

Abstract
Wilson's disease (WD) is a potentially fatal disorder of chronic copper toxicity, primarily affecting the liver and the brain. Judicious treatment can restore health and longevity, even in patients with severe neurological impairment. However, the disease is associated with considerable morbidity and mortality resulting from delay in diagnosis, and difficulty in pacing the medical treatment. In this article, we briefly review the diagnosis and treatment options for WD and share our experience in managing patients with WD. We focus on decoppering (copper chelation) treatment of WD and outline pragmatic strategies for patient management designed to recognize and minimize adverse effects while ensuring treatment compliance and effectiveness.
Keywords: penicillamine, trientine, GAS for WD
Wilson's disease (WD) is a rare inherited multisystemic disorder of chronic copper toxicity, resulting from the inability of the liver to excrete excessive dietary copper into bile. The disease manifests in children and young adults with liver, brain, or osseomuscular impairment. It is relentlessly progressive and without treatment (decoppering or liver transplant [LT]), patients die from progressive liver failure or severe neurological disability.1 The introduction of safe and effective oral copper chelators (penicillamine and trientine), and the demonstration of benefit of treating presymptomatic individuals (those who have WD mutations, but, as yet, have not developed symptoms), has changed the prognosis for the disease.2, 3, 4, 5 Currently, with judicious decoppering, presymptomatic patients with WD can remain symptom free, liver disease can be stabilized, and even patients with neurological disability can recover and live normal and productive lives at par with their peers.6, 7
However, WD continues to be associated with considerable morbidity and mortality as a result of the twin challenges of diagnosing the disease in time, and pacing the treatment to balance effectiveness and treatment‐related adverse effects (AEs).
In the post‐treatment era, failure to diagnose WD in time is the leading cause of death from the disease.8, 9 Often, families witness death of one or more child from WD before the disease is diagnosed in an asymptomatic (presymptomatic) sibling.10 Studies from various parts of the world suggest that diagnosis of WD is often delayed by several years. The major impediments to a timely diagnosis are unfamiliarity with the disease due to its rarity, clinical heterogeneity, innocuous initial symptoms, and lack of an investigation that could reliably confirm or refute its diagnosis.11, 12, 13, 14, 15, 16
The two main approaches to treating WD are decoppering (i.e., removing excess copper deposits from the body and preventing its reaccumulation), and LT, which is performed in cases of rapidly progressive liver failure. Initial reports after introduction of the decoppering agent, penicillamine, established that it was safe and could restore health, as well as reverse even severe neurological disability.17 These clinical outcomes were later replicated for trientine.1,18,19 However, over the years, use of penicillamine and (to a lesser extent) trientine has fallen into disrepute primarily because of the risk of neurological worsening that is sometimes observed after initiation of treatment and because of uncommon drug‐induced immunological AEs.6,7,17,20 A school of thought has emerged that considers the risk of neurological deterioration from decoppering to be greater than that of disease progression, which has encouraged potentially inadequate decoppering. This fear of using decoppering medications in recommended doses is often grounded in lack of individual experience in managing patients with WD (given the rarity of the disease), lack of laboratory or imaging support in guiding the pace of decoppering, and difficulty in rationalizing treatment‐related neurological worsening to anxious parents who have possibly already lost a child to WD.
It can also be a challenge to ensure compliance with medications in the presence of disease‐related cognitive and behavioral problems. Defaulting on medications is especially an issue in presymptomatic patients and those whose symptoms have remitted, for whom the motivation to take medications wanes as missing doses for days, weeks, and then months does not seemingly cause any harm. It is at this phase, however, that the disease may resurface with rapid liver failure with little time for intervention. Therefore, administering treatment to balance effectiveness and AEs, as well as to ensure compliance, requires vigilance and disease monitoring.6,21,22
In this review, we share our experience in managing over 100 patients with WD, including many with severe WD‐related neurological disability. We focus on decoppering treatment and outline techniques for effective monitoring and pacing of treatment.
Recognizing WD
WD is caused by homozygous or compound heterozygous mutation(s) in the ATP7B gene, and genetic analysis is the confirmatory test for the disease. However, until recently, mutational analysis remained an impractical diagnostic tool because there are over 600 mutations described and new ones continue to be reported.23 In the last few years, development of rapid whole‐gene sequencing has made it possible to reliably identify ATP7B mutations in over 98% of patients examined.24,25However, this process is still expensive for confirmatory diagnosis in individual patients. Interestingly, despite marked genetic heterogeneity, population studies show clustering of ATP7B gene mutations within particular subregions of the gene in a given population, with one or a few high‐frequency mutations dominating24–44 (Table 1). Therefore, various research groups are developing DNA chips to detect these high‐frequency, population‐specific WD mutations. Once commercially available, such DNA chips could allow for rapid confirmation of WD in a patient and help screen the patient's siblings for the disease.
Table 1.
Clinical Clues to WD in a Patient With Initial Neurological Symptoms
| General Examination | Child or Young Adult |
| Wilson facies: pseudolaughter, facetious smile, open mouth, dull look, hypersalivation | |
| Kayser‐Fleischer rings Sunflower cataract | |
| Movement disorders | Early‐onset dysarthria or oromandibular dystonia |
| Mixed movement disorder: any combination of dystonia, tremor, parkinsonism, chorea, or myoclonus | |
| Movement disorder with cerebellar signs | |
| Movement disorder with cognitive or behavioral problems | |
| Cognitive or behavioral problems | Behavioral problems: emotional lability, running amok, hypersexuality, impulse control disorders, psychosis, depression, or attempted suicide |
| Drop in scholastic grades or work performance | |
| Accompanying liver disease | History of liver disease in childhood (fleeting jaundice, incidental unexplained abnormal liver function tests, or unexplained hemolytic anemia) |
| Abnormal liver function tests | |
| Liver cirrhosis on abdominal ultrasound | |
| Thrombocytopenia or pancytopenia (from liver‐cirrhosis–related hypersplenism) | |
| Coombs‐negative hemolytic anemia (from sudden release of toxic free copper into blood) | |
| Accompanying osseomuscular involvement | Proximal lower‐limb muscle weakness, bone pains, arthralgias, arthritis, or nontraumatic fractures |
| Family history | Family history of WD |
| Unexplained deaths in the family | |
| Unexplained jaundice in children or young adults in the family | |
| Unexplained neurological disease in children or young adults in the family | |
| Unexplained miscarriage(s) |
Currently, diagnosis of WD is based upon a number of clinical features and laboratory tests. There are two disparate faces of symptomatic WD. The first is that of a child with liver failure who presents in the first two decades of life. The second form of the disease is a young adult who presents with extrapyramidal syndrome or cognitive or behavioral problems with subclinical or clinical liver cirrhosis. Although the initial symptoms of the disease are often innocuous, evidence of unexplained liver, brain, or osseomuscular dysfunction in the patient or their family members helps establish the multisystemic nature of the disorder and greatly narrows the differential diagnosis (Table 2). The characteristic Wilson facies, Kayser‐Fleischer rings (KF), and the familial nature of the ailment are clinical diagnostic clues to the disease.6,45,46
Table 2.
High‐frequency ATP7B mutation spectrum in various world populations
| Country | Sample Size (n) | Nucleotide/Amino Acid Change | Exon | Domain | Allele Frequency (%)a | Reference |
|---|---|---|---|---|---|---|
| Austria | 125 | p.H1069Q | 14 | SEPHL | 34 | Ferenci26 |
| p.G710S | 8 | 6 | ||||
| c.2299insC | 8 | 4 | ||||
| p.R969Q | 13 | 4 | ||||
| Bulgaria | 89 | p.H1069Q | 14 | SEPHL | 59 | Todorov et al.27 |
| c.2304‐2305ins C | 11 | |||||
| c.3400delC | 4 | |||||
| Hungary | 42 | p.H1069Q | 14 | SEPHL | 43 | Firneisz et al.28 |
| Germany | 82 | p.H1069Q | 14 | SEPHL | 63 | Caca et al.29 |
| c.3400delC | 15 | 9 | ||||
| c.2299ins C | 8 | 4 | ||||
| Poland | 142 | p.H1069Q | 14 | SEPHL | 72 | Gromadzka et al.39 |
| c.3402delC | 15 | 8 | ||||
| Brazil | 36 | p.H1069Q | 14 | SEPHL | 37 | Bem et al.44 |
| p.Ala1135Glnfs | 15 | 11 | ||||
| p.Trp779Stop | 8 | 9 | ||||
| p.Met769HisfsX26 | 8 | 9 | ||||
| p.P1273L | 18 | 6 | ||||
| Greece | 24 | p.H1069Q | 14 | SEPHL | 29 | Loudianos et al.30 |
| Sardinia | 46 | c.‐441/‐427del | 5′UTR | Promoter | 61 | Loudianos et al.40 |
| c.2463delC | 10 | Td | 9 | |||
| p.V1146M | 16 | ATP loop | 8 | |||
| Turkey | 46 | p.H1069Q | 14 | SEPHL | 17 | Simsek Papur et al.31 |
| p.G710S | 8 | Tm2 | 5 | |||
| UK | 42 | p.H1069Q | 14 | SEPHL | 17 | Curtis et al.67 |
| p.M769V | 8 | Tm4 | 8 | |||
| 181 | p.H1069Q | 14 | SEPHL | 19 | Coffey et al.25 | |
| p.M769V | 8 | 6 | ||||
| Japan | 47 | c.2871delC | 13 | 16 | Okada et al.43 | |
| p.R778L | 8 | Tm4 | 13 | |||
| China | 40 | p.R778L | 8 | Tm4 | Gu et al.41 | |
| 44 | p.R778L | 8 | Tm4 | Wu et al.32 | ||
| 11 | p.R778L | 8 | Tm4 | 50 | Geng et al.33 | |
| 13 | p.P992L | 13 | 14 | |||
| Korea | 120 | p.R778L | 8 | Tm4 | 40 | Park et al.34 |
| p.A874V | 11 | Td | 8 | |||
| p.N1270S | 18 | ATP hinge | 7 | |||
| India | 52 | p.C271* | 2 | Cu3 | 20 | Aggarwal et al.24 |
| p.E122fs | 2 | Cu1 | 11 | |||
| p.L795F | Tm4/Td | 6 | ||||
| p.T977M | Tm6 | 6 | ||||
| 27 | p.C271* | 2 | Cu3 | ~9 | Santhosh et al.42 | |
| p.G1061E | 14 | ATP N‐binding | ~9 | |||
| 199 | p.C271* | 2 | 22 | Mukherjee et al.35 | ||
| p.G1061E | 14 | ATP N‐binding | 15 | |||
| c.G1708‐1G>C | 4 | Cu6 | 10 | |||
| p.A1241V | 18 | NBD | 7 | |||
| p.E150H‐fs | 2 | Cu2 | 5 | |||
| 664 | c.448_452del5 | 2 | Cu1 | 6 | Gupta et al.36 | |
| 43 | p.I1102T | 15 | ATP loop | 6 | Kumar et al.37 | |
| p.P922H | 13 | Tm6 | 6 | |||
| p.P922* | 13 | Tm6 | 6 | |||
| p.G1010A‐fs | 13 | Tm6 | 6 | |||
| Iran | 65 | c.3061‐1G>A | 14 | Phosphorylation | 6 | Dastooz et al.38 |
| p.M769C‐fsX38 | 8 | 3 | ||||
| p.W779G | 8 | 3 |
Allele frequency is rounded to the nearest percentage for consistency.
Clinical suspicion of WD is supported by a finding of low serum ceruloplasmin, raised 24‐urinary copper excretion, and, in select patients, a high liver‐copper content. However, all of the available laboratory tests have diagnostic limitations and test results need to be interpreted with caution. There is currently no imaging modality to view copper. MRI brain though reveals changes secondary to copper deposition. Whereas the pattern of symmetrical brain stem and basal ganglion involvement is common in WD (Fig. 1), it is not specific to the disease. The American Association of Liver Disease guidelines and the WD diagnostic scoring system proposed by Ferenci et al. provide useful diagnostic algorithms.45,47
Figure 1.
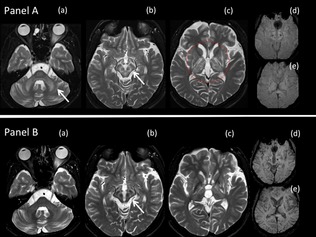
Serial MRI scans of a 22‐year‐old patient with WD after 16 years of irregular treatment, demonstrating typical MRI changes associated with WD and change in MRI abnormalities with supervised decoppering. (A) MRI scan at age 22 years. Axial T2W MRI brain scan through the (a) pons, (b) midbrain, and (c) basal ganglia, showing symmetrical hyperintensities in middle cerebellar peduncles, brainstem, striatum, globus pallidus, and thalami. In the midbrain (b), the hyperintense signal of the white matter tracts shows the gray matter in relief. The midbrain therefore resembles the “face of the panda” with the substantia nigra, red nucleus, and periaqueductal gray matter reminiscent of the ears, eyes, and mouth, respectively, of the panda. Axial susceptibility‐weighted imaging (SWI) through the midbrain (d) and basal ganglia (e) show minimal susceptibility in the corpus striatum. (B) Follow‐up MRI scan taken after 13 months of supervised decoppering and substantial recovery of neurological disability (see Fig. 4). As compared to the initial scan (A), axial T2‐weighted MRI images through the (a) pons, (b) midbrain, and (c) basal ganglia show regression in hyperintense signal and atrophy of cerebellum, brainstem, striatum, globus pallidus, and thalami. Axial SWI through the midbrain (d) and basal ganglia (e) show increased susceptibility and atrophy of the corpus striatum.
Once WD is diagnosed in a patient, it is imperative to screen all of their siblings. Unless WD is reliably confirmed in the siblings, they should be tracked well into adulthood.
Treatment of WD
Goal of Treatment
The goal of WD treatment is to chelate excessive copper that has accumulated over the years as well as prevent ongoing copper deposition. Both presymptomatic and symptomatic patients need lifelong decoppering5 (Table 3).
Table 3.
Goal of treatment of WD
| Symptomatic patient | Restore normalcy (reverse neurological disability + stabilize liver disease) + ensure continued normalcy |
| Presymptomatic patient | Prevent symptom onset |
| First‐degree family members of patient with WD | Rule out WD by mutational analysis. In the absence of genetic testing, track family members well into adulthood (preferably biannually) for clinical or laboratory evidence of WD. |
With adequate decoppering, presymptomatic patients remain symptom free, whereas symptomatic patients recover (see accompanying Videos 1 and 2). Clinical neurological recovery is associated with improvement on brain imaging (Fig. 1). With treatment, KF rings and sunflower cataracts disappear and there is clinical, biochemical, and histological improvement of liver function.6, 46, 47, 48 Well‐treated patients can conceive and normal pregnancy outcomes can be expected.18,49,50 People with one ATP7B mutation and one normal ATP7B allele do not have WD and do not require treatment.
In the initial phase of treatment, decoppering has to be intensive to remove accumulated copper. Once the body copper balance has normalized, decoppering dosage can be significantly lowered to levels sufficient to prevent daily dietary copper gain resulting from impaired biliary copper excretion.6
Treatment Arsenal
Presently, penicillamine (d‐penicillamine) and trientine (triethylenetetramine) are the two oral copper chelators available for treatment of WD. Both drugs are safe and effective and may be used lifelong in patients with WD.1,19,47 Penicillamine has been used worldwide for a long time and has the most associated clinical data. The drug is widely available and affordable. In contrast, trientine is manufactured only in the United States and UK and has to be imported by other countries. Trientine's short shelf life and the need to store it at low temperatures add to the cost of therapy. Though there are no systematic head‐to‐head comparisons between the two drugs, trientine may have some advantage over penicillamine in being less immunogenic.47 Pragmatically, the choice of decoppering drug is dictated by its availability.
Penicillamine and trientine form the mainstay of treatment of WD during both the initial intensive decoppering and the maintenance phases of treatment. Other treatment options include British antilewisite (BAL), ammonium tetrathiomolybdate, zinc, and LT. BAL (dimercaprol) is administered intramuscularly in multiple weekly injections and is associated with considerable toxicity and a rapidly waning decoppering effect on continued use. Therefore, though BAL was the first copper chelator developed for treatment of WD, it has been largely replaced by the two safer, more efficacious oral copper chelators. Recently, there has been some renewed interest in using the drug briefly for initial rapid decoppering in patients with severe neurological WD.6,51 Ammonium tetrathiomolybdate is a promising copper chelator with few known AEs, but it is not, as yet, available commercially. Zinc may be used in presymptomatic patients and during the maintenance phase of treatment of symptomatic patients. However, zinc does not chelate copper and may not be adequate as monotherapy in the initial intensive decoppering phase of treatment in symptomatic patients (see combination therapy below).1,47 LT cures WD by restoring normal biliary copper excretion. Patients with transplanted liver do not require copper chelation, but they do need to take lifelong immunosuppressants to prevent rejection of the transplanted liver. LT is a valued treatment option in patients with rapidly progressing liver disease and may be considered in patients with neurological disease intolerant to medical treatment.52 Catana and Medici52 and Guillaud et al.53 have reviewed the current status of LT for treatment of WD.
Treatment Strategy
The strategy for decoppering is to “start low and go slow” (Tables 4 and 5). In the initial intensive decoppering phase of treatment in symptomatic patients, penicillamine is initiated in doses of 125 or 250 mg/day and doses escalated by 250 mg every 2 to 3 weeks to a maximal dose of up to 1.0 to 2.5 g/day.6,7,54 Because food decreases the bioavailability of penicillamine, the drug must be administered while fasting (shifting drug intake away from meal times).55,56 In our experience, twice‐daily dosing is effective and facilitates compliance. The dosing schedule of trientine is similar to penicillamine, and doses up to 1.0 to 2.4 g/day have been used.18,57 The dosing schedule and AE profile of penicillamine and trientine are outlined in Table 6 1,6,46 (see Figs. 2 and 3; see accompanying Video 3). Penicillamine is usually supplemented with pyridoxine. Zinc supplementation is not required during initial decoppering. Iron or calcium supplements are best avoided during the intensive decoppering phase of treatment.6 Intolerance to both penicillamine and trientine is rare. Treatment options in such patients include brief intensive courses of BAL with drug‐free intervals to prevent tachphylaxis, ammonium tetrathiomolybdate, or LT.
Table 4.
Dos and don'ts of WD treatment
| Intervention | Recommendation |
|---|---|
| Drinking bottled water | Not necessary |
| Strict copper‐free or low‐copper diet | Not necessary |
| Combination of zinc and decoppering (penicillamine/trientine) | To be avoided |
| Large initial or rapid escalation of decoppering | To be avoided |
| Administering penicillamine and trientine away from meal times | Essential |
| Supplementation of zinc during penicillamine or trientine therapy | Not necessary |
| Supplementation of iron or calcium during penicillamine or trientine therapy | To be avoided |
| Lifelong decoppering | Essential |
Table 5.
Measure to aid treatment compliance
| Counseling the patient and the caregivers about the necessity of compliance and warning them about the dangers of noncompliance |
| Designating a caregiver to administer decoppering drugs at a fixed time |
| 1. In all children |
| 2. In all patients with cognitive and behavioral problems |
| 3. In all symptomatic patients during the intensive decoppering phase of treatment |
| 4. In all patients who have repeatedly defaulted on treatment |
| Administering decoppering drug at most twice‐daily to aid compliance with fasting |
| Questioning caregivers and patient about compliance during every visit |
| Tracking the GAS for WD scores for noncompliance related worsening |
| Periodic 24‐hour urinary copper excretion and serum free copper assessments in patients on maintenance treatment |
| Scheduling bi‐ or triannual follow‐up visits in asymptomatic patients to ensure patients are not lost to follow‐up |
| Tracking patients lost to follow‐up |
| Encouraging interaction among patients |
Table 6.
Drugs used in WD: dosage, AEs, and management of AEs
| Drug | Indication | Doses and Monitoring Regimen | AEs | Response to AEs |
|---|---|---|---|---|
| Penicillamine or trientine |
● Symptomatic patients (initial intensive decoppering phase) ● Symptomatic patients (maintenance phase) ● Presymptomatic patients ● Trientine is preferred in patients intolerant to penicillamine. ● Trientine is the drug of choice for patients with renal or autoimmune disease. |
● Symptomatic patients (initial intensive phase): up to 1.0 to 2.5 g/day for penicillamine and up to 1.0 to 2.4 g/day for trientine ● Symptomatic patients (maintenance phase): up to 500 to 1,000 mg/day ● Presymptomatic patients: up to 500 to 1,000 mg/day ● Drug is best administered in two divided doses in a day while fasting; preferably 3 to 4 hours fasting pre‐ and 2 to 3 hours postadministration. ● The first dose may be given on awakening in the morning, and the second dose, midway between lunch and dinner or a few hours after dinner. ● Supplement penicillamine with 20 mg of pyridoxine/day ● Monitoring: during each visit examine for clinical deterioration, fever, and skin rash. After initiation of therapy, check CBC, urine for proteinuria every week for the first month, and then monthly for 3 to 6 months and every 3 months after. During maintenance phase, check CBC and urine protein biannually. ● Check 24‐hour urinary copper excretion and serum free copper periodically to confirm compliance. |
Early reactions (occur within the first few weeks or months of drug treatment): hypersensitivity to drug: fever, skin rash, lymphadenopathy. ● Oberved in 10% to 20% of patients on penicillamine. Infrequent with trientine These are generally dose related. Low incidence if doses are escalated slowly. |
● Treatment: short course of oral steroids (10 to 30 mg prednisolone) and temporary discontinuation or down‐titrating of penicillamine or trientine. The drug can be reintroduced at a lower dose and escalated up at a slower pace. Steroids may be overlapped for a few weeks of rechallenge with penicillamine/trientine. ● Substitution of penicillamine with trientine, or trientine with penicillamine, is usually not required. |
|
Early reactions: hypersensitivity to drug: thrombocytopenia, leucopenia, pancytopenia. Less common with trientine. ● Cytopenias are usually present in patients with WD as part of hypersplenism due to liver cirrhosis and portal hypertension. Rarely, the platelet count and white blood count transiently drop further after introduction of the drug. ● Drug induced bone marrow depression is rare. Less common with trientine |
● A short course of oral steroids (as above) and brief down‐titration of penicillamine or trientine doses. Once counts stabilize, the drug doses can be gradually up‐titrated. Substitution of penicillamine with trientine, or trientine with penicillamine, is usually not required. ● Drug‐induced bone marrow depression necessitates immediate withdrawal of the drug. |
|||
|
Early reaction: penicillamine or trientine‐induced neurological deterioration. ● Observed in 10% to 22% of patients on penicillamine. Less frequent with trientine. |
Down‐titrating drug doses. Once the patient is clinically stable, doses can be gradually up‐titrated (see text and Video 3) | |||
|
Late reactions (occur usually after years of drug treatment): drug‐induced lupus, isolated glomerulonephritis (nephrotic syndrome); these are rare with trientine. ● Reactivation of penicillamine‐induced lupus, isolated glomerulonephritis (nephrotic syndrome) seen with trientine ● Rarely Goodpasture's nephritis with penicillamine |
Administration of steroids and reduction in penicillamine or trientine doses to 0.5 g/day or less. If WD worsens with reduced doses, then replace penicillamine with trientine or vice versa. In some instances, lupus and/or renal impairment may be significant and necessitate immediate drug withdrawal. | |||
| Late reaction: easy bruising, recurrent subcutaneous bleeding occur due to penicillamine induced inhibition of collagen and elastin linking. These are rare if maintenance doses of penicillamine are <1 g/day. |
● Symptomatic treatment; withdrawal of penicillamine not required ● Withdrawal of penicillamine before an elective surgery (to promote wound healing) is not necessary, if penicillamine doses are not large. |
|||
| Late reaction: Penicillamine induced elastosis perforans serpiginosa. Focal lesions, cosmetic concern. Rare and idiosyncratic reaction. Lesions can be reactivated with trientine. | Cryotherapy | |||
| Zinc salts |
● As monotherapy in symptomatic patients (maintenance phase) ● As monotherapy in presymptomatic patients |
● 50 mg of elemental zinc two or three times a day. ● Taken while fasting ● Monitoring: during each visit examine for clinical deterioration, hypersensitivity. Regular CBC |
Gastritis is common. Zinc acetate and zinc gluconate are better tolerated than zinc sulphate. | This may be significant and a dose‐limiting AE; symptomatic treatment |
| Neurological deterioration due to disease progression from inadequate decoppering. |
● Stop zinc ● Reintroduce penicillamine or trientine |
|||
| Hepatitis or worsening liver function resulting from disease progression from inadequate decoppering. |
● Stop zinc ● Reintroduce penicillamine or trientine |
CBC, complete blood count.
Figure 2.
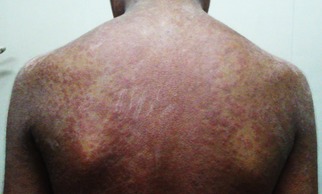
Penicillamine‐induced generalized pruritic erythematous maculopapular rash in a 13‐year‐old boy with WD (GAS for WD tier 1 scores: L3 C3 M2 O0). The rash developed after up‐titrating the penicillamine dose to 750 mg/day, 4 weeks after initiation of treatment.
Figure 3.
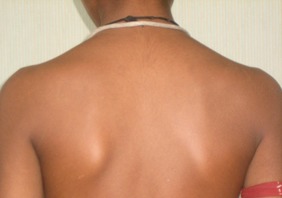
Resolution of rash after 2 weeks of low‐dose oral steroids and withdrawal of penicillamine treatment. The patient subsequently tolerated slow up‐titration of penicillamine to doses of 2 g/day without any AEs.
A worrisome problem encountered in some patients soon after initiation of decoppering is the worsening of neurological signs. This is described most often after treatment with penicillamine, but is also observed with use of alternative treatments, including LT.17,58 The occurrence of decoppering‐induced neurological worsening cannot be predicted and is observed in 10% to 22% of patients receiving penicillamine, with an incidence as high as 52% having been reported. Most studies and our experience have shown that, except in a minority of patients, neurological worsening reverses with continued treatment or temporary interruption of therapy without long‐term consequences 6,7,17,20 (see accompanying Video 3).
The maintenance phase of decoppering in symptomatic patients is initiated once excessive body copper deposits have been depleted. This is heralded by neurological recovery and stabilization of liver impairment. In the maintenance phase of decoppering, dosages of penicillamine or trientine may be reduced to 500 to 1,000 mg/day (administered in one or two divided doses during the day).6,47
Zinc inhibits intestinal absorption of copper and has been used as a stand‐alone drug in the maintenance phase of treatment. Zinc salts containing 50 mg of elemental zinc are administered two to three times a day, while fasting. Gastritis is common and often limits the dose of the drug.46,47 Although zinc salts have no major adverse effects and are inexpensive, they may be insufficient to prevent ongoing copper toxicity. Therefore, patients should be tracked for clinical worsening. Reappearance of KF rings or other signs of WD should prompt discontinuation of zinc therapy and reintroduction of penicillamine or trientine.
The combination of zinc with either penicillamine or trientine presents significant dosing issues, and, in our experience, such a combination should be avoided. Both the decoppering drugs can potentially chelate zinc, reducing the bioavailability of the decoppering drug as well as that of zinc.56,59 Therefore, during combination therapy, the administration of the decoppering drug and zinc have to be spaced widely apart.47 This poses compliance issues because penicillamine and trientine as well as zinc need to be administered in multiple doses and while fasting. Furthermore, the combination of trientine and zinc can induce serious sideroblastic anemia.56
Presymptomatic patients are treated with oral copper chelators in doses similar to those administered in the maintenance phase of therapy in symptomatic patients. Zinc may be used as a stand‐alone drug with careful clinical monitoring (see above).6
Diet and Adjuvant Therapy
Copper is common in water supplies and foods, and dietary copper restriction is not sufficient to prevent copper accumulation in patients with WD. In the initial intensive phases of decoppering in symptomatic patients, it is advisable to avoid foods rich in copper, such as chocolates, nuts, mushrooms, liver, and shell fish. These dietary restrictions may be relaxed once symptoms regress.47 Bottled water is recommended if the copper content of potable water is over 0.1 ppm.60 However, most municipal water supplies have low copper and assessment of copper levels is not typically necessary.46
Patients with impaired swallowing benefit from early elective insertion of a percutaneous endoscopic gastric feeding tube. Hypersalivation improves with anticholinergic drugs and botulinum toxin administered in parotid glands. Control of hypersalivation also helps improve speech and decreases microaspiration.
Use of adjunctive therapy to relieve various movement disorders or behavioral problems associated with WD has not been systematically studied and is based largely on individual experience. Levodopa/carbidopa has been used with varying success in relieving parkinsonism.6 In our experience, in patients for whom l‐dopa/carbidopa is effective, small to moderate doses suffice to alleviate WD‐related parkinsonism and larger doses do not increase effectiveness or afford additional benefit. Because pyridoxine decreases the bioavailability of l‐dopa, the two drug doses need to be spaced apart. Various drugs, such as anticholinergics (often in high doses), l‐dopa/carbidopa, gabapentin, antispasticity drugs, and beta‐blockers, have been used for treatment of dystonia, rigidity, or tremor in patients with WD, but their benefits are limited.6,61,62 Associated parkinsonism and behavioral problems contraindicate use of dopamine blockers and limit the doses of anticholingeric medications. Botulinum toxin injections in areas of focal dystonia often relieve painful fixed dystonic postures or spasms and also help in overall improvement of the generalized dystonia.
As has been reported earlier, and in our experience, antidepressants, antipsychotics, (preferably atypical), and mood stabilizers may be required for patients with serious behavioral problems, especially if they are at risk of self‐harm.6 Symptomatic treatment for movement disorders and behavioral problems can be withdrawn once decoppering has led to adequate clinical improvement. Epilepsy is rare in patients with WD and usually well controlled with standard antiepileptics, with careful monitoring of liver function.63,64
Beta‐blockers, sclerotherapy, or banding for portosystemic varices may be required in patients with significant portal hypertension. Regular review by a liver specialist is essential to prevent and manage complications of liver cirrhosis.
Tracking WD
WD is characterized by a long presymptomatic phase lasting years to decades. Once symptoms appear, patients can deteriorate rapidly over weeks or months, and unattended the disease is fatal. The challenge of WD treatment is to chelate copper at a pace fast enough to prevent disease progression or death. However, (rapid) initiation of decoppering may entail a risk of neurological deterioration, possibly resulting from release of large amounts of copper from the liver into the circulation.17,65 Therefore, patients need to be carefully monitored for disease progression, treatment efficacy, and AEs as well as compliance to therapy.
Tools to Track WD
In our experience, objective clinical assessments are better able to track treatment benefit or disease progression than serum‐free copper and urine copper measurements. We monitor WD therapy using the Global Assessment Scale for WD (GAS for WD), which is a standard WD‐specific scale (Fig. 4). The scale has been shown to be a reliable measure of WD‐related multisystemic disability and to be sensitive to clinical change.11
Figure 4.
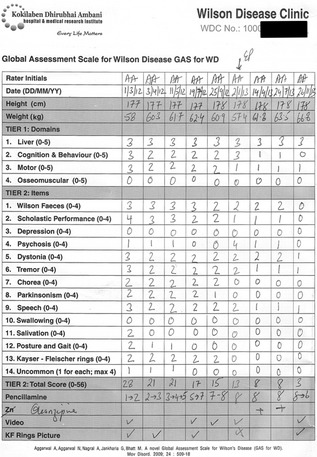
GAS for WD score sheet tracking neurological improvement in a 22‐year‐old man with WD over 20 months of supervised treatment. The patient was diagnosed with WD at 6 years of age and was on irregular treatment until presentation to our center at age 22. Penicillamine was initiated at dosages of 250 mg/day and slowly escalated to 2 g/day. Serial MRI scans of the patient are shown in Figure 1.
GAS for WD is a two‐tier scale that can be administered by the patient's bedside in 20 to 30 minutes (with experience, the time of administration is reduced to 15 minutes). Tier 1 measures WD‐related disability across four domains: liver (L); cognition and behavior (C); motor (M); and osseomuscular (O) systems. Each domain is scored on an ascending six‐point scale (0–5). Tier 2 assesses WD‐related neurological dysfunction using 14 items, including Wilson facies and KF rings. Each item is graded on an ascending five‐point scale (0–4), and the item scores are summed to obtain the total tier 2 score (0–56).11 The GAS for WD scoring sheet with rating instructions is provided as Supporting Information.
Pacing Decoppering
To quote Walshe, treatment of WD involves, “a supreme capacity for taking trouble; there is no place for the philosophy of giving some penicillamine and seeing what will happen.”54 In the initial intensive phase of decoppering in a symptomatic patient, to ensure compliance, we insist on supervised administration of the decoppering medicine twice‐daily at fixed times, by a designated caregiver. During each visit, the decoppering dosing schedule is planned such that the drugs are given while fasting without interfering with meal times. GAS for WD liver scores are tracked for evidence of liver decompensation and, if present, symptomatic treatment of liver failure and the need for fast‐tracked LT discussed with the treating liver specialist. Patients are also assessed for drug‐related AEs.
At the start of treatment, patients are tracked at weekly or biweekly intervals. In most patients with significant neurological disability, there is no change in GAS for WD scores in the first few months of therapy, though, in some patients, scores may improve in the first 2 weeks itself. Worsening of GAS for WD scores after initiation of decoppering suggest disease progression, decoppering‐induced neurological worsening, noncompliance, or an intercurrent illness. Decoppering‐induced neurological worsening usually manifests with abrupt clinical deterioration over days or weeks. This drug‐induced neurological worsening is alleviated with continuation of therapy or with down‐titration of the decoppering dosages. We prefer the latter option, and once decoppering induced neurological worsening has reversed (i.e., GAS for WD scores have improved to predeterioration levels), we gradually re‐escalate the drug doses, but at a slower pace than before (see accompanying Video 3). Patients with disease progression, on the other hand, generally exhibit steady neurological worsening and benefit from perseverance with the decoppering regimen and gradual up‐titration of dosage (see accompanying Video 2, segment 2).
Stabilization of GAS for WD scores after initiation of decoppering, in a symptomatic patient, suggests that the benefit of copper chelation has outpaced natural disease progression. The decoppering doses are gradually escalated to the maximal tolerated levels. Patients and their families are quizzed about compliance and need for compliance reinforced. Once GAS for WD scores start dropping steadily, the patient's visits can be spaced out to once every few months. Decoppering is a slow process and takes 1 to 3 years for a patient with severe neurological disability to recover. The earliest change noted is improvement in WD facies over 3 to 5 months of initiation of treatment. KF rings disappear over 12 to 18 months.11 Significant improvement in GAS for WD scores is accompanied by general well‐being, growth spurt, and onset of puberty (if it was delayed).
Complete clinical recovery in a symptomatic patient heralds that the positive body copper balance has resolved. Patients can now be switched to maintenance therapy and doses of decoppering drugs reduced (Fig. 4). Worsening of GAS for WD scores during the maintenance phase signals noncompliance, if drug‐related AEs and an intercurrent illness are ruled out. Decoppering‐induced neurological worsening is not a concern in patients on steady and small‐dose decoppering treatment, whereas noncompliance is a common and significant problem (Table 5). Maintaining bi‐ or triannual visits and tracing patients lost to follow‐up helps reinforce compliance. During each visit, patients are warned about dangers of defaulting on treatment, including the possibility of sudden deterioration leading to death.21,22 Interaction of patients with one another helps inspire confidence in the benefits of WD treatment and encourages compliance.
Conclusion
In the century since Wilson's original description of a universally fatal familial disease of the young, we have learned much about the disorder.66 We now recognize that the disease results from chronic copper toxicity in (primarily) the liver and brain and have identified the responsible gene.23 Though the search for a simple and reliable diagnostic test is ongoing, genetic analysis may soon provide a practical diagnostic screening tool.23 Remarkably, individual and dedicated group efforts have led to a small, but effective, arsenal of treatment options to vanquish the disease and restore health and longevity.1
However, challenges remain. It is estimated that 75% of patients with WD go undiagnosed and die.19,25 In many other patients, treatment is administered suboptimally and the process and suffering unduly prolonged. We have the means. The challenge is to consistently and effectively employ the available diagnostic, treatment, and monitoring tools to conquer the disease.
Author Roles
(1) Research Project: A. Conception, B. Organization, C. Execution; (2) Statistical Analysis: A. Design, B. Execution, C. Review and Critique; (3) Manuscript: A. Writing of the First Draft, B. Review and Critique.
A.A.: 1A, 1B, 1C, 3A, 3B
M.B.: 1A, 1B, 1C, 3B
Financial Disclosures
A.A. has served on the scientific advisory board of Novartis. A.A. and M.B. have each been awarded research grants from the Seventh Framework Programme, WilsonIndia: People Marie Curie Actions, International Research Staff Exchange Scheme (IRSES).
Supporting information
Supporting Information: GAS for WD scoring sheet with rating instructions.
Video 1. Reversal of neurological disability in an 18‐year‐old patient with cerebellar ataxia‐tremor‐dystonia phenotype of WD. The patient has a homozygous c.813C>A, p.C271X ATP7B mutation. Serial GAS for WD tier 1 scores for liver (L), cognition (C), motor (M), and osseomuscular (0) domains and tier 2 total scores are tabulated (see Supporting Information for GAS WD scoring sheet and rating instructions).
Video 2. Reversal of neurological disability in a 27‐year‐old patient with dystonia‐parkinsonism phenotype of WD. The patient has heterozygous c.2930C>T, p.T977M, and c.4070C>T, p.A1357V ATP7B mutations. Serial GAS for WD tier 1 scores for liver (L), cognition (C), motor (M), and osseomuscular (0) domains and tier 2 total scores are tabulated (see Supporting Information for GAS WD scoring sheet and rating instructions).
Video 3. Neurological worsening with penicillamine and improvement with down‐titration of dosage in a 17‐year‐old patient with WD. The patient has homozygous c.2736_2746delCATTCAGCAGC, p.Ile913fs, ATP7B mutation. Serial GAS for WD tier 1 scores for liver (L), cognition (C), motor (M), and osseomuscular (0) domains and tier 2 total scores are tabulated (see Supporting Information for GAS WD scoring sheet and rating instructions).
References
- 1. Walshe JM. The conquest of Wilson's disease. Brain 2009;132:2289–2295. [DOI] [PubMed] [Google Scholar]
- 2. Walshe JM. Wilson's disease: new oral therapy. Lancet 1956. ;270:25–26. [DOI] [PubMed] [Google Scholar]
- 3. Walshe JM. Copper chelation in patients with Wilson's disease. A comparison of penicillamine and triethylene tetramine dihydrochloride. Q J Med 1973;42:441–452. [PubMed] [Google Scholar]
- 4. Walshe JM. Triethylene tetramine. Lancet 1970;2 :154. [DOI] [PubMed] [Google Scholar]
- 5. Sternlieb I , Scheinberg IH . Prevention of Wilson's disease in asymptomatic patients . N Engl J Med 1968. ; 278 : 352 – 359 . [DOI] [PubMed] [Google Scholar]
- 6. Scheinberg IH , Sternlieb I . Wilson's Disease: Major Problems in Internal Medicine . Philadelphia, PA: : Elsevier Publishing; ; 1984. . [Google Scholar]
- 7. Walshe JM , Yealland M . Chelation treatment of neurological Wilson's disease . Q J Med 1993. ; 86 : 197 – 204 . [PubMed] [Google Scholar]
- 8. Czlonkowska A , Tarnacka B , Litwin T , Gajda J , Rodo M . Wilson's disease‐cause of mortality in 164 patients during 1992–2003 observation period . J Neurol 2005. ; 252 : 698 – 703 . [DOI] [PubMed] [Google Scholar]
- 9. Walshe JM . Cause of death in Wilson disease . Mov Disord 2007. ; 22 : 2216 – 2220 . [DOI] [PubMed] [Google Scholar]
- 10. Walshe JM . Letter: missed Wilson's disease . Lancet 1975. ; 2 : 405 . [DOI] [PubMed] [Google Scholar]
- 11. Aggarwal A , Aggarwal N , Nagral A , Jankharia G , Bhatt M . A novel Global Assessment Scale for Wilson's disease (GAS for WD) . Mov Disord 2009. ; 24 : 509 – 518 . [DOI] [PubMed] [Google Scholar]
- 12. Prashanth LK , Taly AB , Sinha S , Arunodaya GR , Swamy HS . Wilson's disease: diagnostic errors and clinical implications . J Neurol Neurosurg Psychiatry 2004. ; 75 : 907 – 909 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zimbrean PC , Schilsky ML . Psychiatric aspects of Wilson disease: a review . Gen Hosp Psychiatry 2014. ; 36 : 53 – 62 . [DOI] [PubMed] [Google Scholar]
- 14. Walshe JM , Yealland M . Wilson's disease: the problem of delayed diagnosis . J Neurol Neurosurg Psychiatry 1992. ; 55 : 692 – 696 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hu J , Lu D , Wang G . Study on the clinical misdiagnosis of hepatolenticular degeneration . Zhonghua Yi Xue Za Zhi 2001. ; 81 : 642 – 644 . [PubMed] [Google Scholar]
- 16. Gow PJ , Smallwood RA , Angus PW , Smith AL , Wall AJ , Sewell RB . Diagnosis of Wilson's disease: an experience over three decades . Gut 2000. ; 46 : 415 – 419 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Walshe JM . Penicillamine: the treatment of first choice for patients with Wilson's disease . Mov Disord 1999. ; 14 : 545 – 550 . [DOI] [PubMed] [Google Scholar]
- 18. Walshe JM . Treatment of Wilson's disease with trientine (triethylene tetramine) dihydrochloride . Lancet 1982. ; 1 : 643 – 647 . [DOI] [PubMed] [Google Scholar]
- 19. Scheinberg IH . Penicillamine in Wilson's disease . Lancet 1982. ; 1 : 1469 . [DOI] [PubMed] [Google Scholar]
- 20. Brewer GJ , Terry CA , Aisen AM , Hill GM . Worsening of neurologic syndrome in patients with Wilson's disease with initial penicillamine therapy . Arch Neurol 1987. ; 44 : 490 – 493 . [DOI] [PubMed] [Google Scholar]
- 21. Walshe JM , Dixon AK . Dangers of non‐compliance in Wilson's disease . Lancet 1986. ; 1 : 845 – 847 . [DOI] [PubMed] [Google Scholar]
- 22. Emery P , Mackay IR . Compliance and Wilson's disease . Lancet 1986. ; 1 : 1388 . [DOI] [PubMed] [Google Scholar]
- 23. Schmidt HH . Role of genotyping in Wilson's disease . J Hepatol 2009. ; 50 : 449 – 452 . [DOI] [PubMed] [Google Scholar]
- 24. Aggarwal A, Chandhok G, Todorov T, et al. Wilson disease mutation pattern with genotype‐phenotype correlations from Western India: confirmation of p. C271* as a common Indian mutation and identification of 14 novel mutations. Ann Hum Genet 2013;77:299–307. [DOI] [PubMed] [Google Scholar]
- 25. Coffey AJ, Durkie M, Hague S, et al. A genetic study of Wilson's disease in the United Kingdom. Brain 2013;136:1476–1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ferenci P . Regional distribution of mutations of the ATP7B gene in patients with Wilson disease: impact on genetic testing . Hum Genet 2006. ; 120 : 151 – 159 . [DOI] [PubMed] [Google Scholar]
- 27. Todorov T, Savov A, Jelev H, et al. Spectrum of mutations in the Wilson disease gene (ATP7B) in the Bulgarian population. Clin Genet 2005;68:474–476. [DOI] [PubMed] [Google Scholar]
- 28. Firneisz G , Lakatos PL , Szalay F , Polli C , Glant TT , Ferenci P . Common mutations of ATP7B in Wilson disease patients from Hungary . Am J Med Genet 2002. ; 108 : 23 – 28 . [DOI] [PubMed] [Google Scholar]
- 29. Caca K, Ferenci P, Kuhn HJ, et al. High prevalence of the H1069Q mutation in East German patients with Wilson disease: rapid detection of mutations by limited sequencing and phenotype‐genotype analysis. J Hepatol 2001;35:575–581. [DOI] [PubMed] [Google Scholar]
- 30. Loudianos G, Lovicu M, Solinas P, et al. Delineation of the spectrum of Wilson disease mutations in the Greek population and the identification of six novel mutations. Genet Test 2000;4:399–402. [DOI] [PubMed] [Google Scholar]
- 31. Simsek Papur O , Akman SA , Cakmur R , Terzioglu O . Mutation analysis of ATP7B gene in Turkish Wilson disease patients: identification of five novel mutations . Eur J Med Genet 2013. ; 56 : 175 – 179 . [DOI] [PubMed] [Google Scholar]
- 32. Wu Z , Wang N , Murong S , Lin M . Identification and analysis of mutations of the Wilson disease gene in Chinese population . Chin Med J (Engl) 2000. ; 113 : 40 – 43 . [PubMed] [Google Scholar]
- 33. Geng J , Wang J , Yao RE , Liu XQ , Fu QH . Identification of one novel and nine recurrent mutations of the ATP7B gene in 11 children with Wilson disease . World J Pediatr 2013. ; 9 : 158 – 162 . [DOI] [PubMed] [Google Scholar]
- 34. Park S, Park JY, Kim GH, et al. Identification of novel ATP7B gene mutations and their functional roles in Korean patients with Wilson disease. Hum Mutat 2007;28:1108–1113. [DOI] [PubMed] [Google Scholar]
- 35. Mukherjee S, Dutta S, Majumdar S, et al. Genetic defects in Indian Wilson disease patients and genotype‐phenotype correlation. Parkinsonism Relat Disord 2014;20:75–81. [DOI] [PubMed] [Google Scholar]
- 36. Gupta A, Aikath D, Neogi R, et al. Molecular pathogenesis of Wilson disease: haplotype analysis, detection of prevalent mutations and genotype‐phenotype correlation in Indian patients. Hum Genet 2005;118:49–57. [DOI] [PubMed] [Google Scholar]
- 37. Kumar S , Thapa BR , Kaur G , Prasad R . Identification and molecular characterization of 18 novel mutations in the ATP7B gene from Indian Wilson disease patients: genotype . Clin Genet 2005. ; 67 : 443 – 445 . [DOI] [PubMed] [Google Scholar]
- 38. Dastsooz H , Imanieh MH , Dehghani SM , Haghighat M , Moini M , Fardaei M . Multiplex ARMS PCR to detect 8 common mutations of ATP7B gene in patients with Wilson disease . Hepat Mon 2013. ; 13 : e8375 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Gromadzka G, Schmidt HH, Genschel J, et al. Frameshift and nonsense mutations in the gene for ATPase7B are associated with severe impairment of copper metabolism and with an early clinical manifestation of Wilson's disease. Clin Genet 2005;68:524–532. [DOI] [PubMed] [Google Scholar]
- 40. Loudianos G, Dessi V, Lovicu M, et al. Molecular characterization of Wilson disease in the Sardinian population: evidence of a founder effect. Hum Mutat 1999;14:294–303. [DOI] [PubMed] [Google Scholar]
- 41. Gu YH , Kodama H , Du SL , Gu QJ , Sun HJ , Ushijima H . Mutation spectrum and polymorphisms in ATP7B identified on direct sequencing of all exons in Chinese Han and Hui ethnic patients with Wilson's disease . Clin Genet 2003. ; 64 : 479 – 484 . [DOI] [PubMed] [Google Scholar]
- 42. Santhosh S, Shaji RV, Eapen CE, et al. ATP7B mutations in families in a predominantly Southern Indian cohort of Wilson's disease patients. Indian J Gastroenterol 2006;25:277–282. [PubMed] [Google Scholar]
- 43. Okada T, Shiono Y, Hayashi H, et al. Mutational analysis of ATP7B and genotype‐phenotype correlation in Japanese with Wilson's disease. Hum Mutat 2000;15:454–462. [DOI] [PubMed] [Google Scholar]
- 44. Bem RS, Raskin S, Muzzillo DA, et al. Wilson's disease in Southern Brazil: genotype‐phenotype correlation and description of two novel mutations in ATP7B gene. Arq Neuropsiquiatr 2013;71:503–507. [DOI] [PubMed] [Google Scholar]
- 45. Ferenci P, Caca K, Loudianos G, et al. Diagnosis and phenotypic classification of Wilson disease. Liver Int 2003;23:139–142. [DOI] [PubMed] [Google Scholar]
- 46. Pfeiffer RF . Wilson's disease . Handb Clin Neurol 2011. ; 100 : 681 – 709 . [DOI] [PubMed] [Google Scholar]
- 47. Roberts EA , Schilsky ML ; American Association for Study of Liver Diseases . Diagnosis and treatment of Wilson disease: an update . Hepatology 2008. ; 47 : 2089 – 2111 . [DOI] [PubMed] [Google Scholar]
- 48. King AD, Walshe JM, Kendall BE, et al. Cranial MR imaging in Wilson's disease. AJR Am J Roentgenol 1996;167:1579–1584. [DOI] [PubMed] [Google Scholar]
- 49. Scheinberg IH , Sternlieb I . Pregnancy in penicillamine‐treated patients with Wilson's disease . N Engl J Med 1975. ; 293 : 1300 – 1302 . [DOI] [PubMed] [Google Scholar]
- 50. Sinha S , Taly AB , Prashanth LK , Arunodaya GR , Swamy HS . Successful pregnancies and abortions in symptomatic and asymptomatic Wilson's disease . J Neurol Sci 2004. ; 217 : 37 – 40 . [DOI] [PubMed] [Google Scholar]
- 51. Yonetani L , Walshe JM . Surviving Wilson's disease . Clin Med 2001. ; 1 : 72 – 74 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Catana AM , Medici V . Liver transplantation for Wilson disease . World J Hepatol 2012. ; 4 : 5 – 10 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Guillaud O, Dumortier J, Sobesky R, et al. Long term results of liver transplantation for Wilson's disease: experience in France. J Hepatol 2014;60:579–589. [DOI] [PubMed] [Google Scholar]
- 54. Walshe JM. Brief observations on the management of Wilson's disease. Proc R Soc Med 1977;70(suppl 3):1–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Walshe JM . The physiology of copper in man and its relation to Wilson's disease . Brain 1967. ; 90 : 149 – 176 . [DOI] [PubMed] [Google Scholar]
- 56. Walshe JM . Wilson's disease . Lancet 2007. ; 369 : 902 . [DOI] [PubMed] [Google Scholar]
- 57. Scheinberg IH , Jaffe ME , Sternlieb I . The use of trientine in preventing the effects of interrupting penicillamine therapy in Wilson's disease . N Engl J Med 1987. ; 317 : 209 – 213 . [DOI] [PubMed] [Google Scholar]
- 58. Litwin T , Gromadzka G , Czlonkowska A . Neurological presentation of Wilson's disease in a patient after liver transplantation . Mov Disord 2008. ; 23 : 743 – 746 . [DOI] [PubMed] [Google Scholar]
- 59. Scheinberg IH , Sternlieb I . Wilson disease and idiopathic copper toxicosis . Am J Clin Nutr 1996. ; 63 : 842S – 845S . [DOI] [PubMed] [Google Scholar]
- 60. Brewer GJ , Dick RD , Johnson VD , Fink JK , Kluin KJ , Daniels S . Treatment of Wilson's disease with zinc XVI: treatment during the pediatric years . J Lab Clin Med 2001. ; 137 : 191 – 198 . [DOI] [PubMed] [Google Scholar]
- 61. Shoulson I , Goldblatt D , Plassche W , Wilson G . Some therapeutic observations in Wilson's disease . Adv Neurol 1983. ; 37 : 239 – 246 . [PubMed] [Google Scholar]
- 62. Paliwal VK , Gupta PK , Pradhan S . Gabapentin as a rescue drug in D‐penicillamine‐induced status dystonicus in patients with Wilson disease . Neurol India 2010. ; 58 : 761 – 763 . [DOI] [PubMed] [Google Scholar]
- 63. Dening TR , Berrios GE , Walshe JM . Wilson's disease and epilepsy . Brain 1988. ; 111 : 1139 – 1155 . [DOI] [PubMed] [Google Scholar]
- 64. Prashanth LK , Sinha S , Taly AB , Mahadevan A , Vasudev MK , Shankar SK . Spectrum of epilepsy in Wilson's disease with electroencephalographic, MR imaging and pathological correlates . J Neurol Sci 2010. ; 291 : 44 – 51 . [DOI] [PubMed] [Google Scholar]
- 65. Walshe JM . Penicillamine neurotoxicity: an hypothesis . ISRN Neurol 2011. ; 2011 : 464572 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Wilson SAK . Progressive lenticular degeneration: a familial nervous disease associated with cirrhosis of the liver . Brain 1912. ; 34 : 295 – 507 . [DOI] [PubMed] [Google Scholar]
- 67. Curtis D, Durkie M, Balac P, et al. A study of Wilson disease mutations in Britain. Hum Mutat 1999;14:304–311. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information: GAS for WD scoring sheet with rating instructions.
Video 1. Reversal of neurological disability in an 18‐year‐old patient with cerebellar ataxia‐tremor‐dystonia phenotype of WD. The patient has a homozygous c.813C>A, p.C271X ATP7B mutation. Serial GAS for WD tier 1 scores for liver (L), cognition (C), motor (M), and osseomuscular (0) domains and tier 2 total scores are tabulated (see Supporting Information for GAS WD scoring sheet and rating instructions).
Video 2. Reversal of neurological disability in a 27‐year‐old patient with dystonia‐parkinsonism phenotype of WD. The patient has heterozygous c.2930C>T, p.T977M, and c.4070C>T, p.A1357V ATP7B mutations. Serial GAS for WD tier 1 scores for liver (L), cognition (C), motor (M), and osseomuscular (0) domains and tier 2 total scores are tabulated (see Supporting Information for GAS WD scoring sheet and rating instructions).
Video 3. Neurological worsening with penicillamine and improvement with down‐titration of dosage in a 17‐year‐old patient with WD. The patient has homozygous c.2736_2746delCATTCAGCAGC, p.Ile913fs, ATP7B mutation. Serial GAS for WD tier 1 scores for liver (L), cognition (C), motor (M), and osseomuscular (0) domains and tier 2 total scores are tabulated (see Supporting Information for GAS WD scoring sheet and rating instructions).