-Rehner equation [38] (Fig. 2B). PEG-DM did not degrade over the course of the study (Fig. 2A) and remained at a mesh size of 9 ± 3 nm over the study as expected due to relative non-degradability (Fig. 1B). In contrast, PEG-PLA5-DM hydrogels degraded the fastest, over the course of 3 days, followed by PEG-PLA3-DM (6 days) and PEG-PLA1-DM (14 days) (Fig. 2A). Initially, all hydrogels had mesh sizes of ~19 ± 4 nm. By day 3, hydrogels with the greatest numbers of PLA repeats had the largest mesh sizes, with PEG-PLA5-DM, PEG-PLA3-DM, and PEG-PLA1-DM exhibiting mesh sizes of 33 ± 2 nm, 30 ± 1 nm, and 23 ± 1 nm, respectively.
Fig. 2.
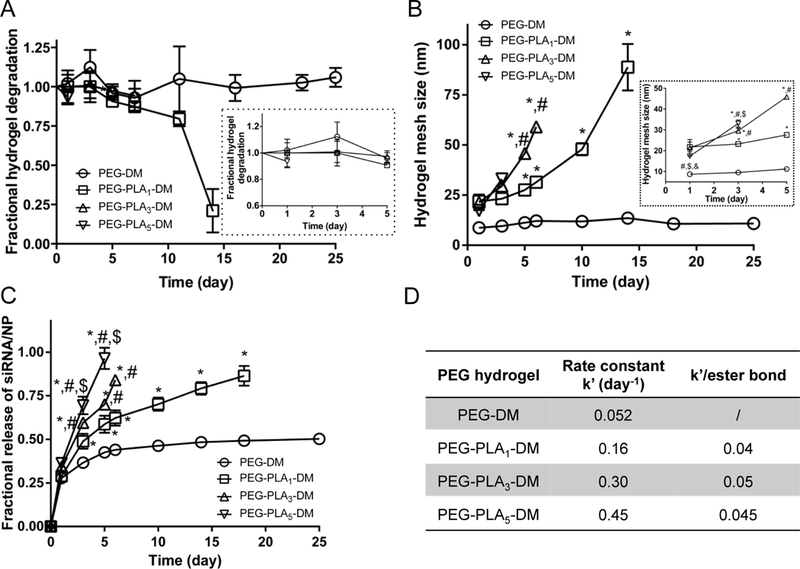
Degradation and swelling behavior of PEG hydrogels with varied numbers of degradable linkers. (A) Degradation behavior represented by mass change of PEG hydrogels normalized to day 1. (B) Mesh size of the hydrogels calculated using the Flory-Rehner equation. Hydrogels were incubated in media for 24 hours to reach swelling equilibrium before the initial measurement at day 1. (C) The fractional release of siRNA-NP from hydrogels increases as the PLA repeats increase. N=3 for all samples except nondegradable gels where N=6, mean ± standard deviation. 2-way ANOVA with Tukey’s post doc analysis. *, #, $, and & indicate significant difference versus PEGs with 0, 1, 3, and 5 PI_A, respectively. (D) Rate constant (k’) fit from first order release models.
Release of siRNA/NP from gels was evaluated over time (Fig. 2C). Non-degradable PEG-DM released only −50% siRNA/NP by day 25. PEG-PLA5-DM release was 100% by day 5 due to rapid degradation. PEG-PLA3-DM and PEG-PLA1-DM reach ~100% release of siRNA/NP by day 6 and 18, respectively. Degradation rate constants (k’) of hydrogels were calculated from fitting to a first order release model (Fig. 2D), with hydrogels with 0, 1, 3, and 5 PLAs showing k’ of 0.052, 0.159, 0.296, and 0.451 day-1. When calculating k’ per ester bond, 1, 3, and 5 PLAs (with 2, 6, and 10 ester bonds) exhibited comparable values of 0.04, 0.05, and 0.045 day−1 per bond, respectively. This indicates and degradation rate is proportional to the number of esters within the PLA blocks. Increasing the number of PLA repeats also increased the release rate of siRNA/NP.
Degradation of hydrogels formed with varied degradable to non-degradable macromers
To prolong hydrogel degradation and sustain siRNA/NP release, another series of hydrogels were synthesized from variable combinations of PEG-DM and PEG-PLA5-DM. PEG-PLA5-DM was chosen as the 100% degradable component as it degrades quickly (~3–5 days). By increasing the proportion of degradable macromer, hydrogels with 0%, 20%, 50%, and 100% were synthesized. The 100% degradable hydrogel degraded the fastest, over 5 days, and achieved the largest mesh size at day 5 of 59 ± 27 nm (Fig. 3A). The 50%, 20% and 0% hydrogels did not fully degrade over the course of 28 days (Fig. 3A), due to the presence of non-degradable macromers. After incubating in media for 24 hours to reach equilibrium swelling ratios, the 0%, 20%, 50% and 100% compositions had initial mesh sizes of 7.3 ± 0.5 nm, 14 ± 4 nm, 21 ±1 nm and 18 ± 0.8 nm, respectively (Fig. 3B). The slight increase in mesh size of degradable hydrogels indicates the rapid onset of degradation. The non-degradable hydrogel maintained a mesh size of 7.7 ± 0.5 nm over the course of 28 days. For 20%, 50%, and 100% degradable hydrogels, there was an increasing trend in mesh size that ranged from ~14 to 83 nm.
Fig. 3.
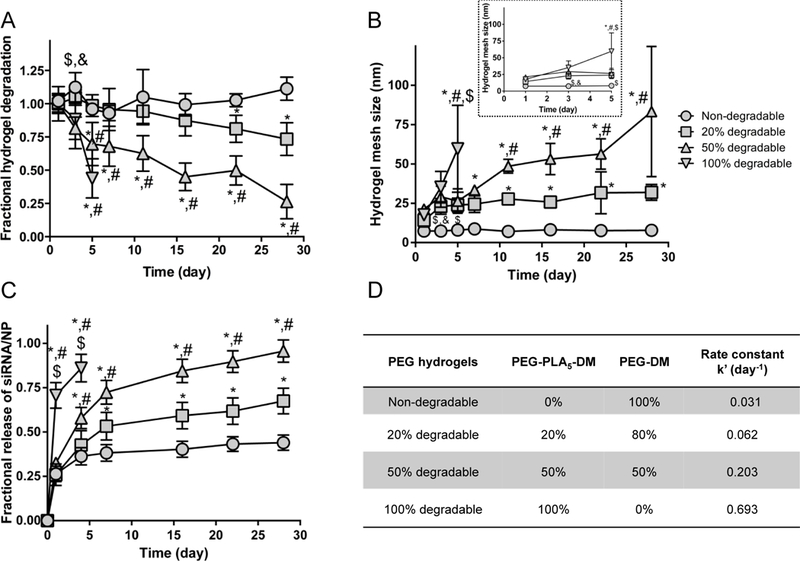
Degradation and swelling behavior of PEG hydrogels synthesized with varied ratio of degradable:non-degradable polymers. (A) Degradation behavior represented by mass change of PEG hydrogels over a course of 28 days normalized to day 1. (B) Mesh size of the hydrogels calculated using the Flory-Rehner equation. (C) The fractional release of siRNA-NP from hydrogels increases as the degradability increases. N=3 for all samples except nondegradable gels where N=6, mean ± standard deviation. 2-way ANOVA with Tukey’s post doc analysis. *, #, $, and & indicate significant difference versus PEGs with 0%, 20%, 50%, and 100% PLA, respectively. (D) Rate constant (k’) fit from a first order release model.
The release profiles of siRNA/NP from hydrogels were studied (Fig. 3C). An initial burst release was observed in all hydrogels, with 100% degradable hydrogels showing the fastest release at day 1. After day 1, 100% degradable hydrogels showed faster release compared to other hydrogels until day 3 when degradation was complete. After day 3, 0%, 20%, 50% still followed the trend where greater degradation led to faster siRNA/NP release. By day 28, the 20% degradable and non-degradable hydrogels had not reached 100% release, which indicates that siRNA/NP release is likely sustained for even longer periods of time. Degradation rate constants (k’) of hydrogels were calculated from fitting to a first order release model (Fig. 3D), with 0%, 20%, 50%, and 100% degradable hydrogels showing k’ of 0.031, 0.062, 0.203, and 0.693 day-1. Overall, the presence of PEG-DM in all hydrogels maintained hydrogel structure longer compared to gels fully formed from degradable macromers, and increased degradability increases the release rate of siRNA/NPs.
Evaluating in vitro gene silencing achieved by controlled siRNA/NP release from hydrogels
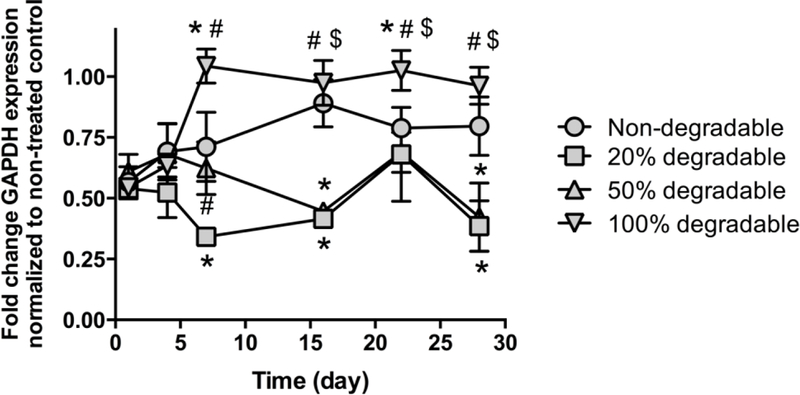
To study gene silencing efficiency with tunable siRNA/NP release (varied degradable to non- degradable polymer ratios), mMSCs were treated by hydrogel-released GAPDH siRNA/NP over time and gene expression evaluated using RT-PCR. Note that gels with varied PLA repeats resulted in rapid siRNA/NP release that were not expected to achieve sustained knockdown, therefore were not included in this study. Fully degradable hydrogel released siRNA/NP resulted in 54% GAPDH knockdown at day 1, which returned to baseline levels due to rapid hydrogel degradation and minimal release beyond day 1 (Fig. 5). MSCs treated with siRNA/NP released from 50% and 20% degradable hydrogels exhibited similar gene expression profiles, where ~50% silencing was maintained over 4 weeks, due to sustained siRNA/NP release (Fig. 5). An initial burst release was also observed in non-degradable hydrogels, likely due to surface diffusion of loosely entrapped NP, which leads to detectable and modest gene silencing at day 1 and 7, which returned to baseline levels at day 14 (Fig. 4).
Fig. 5.
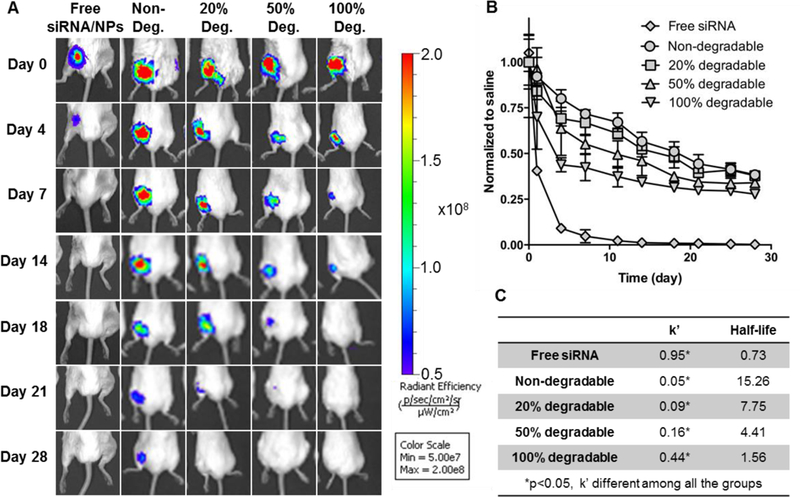
In vivo siRNA-NP localization qualitatively decreased as the hydrogel degradability increased as visualized by MS fluorescent imaging. (A) Representative IVIS images of Cy5- siRNA/NP loaded hydrogels placed around femur fractures over time. (B) Quantification of siRNA/NP localization from IVIS imaging. Data showed the total radiant efficiency of drawn region of interest (ROI) in IVIS images normalized to day 0. Mean ± STDEV, N=6. (C) Rate constant (k’) and half-lives are derived from a one phase decay model.
Fig. 4.
Fractional release of siRNA-NP from hydrogels increases as a function of overall hydrogel degradability. *, #, and $ indicates significant difference between nondegradable and 100%, 50% and 20% degradable hydrogels, respectively. & indicates significant difference between 20% and 100% degradable hydrogels. (p<0.05, N=4, mean ± standard deviation. 2- way ANOVA with Tukey’s post hoc analysis).
In vivo sustained release of siRNA/NP from hydrogels
Temporal Cy5-siRNA/NP localization at murine mid-diaphyseal femur fracture models was evaluated. Hydrogels with varied degradable to non-degradable macromer ratios were used for this evaluation, as these formulations exhibited the greatest differences in siRNA/NP release kinetics, achieving sustained release of siRNA/NP up to 28 days versus ~14 days or less for gels composed of macromers with varied repeats of PLA. Local injection of free siRNA/NP was used as a control and showed rapid dispersal and clearance from the fracture site by day 4 (Fig. 5A). In contrast, 0%, 20%, 50%, and 100% degradable hydrogels showed prolonged localization with half-lives ranging from 14 to 28 days (Fig. 5A). In vivo localization of siRNA/NP follows the degradation trend observed in vitro, with 0% degradable gels exhibiting the greatest persistence. siRNA/NP localization was quantified by normalizing the total radiant efficiency of region of interest (ROI) to day 0 (Fig. 5B). Fitting into a non-linear one phase decay model revealed that the rate constant k’ is significantly different among all the groups (Fig. 5C). Free injection of siRNA/NP has a rapid clearance with half-life of 0.73 days, while hydrogels exhibited half-lives of 1.5 to 15 days, which decreased as a function of overall hydrogel degradability. Furthermore, in vivo k’ trends were comparable with in vitro k’ (Fig. 3). These results indicate that NP/hydrogel depots are able to maintain locally high siRNA/NP concentration in the surrounding tissues over an extended period, and that the in vivo localization can be tuned effectively by varying the degradable to non-degradable macromer ratio during hydrogel formation.
Theoretical modeling predicts siRNA/NP release from hydrogels with varied degradation rates
A model was developed based on Fickian diffusion of siRNA/NP, polymer degradation, and swelling behavior (Fig. 6). Predicted longitudinal release is shown in terms of mass fraction released into the surrounding solution (Mt/Minf). With increases in degradation rate of hydrogels prepared using degradable PEG ranging from 20% to 100%, a decrease in half-lives were observed, ranging from 2 to 28 days. This approximation is appropriate for predicting release rate from hydrogel with varied percentage of degradable PEG as it correlates with the actual data from release study.
Fig. 6.
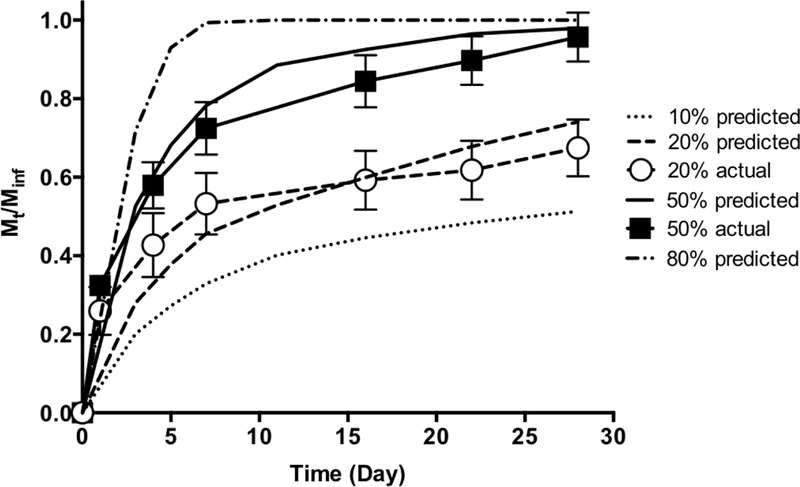
Theoretical prediction of siRNA/NP release from hydrogels synthesized with PEG with varied degradation rates plotted together with actual release from 20% and 50% degradable hydrogels (Fig. 3).
Discussion
Despite successful implementation of hydrogel-mediated siRNA delivery, how various polymer properties can be exploited to control in vitro and in vivo release has yet to be established [27]. This study introduces a system that combines successful siRNA delivery via diblock copolymer NPs and controlled degradation of PEGa-PLAb-DM hydrogels to enable tunable and longitudinal delivery of siRNA in vivo. The work is of significance to the development of hydrogels with well- controlled drug release behaviors. The system developed has great practical utility as it provides up to one month of siRNA/NP delivery, which can be tuned based on application requirements.
Hydrogels are crosslinked networks that contain > 90 wt% water, as polymer chains are hydrophilic. Therefore, hydrogels mimic natural soft tissue, creating a suitable depot for in vivo delivery of therapeutics [39]. Hydrogels have been used to deliver a variety of therapeutics, including small molecules [2], peptides [40,41], as well as siRNA [35]. To date, there is no study that exploits PEG hydrogel compositions to alter siRNA/NP release kinetics. One of the biggest advantages of PEG hydrogels is chemical versatility. Modifying PEG macromer chemistry, including the incorporation of degradable linkers as used here [42], provides myriad possibilities to predict and control drug release. Additionally, PEG molecular weight and weight percentage have been explored to control hydrogel mesh size and release rate of a variety of drugs [41]. Increasing PEG molecular weight and decreasing PEG weight percentage increases hydrogel mesh size, leading to more rapid release. These hydrogel characteristics have also been explored for cell encapsulation and delivery, where cell adhesion, cell growth, and cell viability are all related to hydrogel mesh size and mechanical strength, which are tuned by polymer properties [23, 43, 44].
Here, we show tunable degradation and subsequent siRNA/NP release as a function of macromer chemistry and hydrogel composition. Specifically, hydrogels formed from PEG macromers flanked with lactide repeats of 0, 1, 3 and 5, and of 0%, 20%, 50%, and 100% degradability using combinations of PEG-DM and PEG-PLA5-DM. The results showed that increasing PLA repeats resulted in increased degradation and release rates. However, release was only sustained for 7–10 days using hydrogels with variable PLA repeats. In contrast, changing the ratios of degradable to non-degradable polymers provided greater dynamic range of siRNA/NP release kinetics in vitro and in vivo. In this system, the incorporation of non- degradable PEG maintains the structural integrity of hydrogels and realizes greater control over siRNA/NP diffusion from the gels. In fact, non-degradable, 20%, and 50% degradable hydrogels were still intact after 28 days, indicating siRNA release is likely sustained for longer than 28 days. Finally, hydrogels with varied degradable polymer ratios were tested in vivo and showed localization profiles that corroborates in vitro characterization, indicating that tuning the degradation of hydrogels is a promising strategy for controlled release of siRNA in vivo. Non- degradable hydrogels without PLA still exhibit some hydrolysis due to esters juxtaposed to methacrylate functionalities, although degradation rates are on time scales much greater than studied here. Additionally, tissue resident macrophages recruited to the implant site may expedite degradation via enzyme or acid-catalyzed processes [45].
In hydrogel drug delivery depots, the kinetics of ester bond hydrolysis and diffusion of nanoparticles within hydrogels control release kinetics. PEG-PLA hydrogels undergo bulk degradation upon exposure to water, whereby network crosslinks are broken hydrolytically throughout the hydrogel, leading to decreased crosslinking density and increased mesh sizes [7]. Therefore, greater number of esters result in greater probability of chain scission, which has shown to result in more rapid drug release [46]. Results in this study are in agreement with these findings, where PEG-PLA5-DM, which contains the greatest number of ester bonds, exhibited fastest siRNA/NP release. When the ratio of PEG-DM and PEG-PLAs-DM was varied to adjust degradation kinetics, the presence of PEG-DM led to greater persistence of hydrogels, resulting in prolonged yet differential release and subsequent gene silencing efficacy. Although only a limited number of gels were examined, this approach can be applied to generate hydrogels that degrade beyond the time range of this study by using different degradable macromer chemistries or PLA repeats, or changing the ratio of non-degradable to degradable macromer ratio in the hydrogels.
While gels provide a convenient and tunable depot, some inconsistencies regarding hydrogel properties and siRNA/NP release are observed. As the initial mesh sizes of hydrogels (<20 nm) are smaller than siRNA/NP size (~27 nm), initial release should be limited to loosely-associated siRNA/NP at the gel surface. However, a fairly substantial (up to about 30%) siRNA/NP burst release is realized initially from all gels and at steady state from non-degradable gels whose mesh size is statistically unchanged over times investigated here. This level is much greater than expected based on loosely-associated siRNA/NPs considering the surface area to volume ratios of the gels. In addition to surface-assocated siRNA/NPs, chain-polymerized hydrogels, which are employed here, contain significant network heterogeneities, which underpins variable mesh sizes [47]. Thus, it is likely that some embedded siRNA/NP have lower diffusional hindrance, contributing to this apparent deviation. This likelihood is further supported by sustained siRNA/NP release observed from some degradable gels despite overall gel mesh sizes exceeding siRNA/NP size. Greater consistency between mesh size and release may be afforded by use of alternative crosslinking chemistries including click-based thiol-ene polymerizations which form networks with greater homogeneity [47].
Previous proof-of-concept studies showed that hydrogels with 100% degradable PEG macromers with 2 PLA repeats exhibited half-live of siRNA/NP release of ~ 3.9 days in vivo, with successful augmentation of fracture healing in a mouse femoral fracture model via delivery of a therapeutic siRNA targeting WW domain containing E3 ubiquitin protein ligase 1 (Wwp1) [29]. The mechanical properties of treated fractures showed significant enhancement compared to non-treated and vehicle controls, however, were still below the non-fractured bone controls [29]. This is likely due to insufficient local concentration and longevity of siRNA/NP at fractures. Therefore, the studies herein further explored the tunability of this hydrogel-based drug delivery platform for in vivo siRNA/NP delivery and demonstrated that the half-life can be sustained to ~15 days. The longer half-lives observed here are likely be able to further enhance fracture healing, which will be explored in future studies. In addition, theoretical release profiles based on the model (Fig. 6) showed agreement with experimental data (20% and 50% degradable hydrogel) indicating robust predictions for future experimental approaches. This model is versatile since parameters such as NP size and hydrogel degradation kinetics can be easily adjusted for simulation of NP release over time for selection of optimized hydrogel formulation.
The system developed in this study has broad potential for local, sustained macromolecular drug delivery with respect to drug classes as well as applications. The system developed here can also be applied to release large molecules (proteins, peptides), or other drug-loaded NPs. For direct delivery of small molecules, however, the potential of this system is limited since small molecule size (~10 A) is smaller than gel mesh size. For alternative applications, a prophylactic hydrogel patch has been developed recently for local treatment of colon cancer, where gold nanoparticles/rods were loaded with siRNA and VEGF inhibitor, leading to complete tumor remission [48]. With hydrogels with engineered release kinetics, multi-modal approach can be easily achieved, leading to efficacious drug delivery to the right site with controlled doses and treatment durations. Though not exploited here, in situ polymerization of hydrogels is possible [49]. Furthermore, as the hydrogel is degradable, no secondary surgery is required for implant retrieval.
Conclusions
The hybrid siRNA/NP hydrogel delivery system explored here combines the potent gene silencing efficiency of siRNA/NP complexes and controlled degradation of the PEG-based hydrogel to achieve localized and tunable delivery of siRNA-NP. The hydrogel system synthesized from combinations of PEG-DM and PEG-PLA5-DM showed greater control over release (e.g., greater dynamic range of release kinetics enabling delivery over longer time scales) versus PEGa-PLAb-DM with varying PLA repeats. Faster hydrogel degradation and increase in mesh size were correlated with faster siRNA-NP release. A theoretical model was established for predictability of siRNA/NP release for a wide range of hydrogel and nanoparticle compositions. The combination of PEG-DM and PEG-PLA5-DM in a degradable hydrogel siRNA-NP system shows great potential as a therapeutic tool for a wide variety of applications.
Supplementary Material
Acknowledgments
Funding for this study was provided by National Science Foundation (NSF) DMR1206219 and CBET1450987, National Institutes of Health (NIH) R01 AR064200, AR056696, and DE018023, and New York State Stem Cell Science (NYSTEM) funding N11G-035. Equipment, including the IVIS Live Animal Imaging System, Visiopharm software, and whole-slide scanner were purchased through NIH funds (S10-RR026542, P30-AR069655, and S10-RR027340). The authors would like to thank Dr. James L. McGrath (University of Rochester, Department of Biomedical Engineering) for the use of his equipment. The authors also wish to thank T.J. Sheu and Emma Gira for their assistance with mouse surgeries and IVIS imaging, respectively.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Kanasty R, Dorkin JR, Vegas A, Anderson D, Delivery materials for siRNA therapeutics, Nature materials, 12 (2013) 967. [DOI] [PubMed] [Google Scholar]
- [2].Benoit DS, Nuttelman CR, Collins SD, Anseth KS, Synthesis and characterization of a fuvastatin-releasing hydrogel delivery system to modulate hMSC differentiation and function for bone regeneration, Biomaterials, 27 (2006) 6102–6110. [DOI] [PubMed] [Google Scholar]
- [3].Whitehead KA, Langer R, Anderson DG, Knocking down barriers: advances in siRNA delivery, Nature reviews. Drug discovery, 8 (2009) 129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Whitehead KA, Langer R, Anderson DG, Knocking down barriers: advances in siRNA delivery, Nat Rev Drug Discov, 8 (2009) 129–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Arany S, Benoit DS, Dewhurst S, Ovitt CE, Nanoparticle-mediated gene silencing confers radioprotection to salivary glands in vivo, Mol Ther, 21 (2013) 1182–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Freeberg MAT, Farhat YM, Easa A, Kallenbach JG, Malcolm DW, Buckley MR, Benoit DSW, Awad HA, Serpine1 Knockdown Enhances MMP Activity after Flexor Tendon Injury in Mice: Implications for Adhesions Therapy, Scientific Reports, 8 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Hoffman MD, Van Hove AH, Benoit DS, Degradable hydrogels for spatiotemporal control of mesenchymal stem cells localized at decellularized bone allografts, Acta biomaterialia, 10 (2014) 3431–3441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Urban-Klein B, Werth S, Abuharbeid S, Czubayko F, Aigner A, RNAi-mediated genetargeting through systemic application of polyethylenimine (PEI)-complexed siRNA in vivo, Gene therapy, 12 (2005) 461. [DOI] [PubMed] [Google Scholar]
- [9].Benoit DS, Boutin ME, Controlling mesenchymal stem cell gene expression using polymer-mediated delivery of siRNA, Biomacromolecules, 13 (2012) 3841–3849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Wang Y, Malcolm DW, Benoit DS, Controlled and sustained delivery ofsiRNA/NPs from hydrogels expedites bone fracture healing, Biomaterials, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Benoit DS, Boutin ME, Controlling mesenchymal stem cell gene expression using polymer-mediated delivery of siRNA, Biomacromolecules, 13 (2012) 3841–3849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Convertine AJ, Benoit DS, Duvall CL, Hoffman AS, Stayton PS, Development of a novel endosomolytic diblock copolymer for siRNA delivery, Journal of controlled release, 133 (2009) 221–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Malcolm DW, Freeberg MA, Wang Y, Sims KR Jr, Awad HA, Benoit DS, Diblock copolymer hydrophobicity facilitates efficient gene silencing and cytocompatible nanoparticle- mediated siRNA delivery to musculoskeletal cell types, Biomacromolecules, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Malcolm DW, Varghese JJ, Sorrells JE, Ovitt CE, Benoit DS, The Effects of Biological Fluids on Colloidal Stability and siRNA Delivery of a pH-Responsive Micellar Nanoparticle Delivery System, ACS nano, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Nguyen K, Dang PN, Alsberg E, Functionalized, biodegradable hydrogels for control over sustained and localized siRNA delivery to incorporated and surrounding cells, Acta biomaterialia, 9 (2013) 4487–4495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Krebs MD, Alsberg E, Localized, targeted, and sustained siRNA delivery, Chemistry, 17 (2011) 3054–3062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Shapira K, Dikovsky D, Habib M, Gepstein L, Seliktar D, Hydrogels for cardiac tissue regeneration, Bio-medical materials and engineering, 18 (2008) 309–314. [PubMed] [Google Scholar]
- [18].Peppas NA, Bures P, Leobandung W, Ichikawa H, Hydrogels in pharmaceutical formulations, European journal of pharmaceutics and biopharmaceutics : official journal of Arbeitsgemeinschaft fur Pharmazeutische Verfahrenstechnik e.V, 50 (2000) 27–46. [DOI] [PubMed] [Google Scholar]
- [19].Slaughter BV, Khurshid SS, Fisher OZ, Khademhosseini A, Peppas NA, Hydrogels in regenerative medicine, Advanced materials, 21 (2009) 3307–3329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Lin CC, Metters AT, Hydrogels in controlled release formulations: network design and mathematical modeling, Advanced drug delivery reviews, 58 (2006) 1379–1408. [DOI] [PubMed] [Google Scholar]
- [21].Silviya JBL Zustiak P, Hydrolytically Degradable Poly(Ethylene Glycol) Hydrogel Scaffolds with Tunable Degradation and Mechanical Properties, Biomacromolecules, 11 (2010) 1348–1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].H Van Hove A, Beltejar MJ, Benoit DS, Development and in vitro assessment of enzymatically-responsive polyethylene glycol) hydrogels for the delivery of therapeutic peptides, Biomaterials, 35 (2014) 9719–9730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Hoffman MD, Xie C, Zhang X, Benoit DS, The effect of mesenchymal stem cells delivered via hydrogel-based tissue engineered periosteum on bone allograft healing, Biomaterials, 34 (2013) 8887–8898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Hoffman MD, Van Hove AH, Benoit DS, Degradable Hydrogels for Spatiotemporal Control of Mesenchymal Stem Cells Localized at Decellularized Bone Allografts, Acta Biomater, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].R Nuttelman C, Benoit DS, Tripodi MC, Anseth KS, The effect of ethylene glycol methacrylate phosphate in PEG hydrogels on mineralization and viability of encapsulated hMSCs, Biomaterials, 27(2006) 1377–1386. [DOI] [PubMed] [Google Scholar]
- [26].Benoit DS, Dumey AR, Anseth KS, The effect of heparin-functionalized PEG hydrogels on three-dimensional human mesenchymal stem cell osteogenic differentiation, Biomaterials, 28 (2007) 66–77. [DOI] [PubMed] [Google Scholar]
- [27].Lin CC, Anseth KS, PEG hydrogels for the controlled release of biomolecules in regenerative medicine, Pharmaceutical research, 26(2009)631–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Benoit DS, Dumey AR, Anseth KS, Manipulations in hydrogel degradation behavior enhance osteoblast function and mineralized tissue formation, Tissue engineering, 12 (2006) 1663–1673. [DOI] [PubMed] [Google Scholar]
- [29].Shu L, Zhang H, Boyce BF, Xing L, Ubiquitin E3 ligase Wwpl negatively regulates osteoblast function by inhibiting osteoblast differentiation and migration, Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research, 28 (2013) 1925–1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Benoit DS, Convertine AJ, Duvall CL, Hoffman AS, Stayton PS, Development of a novel endosomolytic diblock copolymer for siRNA delivery, Journal of controlled release : official journal of the Controlled Release Society, 133 (2009)221–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Malcolm DW, Sorrells JE, Van Twisk D, Thakar J, Benoit DS, Evaluating side effects of nanoparticle-mediated siRNA delivery to mesenchymal stem cells using next generation sequencing and enrichment analysis, Bioengineering & translational medicine, 1 (2016) 193–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Fairbanks BD, Schwartz MP, Bowman CN, Anseth KS, Photoinitiated polymerization of PEG-diacrylate with lithium phenyl-2, 4, 6-trimethylbenzoylphosphinate: polymerization rate and cytocompatibility, Biomaterials, 30 (2009) 6702–6707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Flory PJ, Rehner J Jr, Statistical Mechanics of Cross - Linked Polymer Networks Π. Swelling, The Journal of Chemical Physics, 11 (2004)521–526. [Google Scholar]
- [34].Liu W, Saint DA, A new quantitative method of real time reverse transcription polymerase chain reaction assay based on simulation of polymerase chain reaction kinetics, Analytical biochemistry, 302 (2002) 52–59. [DOI] [PubMed] [Google Scholar]
- [35].Wang Y, Malcolm DW, Benoit DS, Controlled and sustained delivery ofsiRNA/NPs from hydrogels expedites bone fracture healing, Biomaterials, 139 (2017) 127–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Amsden B, Solute diffusion within hydrogels. Mechanisms and models, Macromolecules, 31(1998)8382–8395. [Google Scholar]
- [37].Guo Q, Knight PT, Mather PT, Tailored drug release from biodegradable stent coatings based on hybrid polyurethanes, Journal of Controlled Release, 137 (2009) 224–233. [DOI] [PubMed] [Google Scholar]
- [38].Mason MN, Metters AT, Bowman CN, Anseth KS, Predicting controlled-release behavior of degradable PLA-b-PEG-b-PLA hydrogels, Macromolecules, 34 (2001) 4630–4635. [Google Scholar]
- [39].Lin C-C, Anseth KS, PEG hydrogels for the controlled release of biomolecules in regenerative medicine, Pharmaceutical research, 26 (2009) 631–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Van Hove AH, Antonienko E, Burke K, Brown E 3rd, Benoit DS, Temporally tunable, enzymatically responsive delivery of proangiogenic peptides from poly(ethylene glycol) hydrogels, Advanced healthcare materials, 4 (2015) 2002–2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Van Hove AH, Burke K, Antonienko E, Brown E 3rd, Benoit DS, Enzymatically- responsive pro-angiogenic peptide-releasing poly(ethylene glycol) hydrogels promote vascularization in vivo, Journal of controlled release : official journal of the Controlled Release Society, 217 (2015) 191–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Metters A, Anseth K, Bowman C, Fundamental studies of a novel, biodegradable PEG-b- PLA hydrogel, Polymer, 41 (2000) 3993–4004. [Google Scholar]
- [43].Vats K, Benoit DS, Dynamic manipulation of hydrogels to control cell behavior: a review, Tissue engineering. Part B, Reviews, 19 (2013) 455–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Shubin AD, Felong TJ, Schutrum BE, D SLJ, Glogauer CE, D SWB Encapsulation of primary salivary gland cells in enzymatically degradable poly(ethylene glycol) hydrogels promotes acinar cell characteristics, Acta biomaterialia, 8 (2016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Sheikh Z, Brooks PJ, Barzilay O, Fine N, Glogauer M, Macrophages, foreign body giant cells and their response to implantable biomaterials, Materials, 8 (2015) 5671–5701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Lu S, Anseth KS, Release behavior of high molecular weight solutes from poly (ethylene glycol)-based degradable networks, Macromolecules, 33 (2000) 2509–2515. [Google Scholar]
- [47].Vats K, Marsh G, Harding K, Zampetakis I, Waugh RE, Benoit DS, Nanoscale physicochemical properties of chain- and step-growth polymerized PEG hydrogels affect cell- material interactions, J Biomed Mater Res A, 105 (2017) 1112–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Conde J, Oliva N, Zhang Y, Artzi N, Local triple-combination therapy results in tumour regression and prevents recurrence in a colon cancer model, Nature materials, 15 (2016) 11281138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Burdick JA, Anseth KS, Photoencapsulation of osteoblasts in injectable RGD-modified PEG hydrogels for bone tissue engineering, Biomaterials, 23 (2002) 4315–4323. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.