

Abstract
MSA is a progressive neurodegenerative disorder characterized by autonomic failure and a variable combination of poor levodopa‐responsive parkinsonism and cerebellar ataxia (CA). Current therapeutic management is based on symptomatic treatment. Almost one third of MSA patients may benefit from l‐dopa for the symptomatic treatment of parkinsonism, whereas physiotherapy remains the best therapeutic option for CA. Only midodrine and droxidopa were found to be efficient for neurogenic hypotension in double‐blind, controlled studies, whereas other symptoms of autonomic failure may be managed with off‐label treatments. To date, no curative treatment is available for MSA. Recent results of neuroprotective and ‐restorative trials have provided some hope for future advances. Considerations for future clinical trials are also discussed in this review.
Keywords: multiple system atrophy, treatment, atypical parkinsonism, ataxia, autonomic dysfunction
MSA is a progressive neurodegenerative disorder leading to severe disability and impairment in quality of life.1, 2 MSA is characterized by a variable combination of poor levodopa‐responsive parkinsonism, cerebellar ataxia (CA), and autonomic failure.3 The MSA‐parkinsonian type (MSA‐P) predominates in the Western hemisphere, and the MSA‐cerebellar type (MSA‐C) is the major phenotype in the Eastern hemisphere.4
Autonomic failure, depression, and motor symptoms strongly correlate with poor quality of life,2 thus making them the main target for symptomatic therapy. Median survival ranges between 6 and 9 years after symptom onset,5 with nocturnal sudden death and aspiration pneumonia being major causes of death, as indicated by one small study.6 No treatment has yet been shown to improve survival in MSA, creating a strong need for new therapeutic approaches.
The pathological hallmark of MSA is the accumulation of alpha‐synuclein (α‐Syn) within glial cytoplasmic inclusions (GCIs).7 GCIs also contain many other proteins, suggesting that additional mechanisms are contributing to the progressive neurodegenerative process.8 The most consistently and severely affected brain regions are the olivopontocerebellar and striatonigral systems. In most cases, cell loss predominates in one of these systems, which is consistent with the clinical distinction between MSA‐P and MSA‐C.
In this article, we aimed at updating data on symptomatic and preventive/curative approaches for the management of MSA, based on our previous review article published in 2010.9 Some considerations will also be discussed regarding clinical trials in this population.
Symptomatic Therapies
In this section, currently available symptomatic treatments for MSA patients will be discussed. A summary of current therapies is presented in Table 1.
Table 1.
Symptomatic treatments for MSA patients
| Feature | Current First‐Line Treatment | Alternative Treatments |
|---|---|---|
| Parkinsonism | Levol‐dopa up to 1 g/day | DAs, amantadine (positive trend in small trial)*, paroxetine (positive trend in small trial)*, physiotherapy |
| Ataxia | Physiotherapy | Clonazepam, vitamin E, propanolol, baclofen, or amantadine, buspirone, gabapentin |
| OH | Nonpharmacological measures (custom‐fitted elastic stockings, raising the head of the bed when sleeping, water drinking, small meals). Midodrine*, droxidopa* | Fludrocortisone, pyridostigmine*, desmopressin at bedtime |
| Neurogenic lower urinary tract dysfunction |
Postvoid residual volume ≥100 mL: clean intermittent self‐catheterization. Postvoid residual volume <100 mL: anticholinergic agents for detrusor hyperactivity, (alpha‐adrenergic blockers for detrusor sphincter) dyssynergia. |
BoNT‐A in the detrusor muscle or urethral sphincter, surgery (sphincterotomy or sphincteric wall stenting), permanent catheterization |
| Constipation | High fluid and fiber intake, classical laxative therapy, polycarbophil 70, macrogol | Lubiprostone |
| Erectile dysfunction | Intracavernosal injection of papaverine or prostaglandin E1 | Sildenafil (not recommended in patients with OH)*, SC apomorphine injections |
| Drooling | Anticholinergic agents, injection of BoNT‐A into the salivary glands | |
| Breathing disorders | Continuous positive air pressure | Tracheotomy (life‐threatening and/or daytime stridor, abnormal vocal cord, mobility on laryngoscopy), laryngeal surgery or BoNT therapy, adaptative servoventilation |
| Dystonia | BoNT injection (focal dystonia) | Anticholinergics, amantadine, DAs, muscle relaxants, or tetrabenazine |
| Camptocormia | Physiotherapy in combination with specific orthosis, wearing a backpack | BoNT injections, proterelin |
| RBD | Clonazepam, melatonin | |
| RLS | Pregabalin, pramipexole, ropinirole, and rotigotine | Gabapentin, enacarbil, pramipexole, ropinirole, l‐dopa |
| Depression | Psychotherapy, SSRIs | Electroconvulsive therapy, repetitive TMS |
Almost all of the therapeutics currently being used are based on expert opinion and do not meet scientific evidence standards. Recommendations based on double‐blind, randomized, placebo‐controlled trials in MSA patients are indicated with asterisks (*).
Parkinsonism
A poor l‐dopa response is one of the consensus criteria for the diagnosis of MSA3 and helps to differentiate MSA from Parkinson's disease (PD).10 l‐dopa unresponsiveness should only be accepted after a treatment with at least 1 g of l‐dopa per day for at least 3 months.3 Notwithstanding, clinical experience suggests that approximately one third of patients may experience some benefit, especially if they have MSA‐P.11 Indeed, in series with pathological confirmation, a positive l‐dopa response was reported in 28% to 65% of patients.12, 13, 14, 15, 16 However, this beneficial effect persists for several years in only 13% of patients.17 In some cases, benefits might be subjective, and not apparent, in a standardized motor examination, whereas others convey subsequent motor degradation after l‐dopa discontinuation.17 In a recent study from the European MSA Study Group, including 141 patients with MSA, a positive l‐dopa response was found in 31% of subjects, with a mean response duration of 3.5 years.18
In agreement with these results, a recent drug utilization study found a similar l‐dopa intake in PD and MSA‐P patients, whereas it was less frequently used in MSA‐C patients.19
Although l‐dopa may induce less delirium and hallucinations in MSA than in PD, according to the results of a single, small study,10 it can lead to adverse effects, such as worsening of orthostatic hypotension (OH) or pathological hypersexuality.17, 20 Moreover, early orofacial l‐dopa‐induced dyskinesias occur in MSA patients frequently in the absence of any motor benefit.17, 21 As in PD, domperidone may be added to prevent dopaminergic side effects, such as nausea and vomiting. Owing to possible cardiovascular adverse reactions, including prolongation of the QT interval, the European Medicines Agency has recently recommended that domperidone should be administered in doses of 10 mg no more than three times daily.22 Moreover, an electrocardiogram might be performed before it is used and the patients should be controlled frequently.
Efficacy of dopamine agonists (DAs) in MSA has not been yet tested by controlled trials. In a retrospective case‐record analysis, only 10% of patients experienced a benefit with DAs.17 Furthermore, given the higher rate of side effects compared with l‐dopa, particularly worsening of OH, DAs cannot be recommended as a first‐line treatment. Notwithstanding, in our drug utilization study, they were consumed by 29% of MSA‐P and 9% of MSA‐C patients versus 82% of PD patients.19 Recently, the effects of rotigotine transdermal patch have been studied in an uncontrolled, open‐label trial in 61 patients with atypical parkinsonism, of whom 13 had MSA‐P and 4 MSA‐C.23 In general, a decrease of UPDRS III scores without increasing behavioral disturbances was observed in the group of patients as a whole. Regrettably, results for each syndrome were not disclosed, and therefore it is not clear whether these results are applicable to MSA patients. Furthermore, the design of the study precludes any conclusion about the safety and efficacy of the drug in MSA.
Amantadine may be an alternative symptomatic treatment. Whereas a small, double‐blind, placebo‐controlled, crossover trial failed to demonstrate any significant motor benefit in 8 MSA patients,24 a retrospective study disclosed good or excellent responsiveness in 15%.17 Amantadine was equally consumed by approximately 15% of MSA and PD patients in our French cohort.19
Other antiparkinsonian agents, such as entacapone (added to l‐dopa) or monoamine oxidase B inhibitors are also used in some MSA patients,19 without any evidence supporting their efficacy.
The efficacy of the selective serotonin reuptake inhibitor, paroxetine, has been assessed for 2 weeks in a double‐blind, placebo‐controlled, randomized study including 19 MSA patients.25 Motor improvement was slight, but significant, in comparison to placebo. Further investigations are needed before any conclusion can be drawn on the efficacy of selective serotonin reuptake inhibitors (SSRIs) on motor, mood, and other symptoms in MSA. A double‐blind, multicenter trial with fluoxetine in France has been recently completed and final results are expected soon.26
Medical rehabilitation seems to improve balance and motor impairment27, 28, 29 as does the practice of tai chi.30 A recent randomized, controlled trial (RCT) demonstrated that tai chi training reduces balance impairments in patients with mild‐to‐moderate PD, with additional benefits of improved functional capacity and reduced falls.31 Such an approach may also prove beneifical in MSA patients.
Although bilateral subthalamic DBS may have been beneficial in few MSA patients,32 especially in those with preserved l‐dopa‐response,33 a review of the literature highlighted the poor efficacy of DBS.34 Moreover, more than one quarter of patients died within 7 months of surgery. Owing to the limited number of reports, the poor outcome, and the possibility of a harmful effect, DBS is currently not recommended in MSA.35, 36, 37, 38
CA
Physiotherapy remains the best therapeutic option for CA.39 In MSA, clinical effects of occupational therapy were tested in a controlled, nonrandomized, open‐label trial in a small number of MSA patients.27 Results suggested positive effects on UPDRS and 39‐item Parkinson Disease Questionnaire scores. Data on ataxia symptoms were not specifically collected.
Off‐label use of clonazepam, vitamin E, propanolol, baclofen, or amantadine have shown modest and transient efficacy.40 Buspirone (off‐label) improved upper‐limb ataxia in an open‐label trial including 9 MSA‐C patients.41 Gabapentin (off‐label) may have symptomatic benefits on ataxia, oscillopsia, and dysarthria according to a case report in 2 MSA‐C patients.42 Finally, protirelin tartrate (off‐label) has been used for ataxic diseases in Japan, but without any apparent effect in MSA.43 In concordance with these results, the French cohort of MSA‐C patients consumed vitamin E and buspirone more frequently than MSA‐P or PD patients.19 N‐acetylcysteine, which acts as a precursor to the antioxidant, glutathione, and modulates glutamatergic, neurotrophic, and inflammatory pathways, might also be effective for this indication according to recent data.44
Autonomic Failure
OH
OH is one of the major criteria for clinical MSA diagnosis3 and appears to affect equally MSA‐P and ‐C patients.45 It appears to be related to the loss of sympathetic preganglionic neurons in the intermediolateral column of the thoracolumbar spinal cord as well as in some supraspinal areas, including the Edinger‐Westphal nucleus, posterior hypothalamus, brainstem reticular formation, and ventrolateral medulla.46, 47 Functional neuroimaging PET studies also identified widespread neurodegeneration in noradrenergic brain regions, including the locus coeruleus.48 Sympathetic postganglionic neurons appear to be spared in MSA, in contrast to PD.
Identification of the mechanism of OH (disease, drug, or other causes) is the first step in the treatment, followed by nonpharmacological measures. Exposure to drugs potentially related to hypotension, including antihypertensive agents, diuretics, amantadine, l‐dopa, DAs, inhibitors of phosphodiesterase type 5 (e.g., sildenafil, tadalafil, and vardenafil), alpha‐adrenergic blockers used for prostatic hyperplasia, clonidine, and several antidepressants should be reconsidered.49, 50, 51 Patients should also avoid precipitating factors, such as sudden postural change, large meals, hot baths, and alcohol.52 Other nonpharmacological methods for treating OH include the addition of salt to the diet, drinking at least 240 mL of tap water,53 raising the head of the bed by 30 degrees at night, using compression stockings, or physical maneuvers that help to raise blood pressure.
In patients with insufficient or absent response to nonpharmacological measures, pharmacological options may be considered. A broad range of drugs has been used in OH, such as fludrocortisone, midodrine, ephedrine, octreotide,40 or droxidopa.54 Notwithstanding, there is a paucity of well‐done clinical trials to support the efficacy of such drugs. For example, the efficacy of midodrine has only been investigated in a few double‐blind, placebo‐controlled trials, which included patients with neurogenic OH, some of whom suffered from MSA.55, 56, 57 This α1‐adrenoceptor agonist alleviated moderate‐to‐severe OH with oral doses that ranged from 2.5 to 30 mg daily. Blood pressure should be regularly monitored in patients receiving midodrine to detect supine or nocturnal hypertension, which may be prevented by taking the evening dose more than 4 hours before bedtime.58
Droxidopa is an oral prodrug that is converted to norepinephrine through decarboxylation by the action of l‐aromatic amino acid decarboxylase, both at the peripheral tissues and the central nervous system.59 Droxidopa is currently marketed in Japan60 and has been recently approved by the U.S. Food and Drug Administration (FDA) for the treatment of symptomatic neurogenic OH.61 Generally speaking, clinical trials suggest that droxidopa prevents postprandial fall in blood pressure, increases blood pressure in supine and standing positions, and reduces the intensity of OH symptoms.54 It must be mentioned that, as happens for other drugs, trials with droxidopa included patients with neurogenic OH, suffering either from MSA or other pathologies. Notwithstanding, a study focusing on MSA patients is underway (clinicaltrials.gov NCT02071459).
Fludrocortisone increases renal sodium reabsorption and expands plasma volume, thus increasing blood pressure.62 Effects last longer than those of midodrine, thus making it more difficult to avoid nocturnal hypertension. Most common adverse include nausea, chest discomfort and morning headache. With prolonged treatment, nocturnal hypertension, hypokalemia, and postural edemas can also be observed.
Pyridostigmine modestly, but significantly, improved OH without supine hypertension in a double‐blind, randomized, crossover study,63 probably by increasing sympathetic tone by stimulating ganglionic neurons. Notwithstanding, only 17 of the 58 randomized patients suffered from MSA, making firm conclusions difficult. A treatment algorithm is presented in Figure 1.
Figure 1.
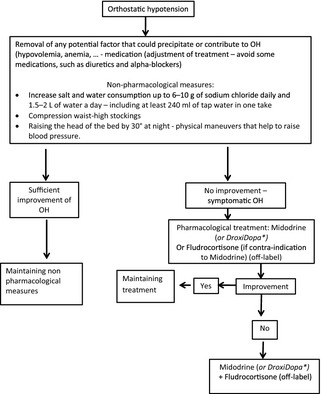
Treatment algorithm for OH in MSA. *Not labeled in Europe—labeled in the United States (FDA approval in February 2014).
Neurogenic Lower Urinary Tract Dysfunction
Urinary urgency, frequency, or incomplete bladder emptying as well as urinary incontinence remain key features of the consensual diagnosis criteria.3 Urinary disorders severely affect quality of life in MSA patients and may lead to repetitive lower urinary tract and kidney infection as major causes of morbidity in MSA.64 Clean intermittent self‐catheterization is recommended as the first‐line treatment when the postvoid residual is above 100 mL.65 Thus, the residual volume should be regularly monitored, for example, with a portable ultrasound device. The critical threshold of 100 mL is reached in a mean of 2 years after disease diagnosis.64
If the postvoid residual is below 100 mL, anticholinergic agents inhibiting the activity of the detrusor bladder muscle may be prescribed in the case of detrusor hyperactivity (e.g., urinary urgency, frequency, and urge incontinence), but with a risk of worsening urinary retention.66 Poor detrusor contractility, a frequent feature in later stages of MSA,67 reduces the efficacy of these drugs and carries the risk for elevated postmicturition residuals. Alpha‐adrenergic blockers may have some usefulness in urinary tract dysfunction in MSA,68 given that they may help reducing postmicturition residuals in patients with detrusor sphincter dyssynergia.69 However, alpha‐adrenergic blockers may aggravate OH69 and should thus be used carefully. In our French cohort of MSA patients, more‐frequent exposure to alpha‐adrenergic blockers was noted in patients with disease duration over 5 years.19 Conversely, patients with a “probable” MSA diagnosis (i.e., patients with a higher diagnostic certitude) were less frequently exposed to these agents.
The synthetic antidiuretic hormone desmopressin (off‐label) decreases urinary volumes.70 When given at bedtime, desmopressine reduces nocturia and improves morning hypotension. Regular monitoring is needed to detect water intoxication and hyponatremia as early as possible. Injection of botulinum toxin (BoNT) A (BoNT‐A) in the detrusor muscle may be an alternative treatment for detrusor overactivity, but the usefulness of this strategy only relying on a case report of 2 MSA patients needs confirmation in a larger number of patients,71 as well as urethral sphincter injections for urethral hypertonia when alpha‐blockers are not tolerated.72
More‐radical surgical procedures, such as sphincterotomy or sphincteric wall stenting, may be considered as a last option in MSA to reduce the risk of infection resulting from permanent catheterization.64, 65 Notwithstanding, both treatments may lead to incontinence, and thus caution is advisable. A treatment algorithm is presented in Figure 2.
Figure 2.
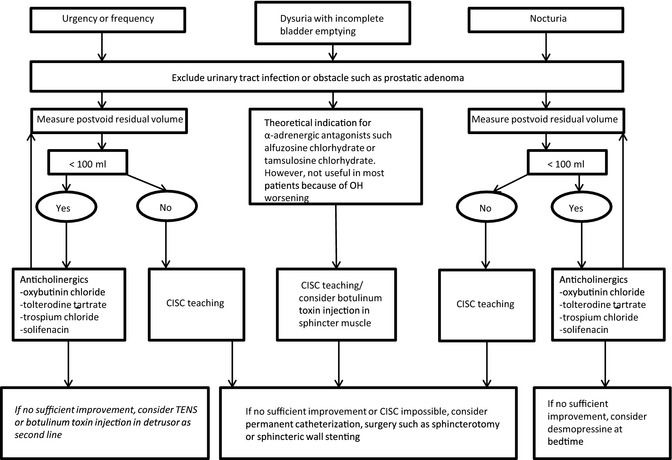
Treatment algorithm for lower urinary tract dysfunction in MSA. Detrusor hyperactivity or urethral hypertonia may be differentiated by urodynamic assessment. The postvoid residual volume should be regularly monitored, for example, with a portable ultrasound device, within 1 month after introduction of anticholinergics. CISC, clean intermittent self‐catheterization; TENS, transcutaneous electrical nerve stimulation.
Constipation
Laxative therapy is often necessary to relieve constipation in MSA, in addition to exercise and high fluid and fiber intake. Classical laxative treatment may be used. The efficacy of polycarbophil73 and mosapride citrate74 to reduce the time of transit in MSA patients has been assessed in two small, open trials. Mosapride has been withdrawn from the United States and Europe because of safety issues.75 Macrogol, which has been found to be effective in PD,76 was subjectively efficient in 2 MSA patients.77 In our French cohort, laxatives were more frequently administered to MSA, as compared to PD, patients and, particularly, to MSA patients with more severe disease.19
Erectile Dysfunction
The use of sildenafil in MSA may increase the risk of severe blood pressure drop. Indeed, in a randomized, double‐blind, placebo‐controlled, crossover study, sildenafil enhanced erectile function in the 6 MSA patients studied, but half of them experienced a severe drop in blood pressure 1 hour after drug ingestion.78 Therefore, its administration may not be recommended in patients with symptomatic OH. Notwithstanding, given that sildenafil has a half‐life of 3 to 4 hours, patients with OH may take it as long as they remain in the supine position during intercourse and 4 hours afterward. Sildenafil may be more useful in MSA patients than vardenafil and tadalafil given that they have half‐lives of 5 and 17 hours, respectively.
Intracavernosal injection of papaverine or prostaglandin E1 may be used for erectile dysfunction in MSA.68 A more convenient option might be the use of subcutaneous (SC) apomorphine, as in PD.79, 80 However, it has not been tested in MSA and carries the risk of increasing OH.
Drooling
Anticholinergic drugs, such as glycopirrolate, may be efficient, but adverse effects are frequent, including dry mouth, cognitive impairment, constipation, blurred vision, or urinary retention.81 Such adverse events might be more frequent with other antimuscarinic agents, such as atropine, as has been observed in PD.82 Injection of BoNT‐A into the salivary glands is the treatment of choice in excessive drooling because central side effects can be avoided.83 However, it may aggravate swallowing problems, which may require transient nasogastric tube feeding, and should not be considered in patients with marked impairment of swallowing. In patients with marked impairment of swallowing, percutaneous endoscopic gastrostomy (PEG) may be considered.
Breathing Disorders
A plethora of breathing disorders can be found in MSA, including central and obstructive sleep apnea, Cheyne‐Stokes respiration, dyspnea, hypoxemia, inspiratory sighs, and laryngeal stridor.3, 84, 85, 86, 87 These breathing disorders reflect the extension of the neurodegenerative process to the pontomedullary respiratory nuclei.88 They are key aspects of the management of MSA patients. Respiratory disorders may manifest in the early stages of MSA89 and may explain sudden death.90 Stridor is usually the consequence of vocal cord dystonia91, 92 and its occurrence is associated with poor outcome.93, 94 Noninvasive ventilation, such as continuous positive air pressure (CPAP) or bilevel positive airway pressure, seems the first‐line treatment, but no consensus currently exists.95, 96, 97 More‐invasive procedures, such as tracheotomy, may be considered in case of severe life‐threatening stridor.93, 98 Of note, tracheostomy does not prevent sudden death related to central apnea, which might even be worse after the intervention.6, 99 The use of laryngeal surgery or BoNT therapy is sometimes performed with mixed results in small case series.91, 95 Injecting BoNT in advanced disease carries the risk for worsening of choking, sometimes requiring nasogastric tube feeding for several weeks or even PEG.
Sleep‐disordered breathing might be effectively treated by CPAP,100, 101 but with a low degree of acceptability when disease progresses according to results from our group.97 Moreover, the mean survival does not seem to be modified, although no prospective, controlled studies have been performed.6, 97 Alternatively, adaptive servoventilation may be considered in MSA patients with central sleep apnea.102
Other Movement Disorders
Orofacial dystonia, antecollis, and camptocormia are features supporting the consensus diagnosis of MSA and constitute “red flags” for the differential diagnosis with PD.3, 103
Dystonia
It may affect more than 40% of patients, according to a prospective trial including 24 l‐dopa‐naïve patients with probable MSA.21 BoNT injection is the most commonly used treatment for focal dystonia.104 Symptomatic relief with anticholinergics, amantadine, DAs, muscle relaxants, or tetrabenazine has been reported in some MSA patients when used off label,104, 105 but no controlled trials are available.
Camptocormia
It is an abnormal forward flexion of the trunk that manifests when standing or walking and relieves in the supine position,105 poorly responsive to l‐dopa.105, 106, 107 Camptocormia (CC) might also be observed in later stages of PD as well in other neurological diseases.108 Whether CC is a dystonic posture, a consequence of axial rigidity, or a focal myopathy is still debated.105, 107, 109 Protirelin tartrate improved CC dramatically in one case report, presumably by enhancing motoneuronal excitability of the paraspinal muscles.43 As in focal dystonia, BoNT injections may be effective.105 Theoretically speaking, injections may be more appropriate in rectus abdominus muscles than in lumbar paraspinal muscles because of the risk of transient aggravation of CC, but placebo‐controlled trials are lacking. Physiotherapy, in combination with specific orthosis, may also improve CC and quality of life.110 Some clinical evidence suggests that wearing a backpack may also alleviate CC in PD.111 Although the efficacy has not been specifically tested in MSA, such noninvasive options might be considered. Recent results suggest that SC apomorphine infusion or spinal magnetic stimulation might be effective for this indication, at least in PD.112, 113
Sleep Disorders
Rapid Eye Movement Sleep Behavior Disorder
Rapid eye movement sleep behavior disorder (RBD) may be the presenting symptom of MSA114 and is observed in 69% to 100% of systematic polysomnography recordings in MSA patients.87, 115, 116
Securing the bed environment by physically removing hazards and lowering the bed has been recommended as the first measure to be taken.117 Clonazepam (off‐label) or melatonin (off‐label) may be used for further relief. Regrettably, evidence about the efficacy and safety of these drugs comes only from uncontrolled or very small, controlled trials, so further research is needed. Clonazepam and other benzodiazepines may aggravate existing obstructive sleep apnea and increase the risk of falls when the patient stands up at night because of nycturia. Melatonin is a hormone secreted by the pineal gland that modulates sleep initiation and circadian rhythms in humans; it has few side effects and is very well tolerated.
RLS
RLS affects between 4.7% and 28% of MSA patients.118, 119 Recently, a task force was established by the International Restless Legs Syndrome Study Group to develop evidence‐ and consensus‐based recommendations for the long‐term pharmacological treatment of RLS.120 Level A recommendations from this task force were as follows: Pregabalin has been established as effective for up to 1 year and pramipexole, ropinirole, and rotigotine for up to 6 months. Level B (i.e., probably effective) recommendations have been issued for the treatment of RLS ranging from 1 to 5 years: gabapentin, enacarbil, pramipexole, and ropinirole (1 years); l‐dopa (2 years); and rotigotine (5 years). Pergolide and cabergoline are not recommended owing to safety issues. None of these trials included MSA patients; thus, their utility in this pathology remains controversial.
Other Comorbidities
Depression
Depression is more prevalent in MSA than in PD121 and correlates with poor quality of life.2 In line with this, we found that exposure to antidepressants was significantly more frequent in MSA than in PD.19 SSRIs are the most prescribed antidepressive treatment, with less expected risk of inducing orthostatic hypotension than tricyclic drugs, but this remains to be proven in MSA. Interestingly, in the French cohort of MSA patients, exposure to antidepressants was related to higher frequency of autonomic symptoms and worse disease severity.19
Electroconvulsive therapy may be considered as a last‐line option in MSA patients with severe treatment‐refractory depression.122 Repetitive transcranial magnetic stimulation (TMS) may have antidepressant efficacy in PD, but the effects in MSA are unknown.123 Psychological support is necessary, as well as nursing care and family education.124 Finally, l‐dopa or DAs may slightly improve mood disorders in MSA,125 but this remains to be proven in well‐designed clinical trials.
Cognitive Impairment
Dementia is currently an exclusion criterion for a clinical diagnosis of MSA. However, cognitive disturbances in MSA are very frequent. They range from mild single‐domain deficits to impairments in multiple domains and even to frank dementia in 10% to 15%.126 Executive dysfunction is the most common presentation, followed by impairment in visuospatial functions. There is no evidence about the efficacy of any therapeutic strategy.
Preventive and Restorative Therapies
Clinical trials have failed to show disease‐modifying properties, contrasting with the effects observed in a variety of animal models of MSA.127 This highlights a significant gap between preclinical MSA models and human disease. Although toxin‐based models can reproduce various extents of neurodegeneration in the nigrostriatal system, they fail to reproduce the cytopathological hallmark of MSA, as shown by a lack of oligodendroglial α‐Syn accumulation. Conversely, transgenic (Tg) mouse models based on expression of α‐Syn in oligodendrocytes do reproduce the cytopathological hallmark of MSA, but display a modest magnitude of neurodegeneration and a mild progression over time (for review, see a previous work128). In addition, constitutive expression of α‐Syn in Tg animals may result in a number of adaptations and compensations that can further limit the translational potential of these models. Albeit imperfect and requiring improvements, currently available models are useful as a first screen for candidate disease‐modifying strategies, even though their predictive value is not firmly established (as is the case for most models of neurodegenerative disorders). A summary of the results from recent studies with putative neuroprotective or restorative therapies is presented in Table 2.
Table 2.
Summary of trials with putative neuroprotective and ‐restorative therapies in MSA
| Drugs | Trial Design | Main Results |
|---|---|---|
| Neuroprotective therapies | ||
| Riluzole |
RCT, DB, PC, CO in 10 MSA patients for 4 weeks140
RCT, DB, PC in 398 MSA patients for 36 months141 |
No effects on survival time or disease severity |
| Minocycline | RCT, DB, PC in 63 MSA‐P patients for 48 weeks142 | No change in clinical measures of motor impairment or health‐related quality of life |
| Estrogen | OL, RCT, add‐on to buspirone in 18 MSA patients41 | Negative results |
| Lithium | RCT, DB, PC in 10 MSA patients for 1 year143 | An interim analysis led to the interruption of the trial owing to higher proportion of trial abandon and a higher number of adverse effects in the lithium group |
| Rasagiline | RCT, DB, PC for 48 weeks in 174 patients with MSA‐P for 1 year144 | No significant difference in progression in UMSARS I+II scores |
| Rifampicin | RCT, DC, PC futility trial in 100 MSA patients for 1 year145 | No significant difference in progression of UMSARS I score |
| Growth hormone | RCT, DB, PC of 1 year in 43 MSA patients for 6 months146 | A trend to a smaller increase in disease severity (UPDRS and UMSARS) in treated patients |
| Intravenous immunoglobulins | OL, UC, pilot trial in 7 MSA patients for 6 months147 | UMSARS I and II improved post‐treatment |
| Neurorestorative therapies | ||
| Autologous mesenchymal stem cells | RCT, DB, PC in 34 MSA‐C patients for 1 year148, 149 | Significantly lower increase in disease severity (UMSARS) in treated patients |
| Granulocyte colony‐stimulating factor | OL, UC, in 11 patients with atypical parkinsonism (MSA = 4) for 3 months150 | No major side effects. Some efficacy was noted (UMSARS). |
CO, crossover; DB, double‐blind; OL, open‐label; PC, placebo‐controlled; UC, uncontrolled.
Considerations for Future Clinical Trials
For a better comparison of outcome results, clinical trials should apply consensus criteria for the diagnosis of MSA3 and assess disease severity by validated clinical outcome measures such as Unified MSA Rating Scale (UMSARS), which has four parts.129 The first part deals with limitations in patients' activities of daily living and symptoms of autonomic dysfunction, the second part assesses motor impairment during a neurological examination, the third part is based on blood pressure recording in supine and upright position, and the fourth part evaluates global disability. Autonomic dysfunction can be evaluated in MSA by the Scales for Outcomes in Parkinson's disease‐Autonomic (SCOPA‐Aut) scale or the different versions of the Composite Autonomic Symptom Scale (COMPASS).130, 131 SCOPA‐Aut is insensitive to change over time and seems therefore not to be useful for neuroprotection trials.132 By contrast, COMPASS change scale scores worsened in a preliminary study during follow‐up, suggesting that this scale may be more helpful for detecting neuroprotective effects on autonomic outcomes.133
Health‐related quality of life can be reliably assessed by the MSA‐QoL questionnaire.134 This questionnaire consists in threeindependent subscores measuring motor, nonmotor, and emotional/social functioning. Sensitivity to change over time is limited, thus precluding the use in its current form in neuroprotective trials.130 Several natural history studies have allowed the calculation of the required number of MSA patients in each treatment group for various outcomes, including UMSARS, MSA‐Qol, and survival.8 Notwithstanding, objective outcome measures are needed to complement subjective rating scales.
The choice of the right study design is of critical importance, owing to the fact that the classic randomized, double‐blind, placebo‐controlled type might need large samples of patients, which might be impractical, especially for early stages of drug development. Futility trials have been purposed as a means to detect “signals” of drug inefficacy.135 Such design has been used in other areas of clinical pharmacology, including the development of drugs for treatment of cancer or PD.
As in other neurodegenerative disorders, a putative neuroprotective drug should be given as early as possible. Therefore, sensitivity for the diagnosis of MSA needs further improvement (41% with current diagnosis criteria) in early disease stages.136 In an ideal world, patients at risk of developing MSA should be identified before the onset of core features. Early autonomic symptoms or preceding RBD may prove useful for early MSA detection,114, 137 but whether a patient with RBD will develop MSA, PD, or no neurodegenerative disorder remains currently unpredictable.138 Another argument to enroll patients in early disease comes from the latest natural history study of the European MSA Study Group showing that progression of UMSARS scores is highest within the first years of disease.18 Sensitive progression biomarkers are needed to assess the small effect of a putative neuroprotective drug in trials. Assessment of structural and functional neuroimaging abnormalities shows promising results.139
Conclusion
Symptomatic management in MSA should target primarily motor impairment, autonomic failure, and depression, given that these features are associated with a poor quality of life.2 l‐dopa remains the most frequently used treatment for parkinsonian symptoms in MSA, despite its modest, transient effect. Midodrine and droxidopa are the drugs of choice for treatment of OH, in addition to exhaustive nonpharmacological management. Strategies for urinary disorders are well standardized, whereas other symptoms, such as breathing disorders, RBD, depression, or dystonia, remain out of consensus.
Although there have been major advances in our understanding of the cellular pathology and in performing prospective trials designed for putative neuroprotective drugs, no curative treatment is yet available. Results with recombinant human growth hormone, intravenous immunoglobulins, and autologous mesenchymal stem cells deserve further research. Synergistic efforts of multicentric networks in Europe, North America, and Asia under the umbrella of the International Parkinson and Movement Disorder Society–sponsored MSA Study Group will catalyze the conduction of future clinical trials.
Author Roles
(1) Research Project: A. Conception, B. Organization, C. Execution; (2) Statistical Analysis: A. Design, B. Execution, C. Review and Critique; (3) Manuscript: A. Writing of the First Draft, B. Review and Critique.
S. P.‐L.: 1A, 1B, 1C, 3A, 3B
A.P.‐L.T.: 3B
O.F.: 3B
P.‐O.F.: 3B
M.V.R.: 3B
A.F.‐S.: 3B
F.T.: 3B
O.R.: 1A, 1B, 1C, 3B
W.G.M.: 1A, 1B, 1C, 3B
Disclosures
Funding Sources and Conflicts of Interest: The authors report no sources of funding and no conflicts of interest.
Financial Disclosures for previous 12 months: S.P.‐L. consulted for Aguettant Laboratories. A.P.‐L.T. received teaching honoraria and travel grants from Teva and UCB‐Pharma. F.T. consulted for Novartis, Addex, and Abott; participated in advisory boards for Novartis, Lundbeck, and UCB; received honoraria from UCB, GlaxoSmithKline (GSK), grants from the Michael J. Fox Foundation (MJFF), and travel support from Novartis, Lundbeck, and UCB. O.R. received honoraria and grants from Boehringer Ingelheim, Eisai, GSK, Novartis, Schering, Solvay, Teva Neurosciences, Lundbeck, and UCB. W.G.M. participated in advisory boards for ANM and Novartis France and received honoraria from Novartis Pharma, UCB, and Teva/Lundbeck, grants from the European Community, the MJFF, University Hospital Bordeaux, the French Ministry of Health, ANR, APTES, and PSP France, and travel grants from Affiris, Novartis, and Teva/Lundbeck.
Relevant disclosures and conflicts of interest are listed at the end of this article.
References
- 1. Tison F, Yekhlef F, Chrysostome V, et al. Parkinsonism in multiple system atrophy: natural history, severity (UPDRS‐III), and disability assessment compared with Parkinson's disease. Mov Disord 2002;17:701–709. [DOI] [PubMed] [Google Scholar]
- 2. Schrag A, Geser F, Stampfer‐Kountchev M, et al. Health‐related quality of life in multiple system atrophy. Mov Disord 2006;21:809–815. [DOI] [PubMed] [Google Scholar]
- 3. Gilman S, Wenning GK, Low PA, et al.Second consensus statement on the diagnosis of multiple system atrophy. Neurology 2008;71:670–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Watanabe H, Saito Y, Terao S, et al. Progression and prognosis in multiple system atrophy: an analysis of 230 Japanese patients. Brain 2002;125(Pt 5):1070–1083. [DOI] [PubMed] [Google Scholar]
- 5. Schrag A, Wenning GK, Quinn N, Ben‐Shlomo Y. Survival in multiple system atrophy. Mov Disord 2008;23:294–296. [DOI] [PubMed] [Google Scholar]
- 6. Shimohata T, Ozawa T, Nakayama H, Tomita M, Shinoda H, Nishizawa M. Frequency of nocturnal sudden death in patients with multiple system atrophy. J Neurol 2008;255:1483–1485. [DOI] [PubMed] [Google Scholar]
- 7. Ahmed Z, Asi YT, Sailer A, et al. The neuropathology, pathophysiology and genetics of multiple system atrophy. Neuropathol Appl Neurobiol 2012;38:4–24. [DOI] [PubMed] [Google Scholar]
- 8. Fernagut PO, Dehay B, Maillard A, et al. Multiple system atrophy: a prototypical synucleinopathy for disease‐modifying therapeutic strategies. Neurobiol Dis 2014;67:133–139. [DOI] [PubMed] [Google Scholar]
- 9. Flabeau O, Meissner WG, Tison F. Multiple system atrophy: current and future approaches to management. Ther Adv Neurol Disord 2010;3:249–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wenning GK, Ben‐Shlomo Y, Hughes A, Daniel SE, Lees A, Quinn NP. What clinical features are most useful to distinguish definite multiple system atrophy from Parkinson's disease? J Neurol Neurosurg Psychiatry 2000;68:434–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Constantinescu R, Richard I, Kurlan R. Levodopa responsiveness in disorders with parkinsonism: a review of the literature. Mov Disord 2007;22:2141–2148; quiz 2295. [DOI] [PubMed] [Google Scholar]
- 12. Colosimo C, Albanese A, Hughes AJ, de Bruin VM, Lees AJ. Some specific clinical features differentiate multiple system atrophy (striatonigral variety) from Parkinson's disease. Arch Neurol 1995;52:294–298. [DOI] [PubMed] [Google Scholar]
- 13. Fearnley JM, Lees AJ. Striatonigral degeneration. A clinicopathological study. Brain 1990;113(Pt 6):1823–1842. [DOI] [PubMed] [Google Scholar]
- 14. Rajput AH, Rozdilsky B, Rajput A, Ang L. Levodopa efficacy and pathological basis of Parkinson syndrome. Clin Neuropharmacol 1990;13:553–558. [DOI] [PubMed] [Google Scholar]
- 15. Wenning GK, Tison F, Ben Shlomo Y, Daniel SE, Quinn NP. Multiple system atrophy: a review of 203 pathologically proven cases. Mov Disord 1997;12:133–147. [DOI] [PubMed] [Google Scholar]
- 16. Hughes AJ, Daniel SE, Kilford L, Lees AJ. Accuracy of clinical diagnosis of idiopathic Parkinson's disease: a clinico‐pathological study of 100 cases. J Neurol Neurosurg Psychiatry 1992;55:181–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wenning GK, Ben Shlomo Y, Magalhaes M, Daniel SE, Quinn NP. Clinical features and natural history of multiple system atrophy. An analysis of 100 cases. Brain 1994;117(Pt 4):835–845. [DOI] [PubMed] [Google Scholar]
- 18. Wenning GK, Geser F, Krismer F, et al. The natural history of multiple system atrophy: a prospective European cohort study. Lancet Neurol 2013;12:264–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Rey MV, Perez‐Lloret S, Pavy‐Le Traon A, Meissner WG, Tison F, Rascol O. A cross‐sectional study on drug use in multiple system atrophy. CNS Drugs 2014;28:483–490. [DOI] [PubMed] [Google Scholar]
- 20. Klos KJ, Bower JH, Josephs KA, Matsumoto JY, Ahlskog JE. Pathological hypersexuality predominantly linked to adjuvant dopamine agonist therapy in Parkinson's disease and multiple system atrophy. Parkinsonism Relat Disord 2005;11:381–386. [DOI] [PubMed] [Google Scholar]
- 21. Boesch SM, Wenning GK, Ransmayr G, Poewe W. Dystonia in multiple system atrophy. J Neurol Neurosurg Psychiatry 2002;72:300–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. European Medicines Agency . CMDh confirms recommendations on restricting use of domperidone‐containing medicines. 2014.
- 23. Moretti DV, Binetti G, Zanetti O, Frisoni GB. Rotigotine is safe and efficacious in Atypical Parkinsonism Syndromes induced by both alpha‐synucleinopathy and tauopathy. Neuropsychiatr Dis Treat 2014;10:1003–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wenning GK. Placebo‐controlled trial of amantadine in multiple‐system atrophy. Clin Neuropharmacol 2005;28:225–227. [DOI] [PubMed] [Google Scholar]
- 25. Friess E, Kuempfel T, Modell S, et al. Paroxetine treatment improves motor symptoms in patients with multiple system atrophy. Parkinsonism Relat Disord 2006;12:432–437. [DOI] [PubMed] [Google Scholar]
- 26. Rascol O; French MSA‐Fluoxetine Study Group . Preliminary results of the French MSA‐fluoxetine study. Eur J Neurol 2012;19(suppl 1):262. [Google Scholar]
- 27. Jain S, Dawson J, Quinn NP, Playford ED. Occupational therapy in multiple system atrophy: a pilot randomized controlled trial. Mov Disord 2004;19:1360–1364. [DOI] [PubMed] [Google Scholar]
- 28. Landers M, Adams M, Acosta K, Fox A. Challenge‐oriented gait and balance training in sporadic olivopontocerebellar atrophy: a case study. J Neurol Phys Ther 2009;33:160–168. [DOI] [PubMed] [Google Scholar]
- 29. Wedge F. The impact of resistance training on balance and functional ability of a patient with multiple system atrophy. J Geriatr Phys Ther 2008;31:79–83. [DOI] [PubMed] [Google Scholar]
- 30. Venglar M. Case report: tai chi and parkinsonism. Physiother Res Int 2005;10:116–121. [DOI] [PubMed] [Google Scholar]
- 31. Li F, Harmer P, Fitzgerald K, et al. Tai chi and postural stability in patients with Parkinson's disease. N Engl J Med 2012;366:511–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Visser‐Vandewalle V, Temel Y, Colle H, van der Linden C. Bilateral high‐frequency stimulation of the subthalamic nucleus in patients with multiple system atrophy–parkinsonism. Report of four cases. J Neurosurg 2003;98:882–887. [DOI] [PubMed] [Google Scholar]
- 33. Ullman M, Vedam‐Mai V, Resnick AS, et al. Deep brain stimulation response in pathologically confirmed cases of multiple system atrophy. Parkinsonism Relat Disord 2012;18:86–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Shih LC, Tarsy D. Deep brain stimulation for the treatment of atypical parkinsonism. Mov Disord 2007;22:2149–2155. [DOI] [PubMed] [Google Scholar]
- 35. Santens P, Vonck K, De Letter M, et al. Deep brain stimulation of the internal pallidum in multiple system atrophy. Parkinsonism Relat Disord 2006;12:181–183. [DOI] [PubMed] [Google Scholar]
- 36. Tarsy D, Apetauerova D, Ryan P, Norregaard T. Adverse effects of subthalamic nucleus DBS in a patient with multiple system atrophy. Neurology 2003;61:247–249. [DOI] [PubMed] [Google Scholar]
- 37. Wenning GK, Stefanova N. Recent developments in multiple system atrophy. J Neurol 2009;256:1791–1808. [DOI] [PubMed] [Google Scholar]
- 38. Lambrecq V, Krim E, Meissner W, Guehl D, Tison F. Deep‐brain stimulation of the internal pallidum in multiple system atrophy. Rev Neurol (Paris) 2008;164:398–402. [DOI] [PubMed] [Google Scholar]
- 39. Ilg W, Bastian AJ, Boesch S, et al. Consensus paper: management of degenerative cerebellar disorders. Cerebellum 2014;13:248–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wenning GK, Geser F, Stampfer‐Kountchev M, Tison F. Multiple system atrophy: an update. Mov Disord 2003;18(suppl 6):S34–S42. [DOI] [PubMed] [Google Scholar]
- 41. Heo JH, Lee ST, Chu K, Kim M. The efficacy of combined estrogen and buspirone treatment in olivopontocerebellar atrophy. J Neurol Sci 2008;271:87–90. [DOI] [PubMed] [Google Scholar]
- 42. Gazulla J, Benavente MI. Improvements in the symptoms of olivopontocerebellar atrophy with gabapentin. Rev Neurol 2005;40:285–288. [PubMed] [Google Scholar]
- 43. Takei A, Hamada S, Homma S, Hamada K, Tashiro K, Hamada T. Amelioration of subacute camptocormia in multiple system atrophy by protirelin tartrate. Mov Disord 2009;24:2022–2023. [DOI] [PubMed] [Google Scholar]
- 44. Bavarsad Shahripour R, Harrigan MR, Alexandrov AV. N‐acetylcysteine (NAC) in neurological disorders: mechanisms of action and therapeutic opportunities. Brain Behav 2014;4:108–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Roncevic D, Palma JA, Martinez J, Goulding N, Norcliffe‐Kaufmann L, Kaufmann H. Cerebellar and parkinsonian phenotypes in multiple system atrophy: similarities, differences and survival. J Neural Transm 2014;121:507–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Iodice V, Low DA, Vichayanrat E, Mathias CJ. Cardiovascular autonomic dysfunction in MSA and Parkinson's disease: similarities and differences. J Neurol Sci 2011;310:133–138. [DOI] [PubMed] [Google Scholar]
- 47. Wenning GK, Granata R, Krismer F, et al. Orthostatic hypotension is differentially associated with the cerebellar versus the parkinsonian variant of multiple system atrophy: a comparative study. Cerebellum 2012;11:223–226. [DOI] [PubMed] [Google Scholar]
- 48. Lewis SJ, Pavese N, Rivero‐Bosch M, et al. Brain monoamine systems in multiple system atrophy: a positron emission tomography study. Neurobiol Dis 2012;46:130–136. [DOI] [PubMed] [Google Scholar]
- 49. Maule S, Papotti G, Naso D, Magnino C, Testa E, Veglio F. Orthostatic hypotension: evaluation and treatment. Cardiovasc Hematol Disord Drug Targets 2007;7:63–70. [DOI] [PubMed] [Google Scholar]
- 50. Perez‐Lloret S, Rey MV, Fabre N, et al. Factors related to orthostatic hypotension in Parkinson's disease. Parkinsonism Relat Disord 2012;18:501–505. [DOI] [PubMed] [Google Scholar]
- 51. Poon IO, Braun U. High prevalence of orthostatic hypotension and its correlation with potentially causative medications among elderly veterans. J Clin Pharm Ther 2005;30:173–178. [DOI] [PubMed] [Google Scholar]
- 52. Thompson P, Wright J, Rajkumar C. Non‐pharmacological treatments for orthostatic hypotension. Age Ageing 2011;40:292–293. [DOI] [PubMed] [Google Scholar]
- 53. Jordan J, Shannon JR, Black BK, et al. The pressor response to water drinking in humans: a sympathetic reflex? Circulation 2000;101:504–509. [DOI] [PubMed] [Google Scholar]
- 54. Perez‐Lloret S, Rey MV, Pavy‐Le Traon A, Rascol O. Droxidopa for the treatment of neurogenic orthostatic hypotension and other symptoms of neurodegenerative disorders. Expert Opin Orphan Drugs 2014;2:509–522. [Google Scholar]
- 55. Jankovic J, Gilden JL, Hiner BC, et al. Neurogenic orthostatic hypotension: a double‐blind, placebo‐controlled study with midodrine. Am J Med 1993;95:38–48. [DOI] [PubMed] [Google Scholar]
- 56. Low PA, Gilden JL, Freeman R, Sheng KN, McElligott MA. Efficacy of midodrine vs placebo in neurogenic orthostatic hypotension. A randomized, double‐blind multicenter study. Midodrine Study Group. JAMA 1997;277:1046–1051. [PubMed] [Google Scholar]
- 57. Wright RA, Kaufmann HC, Perera R, et al. A double‐blind, dose‐response study of midodrine in neurogenic orthostatic hypotension. Neurology 1998;51:120–124. [DOI] [PubMed] [Google Scholar]
- 58. Perez‐Lloret S, Rey MV, Pavy‐Le Traon A, Rascol O. Emerging drugs for autonomic dysfunction in Parkinson's disease. Expert Opin Emerg Drugs 2013;18:39–53. [DOI] [PubMed] [Google Scholar]
- 59. Kaufmann H. L‐dihydroxyphenylserine (Droxidopa): a new therapy for neurogenic orthostatic hypotension: the US experience. Clin Auton Res 2008;18(suppl 1):19–24. [DOI] [PubMed] [Google Scholar]
- 60. Saito S, Shioda K, Nishijima K. Dopamine dysregulation syndrome including mania related to coadministration of droxidopa. J Clin Psychopharmacol 2012;32:428–429. [DOI] [PubMed] [Google Scholar]
- 61. Food and Drug Administration . FDA Approves Northera to Treat Neurogenic Orthostatic Hypotension. Washington DC: 2014. http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm386311.htm. [Google Scholar]
- 62. Shibao C, Okamoto L, Biaggioni I. Pharmacotherapy of autonomic failure. Pharmacol Ther 2012;134:279–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Singer W, Sandroni P, Opfer‐Gehrking TL, et al. Pyridostigmine treatment trial in neurogenic orthostatic hypotension. Arch Neurol 2006;63:513–518. [DOI] [PubMed] [Google Scholar]
- 64. Ito T, Sakakibara R, Yasuda K, et al. Incomplete emptying and urinary retention in multiple‐system atrophy: when does it occur and how do we manage it? Mov Disord 2006;21:816–823. [DOI] [PubMed] [Google Scholar]
- 65. Fowler CJ, O'Malley KJ. Investigation and management of neurogenic bladder dysfunction. J Neurol Neurosurg Psychiatry 2003;74(suppl 4):iv27–iv31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Winge K, Fowler CJ. Bladder dysfunction in parkinsonism: mechanisms, prevalence, symptoms, and management. Mov Disord 2006;21:737–745. [DOI] [PubMed] [Google Scholar]
- 67. Wenning GK, Colosimo C, Geser F, Poewe W. Multiple system atrophy. Lancet Neurol 2004;3:93–103. [DOI] [PubMed] [Google Scholar]
- 68. Papatsoris AG, Papapetropoulos S, Singer C, Deliveliotis C. Urinary and erectile dysfunction in multiple system atrophy (MSA). Neurourol Urodyn 2008;27:22–27. [DOI] [PubMed] [Google Scholar]
- 69. Sakakibara R, Hattori T, Uchiyama T, et al. Are alpha‐blockers involved in lower urinary tract dysfunction in multiple system atrophy? A comparison of prazosin and moxisylyte. J Auton Nerv Syst 2000;79:191–195. [DOI] [PubMed] [Google Scholar]
- 70. Sakakibara R, Matsuda S, Uchiyama T, Yoshiyama M, Yamanishi T, Hattori T. The effect of intranasal desmopressin on nocturnal waking in urination in multiple system atrophy patients with nocturnal polyuria. Clin Auton Res 2003;13:106–108. [DOI] [PubMed] [Google Scholar]
- 71. Giannantoni A, Rossi A, Mearini E, Del Zingaro M, Porena M, Berardelli A. Botulinum toxin A for overactive bladder and detrusor muscle overactivity in patients with Parkinson's disease and multiple system atrophy. J Urol 2009;182:1453–1457. [DOI] [PubMed] [Google Scholar]
- 72. Apostolidis A, Dasgupta P, Denys P, et al. Recommendations on the use of botulinum toxin in the treatment of lower urinary tract disorders and pelvic floor dysfunctions: a European consensus report. Eur Urol 2009;55:100–119. [DOI] [PubMed] [Google Scholar]
- 73. Sakakibara R, Yamaguchi T, Uchiyama T, et al. Calcium polycarbophil improves constipation in primary autonomic failure and multiple system atrophy subjects. Mov Disord 2007;22:1672–1673. [DOI] [PubMed] [Google Scholar]
- 74. Liu Z, Sakakibara R, Odaka T, et al. Mosapride citrate, a novel 5‐HT4 agonist and partial 5‐HT3 antagonist, ameliorates constipation in parkinsonian patients. Mov Disord 2005;20:680–686. [DOI] [PubMed] [Google Scholar]
- 75. Tack J, Camilleri M, Chang L, et al. Systematic review: cardiovascular safety profile of 5‐HT(4) agonists developed for gastrointestinal disorders. Aliment Pharmacol Ther 2012;35:745–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Zangaglia R, Martignoni E, Glorioso M, et al. Macrogol for the treatment of constipation in Parkinson's disease. A randomized placebo‐controlled study. Mov Disord 2007;22:1239–1244. [DOI] [PubMed] [Google Scholar]
- 77. Eichhorn TE, Oertel WH. Macrogol 3350/electrolyte improves constipation in Parkinson's disease and multiple system atrophy. Mov Disord 2001;16:1176–1177. [DOI] [PubMed] [Google Scholar]
- 78. Hussain IF, Brady CM, Swinn MJ, Mathias CJ, Fowler CJ. Treatment of erectile dysfunction with sildenafil citrate (Viagra) in parkinsonism due to Parkinson's disease or multiple system atrophy with observations on orthostatic hypotension. J Neurol Neurosurg Psychiatry 2001;71:371–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. O'Sullivan JD. Apomorphine as an alternative to sildenafil in Parkinson's disease. J Neurol Neurosurg Psychiatry 2002;72:681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. O'Sullivan JD, Hughes AJ. Apomorphine‐induced penile erections in Parkinson's disease. Mov Disord 1998;13:536–539. [DOI] [PubMed] [Google Scholar]
- 81. Seppi K, Weintraub D, Coelho M, et al. The Movement Disorder Society Evidence‐Based Medicine Review Update: treatments for the non‐motor symptoms of Parkinson's disease. Mov Disord 2011;26(suppl 3):S42–S80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Hyson HC, Johnson AM, Jog MS. Sublingual atropine for sialorrhea secondary to parkinsonism: a pilot study. Mov Disord 2002;17:1318–1320. [DOI] [PubMed] [Google Scholar]
- 83. Mancini F, Zangaglia R, Cristina S, et al. Double‐blind, placebo‐controlled study to evaluate the efficacy and safety of botulinum toxin type A in the treatment of drooling in parkinsonism. Mov Disord 2003;18:685–688. [DOI] [PubMed] [Google Scholar]
- 84. Geser F, Seppi K, Stampfer‐Kountchev M, et al. The European Multiple System Atrophy‐Study Group (EMSA‐SG). J Neural Transm 2005;112:1677–1686. [DOI] [PubMed] [Google Scholar]
- 85. Meissner WG, Vital A, Ghorayeb I, Guehl D, Tison F. Dyspnea as first sign of autonomic failure in postmortem confirmed multiple system atrophy. Mov Disord 2010;25:1997–1998. [DOI] [PubMed] [Google Scholar]
- 86. Shimohata T, Shinoda H, Nakayama H, et al. Daytime hypoxemia, sleep‐disordered breathing, and laryngopharyngeal findings in multiple system atrophy. Arch Neurol 2007;64:856–861. [DOI] [PubMed] [Google Scholar]
- 87. Vetrugno R, Provini F, Cortelli P, et al. Sleep disorders in multiple system atrophy: a correlative video‐polysomnographic study. Sleep Med 2004;5:21–30. [DOI] [PubMed] [Google Scholar]
- 88. Benarroch EE. Brainstem respiratory control: substrates of respiratory failure of multiple system atrophy. Mov Disord 2007;22:155–161. [DOI] [PubMed] [Google Scholar]
- 89. Glass GA, Josephs KA, Ahlskog JE. Respiratory insufficiency as the primary presenting symptom of multiple‐system atrophy. Arch Neurol 2006;63:978–981. [DOI] [PubMed] [Google Scholar]
- 90. Tada M, Kakita A, Toyoshima Y, et al. Depletion of medullary serotonergic neurons in patients with multiple system atrophy who succumbed to sudden death. Brain 2009;132(Pt 7):1810–1819. [DOI] [PubMed] [Google Scholar]
- 91. Merlo IM, Occhini A, Pacchetti C, Alfonsi E. Not paralysis, but dystonia causes stridor in multiple system atrophy. Neurology 2002;58:649–652. [DOI] [PubMed] [Google Scholar]
- 92. Vetrugno R, Liguori R, Cortelli P, et al. Sleep‐related stridor due to dystonic vocal cord motion and neurogenic tachypnea/tachycardia in multiple system atrophy. Mov Disord 2007;22:673–678. [DOI] [PubMed] [Google Scholar]
- 93. Silber MH, Levine S. Stridor and death in multiple system atrophy. Mov Disord 2000;15:699–704. [DOI] [PubMed] [Google Scholar]
- 94. Yamaguchi M, Arai K, Asahina M, Hattori T. Laryngeal stridor in multiple system atrophy. Eur Neurol 2003;49:154–159. [DOI] [PubMed] [Google Scholar]
- 95. Iranzo A. Management of sleep‐disordered breathing in multiple system atrophy. Sleep Med 2005;6:297–300. [DOI] [PubMed] [Google Scholar]
- 96. Ghorayeb I, Bioulac B, Tison F. Sleep disorders in multiple system atrophy. J Neural Transm 2005;112:1669–1675. [DOI] [PubMed] [Google Scholar]
- 97. Ghorayeb I, Yekhlef F, Bioulac B, Tison F. Continuous positive airway pressure for sleep‐related breathing disorders in multiple system atrophy: long‐term acceptance. Sleep Med 2005;6:359–362. [DOI] [PubMed] [Google Scholar]
- 98. Silber MH. Tracheostomy can fatally exacerbate sleep‐disordered breathing in multiple system atrophy. Neurology 2008;70:980; author reply 981–982. [DOI] [PubMed] [Google Scholar]
- 99. Jin K, Okabe S, Chida K, et al. Tracheostomy can fatally exacerbate sleep‐disordered breathing in multiple system atrophy. Neurology 2007;68:1618–1621. [DOI] [PubMed] [Google Scholar]
- 100. Iranzo A, Santamaria J, Tolosa E. Continuous positive air pressure eliminates nocturnal stridor in multiple system atrophy. Barcelona Multiple System Atrophy Study Group. Lancet 2000;356:1329–1330. [DOI] [PubMed] [Google Scholar]
- 101. Iranzo A, Santamaria J, Tolosa E, et al. Long‐term effect of CPAP in the treatment of nocturnal stridor in multiple system atrophy. Neurology 2004;63:930–932. [DOI] [PubMed] [Google Scholar]
- 102. Suzuki M, Saigusa H, Shibasaki K, Kodera K. Multiple system atrophy manifesting as complex sleep‐disordered breathing. Auris Nasus Larynx 2010;37:110–113. [DOI] [PubMed] [Google Scholar]
- 103. Kollensperger M, Geser F, Seppi K, et al. Red flags for multiple system atrophy. Mov Disord 2008;23:1093–1099. [DOI] [PubMed] [Google Scholar]
- 104. Papapetropoulos S, Tuchman A, Sengun C, Russell A, Mitsi G, Singer C. Anterocollis: clinical features and treatment options. Med Sci Monit 2008;14:Cr427–Cr430. [PubMed] [Google Scholar]
- 105. Azher SN, Jankovic J. Camptocormia: pathogenesis, classification, and response to therapy. Neurology 2005;65:355–359. [DOI] [PubMed] [Google Scholar]
- 106. Bloch F, Houeto JL, Tezenas du Montcel S, et al. Parkinson's disease with camptocormia. J Neurol Neurosurg Psychiatry 2006;77:1223–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Diederich NJ, Goebel HH, Dooms G, et al. Camptocormia associated with focal myositis in multiple‐system atrophy. Mov Disord 2006;21:390–394. [DOI] [PubMed] [Google Scholar]
- 108. Finsterer J, Strobl W. Presentation, etiology, diagnosis, and management of camptocormia. Eur Neurol 2010;64:1–8. [DOI] [PubMed] [Google Scholar]
- 109. Slawek J, Derejko M, Lass P, Dubaniewicz M. Camptocormia or Pisa syndrome in multiple system atrophy. Clin Neurol Neurosurg 2006;108:699–704. [DOI] [PubMed] [Google Scholar]
- 110. de Seze MP, Creuze A, de Seze M, Mazaux JM. An orthosis and physiotherapy programme for camptocormia: a prospective case study. J Rehabil Med 2008;40:761–765. [DOI] [PubMed] [Google Scholar]
- 111. Gerton BK, Theeler B, Samii A. Backpack treatment for camptocormia. Mov Disord 2010;25:247–248. [DOI] [PubMed] [Google Scholar]
- 112. Arii Y, Sawada Y, Kawamura K, et al. Immediate effect of spinal magnetic stimulation on camptocormia in Parkinson's disease. J Neurol Neurosurg Psychiatry 2014;85:1221–1226. [DOI] [PubMed] [Google Scholar]
- 113. Katerina M, Michaela K, Miroslav V, Sandra K, Petr K. Treatment of camptocormia with continuous subcutaneous infusions of apomorphine: 1‐year prospective pilot study. J Neural Transm 2014;[Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 114. Tison F, Wenning GK, Quinn NP, Smith SJ. REM sleep behaviour disorder as the presenting symptom of multiple system atrophy. J Neurol Neurosurg Psychiatry 1995;58:379–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Manni R, Morini R, Martignoni E, Pacchetti C, Micieli G, Tartara A. Nocturnal sleep in multisystem atrophy with autonomic failure: polygraphic findings in ten patients. J Neurol 1993;240:249–250. [DOI] [PubMed] [Google Scholar]
- 116. Plazzi G, Corsini R, Provini F, et al. REM sleep behavior disorders in multiple system atrophy. Neurology 1997;48:1094–1097. [DOI] [PubMed] [Google Scholar]
- 117. Coeytaux A, Wong K, Grunstein R, Lewis SJ. REM sleep behaviour disorder – more than just a parasomnia. Aust Fam Physician 2013;42:785–788. [PubMed] [Google Scholar]
- 118. Bhalsing K, Suresh K, Muthane UB, Pal PK. Prevalence and profile of Restless Legs Syndrome in Parkinson's disease and other neurodegenerative disorders: a case‐control study. Parkinsonism Relat Disord 2013;19:426–430. [DOI] [PubMed] [Google Scholar]
- 119. Ghorayeb I, Dupouy S, Tison F, Meissner WG. Restless legs syndrome in multiple system atrophy. J Neural Transm 2014;121:1523–1527. [DOI] [PubMed] [Google Scholar]
- 120. Garcia‐Borreguero D, Kohnen R, Silber MH, et al. The long‐term treatment of restless legs syndrome/Willis‐Ekbom disease: evidence‐based guidelines and clinical consensus best practice guidance: a report from the International Restless Legs Syndrome Study Group. Sleep Med 2013;14:675–684. [DOI] [PubMed] [Google Scholar]
- 121. Tison F, Yekhlef F, Chrysostome V. Depression and self‐reported depressive symptoms in multiple system atrophy compared to Parkinson's disease. Mov Disord 2006;21:1056–1057. [DOI] [PubMed] [Google Scholar]
- 122. Shioda K, Nisijima K, Kato S. Electroconvulsive therapy for the treatment of multiple system atrophy with major depression. Gen Hosp Psychiatry 2006;28:81–83. [DOI] [PubMed] [Google Scholar]
- 123. Fregni F, Santos CM, Myczkowski ML, et al. Repetitive transcranial magnetic stimulation is as effective as fluoxetine in the treatment of depression in patients with Parkinson's disease. J Neurol Neurosurg Psychiatry 2004;75:1171–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Hardy J. Multiple system atrophy: pathophysiology, treatment and nursing care. Nurs Stand 2008;22:50–56; quiz 58. [DOI] [PubMed] [Google Scholar]
- 125. Fetoni V, Soliveri P, Monza D, Testa D, Girotti F. Affective symptoms in multiple system atrophy and Parkinson's disease: response to levodopa therapy. J Neurol Neurosurg Psychiatry 1999;66:541–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Stankovic I, Krismer F, Jesic A, et al. Cognitive impairment in multiple system atrophy: a position statement by the Neuropsychology Task Force of the MDS Multiple System Atrophy (MODIMSA) study group. Mov Disord 2014;29:857–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Kuzdas‐Wood D, Stefanova N, Jellinger KA, et al. Towards translational therapies for multiple system atrophy. Prog Neurobiol 2014;118:19–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Fernagut PO, Tison F. Animal models of multiple system atrophy. Neuroscience 2012;211:77–82. [DOI] [PubMed] [Google Scholar]
- 129. Wenning GK, Tison F, Seppi K, et al. Development and validation of the Unified Multiple System Atrophy Rating Scale (UMSARS). Mov Disord 2004;19:1391–1402. [DOI] [PubMed] [Google Scholar]
- 130. Meissner WG, Foubert‐Samier A, Dupouy S, et al. Assessment of quality of life with the multiple system atrophy health‐related quality of life scale. Mov Disord 2012;27:1574–1577. [DOI] [PubMed] [Google Scholar]
- 131. Sletten DM, Suarez GA, Low PA, Mandrekar J, Singer W. COMPASS 31: a refined and abbreviated Composite Autonomic Symptom Score. Mayo Clin Proc 2012;87:1196–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Damon‐Perriere N, Foubert‐Samier A, De Cock VC, et al. Assessment of the Scopa‐Aut questionnaire in multiple system atrophy: relation to UMSARS scores and progression over time. Parkinsonism Relat Disord 2012;18:612–615. [DOI] [PubMed] [Google Scholar]
- 133. Kollensperger M, Stampfer‐Kountchev M, Seppi K, et al. Progression of dysautonomia in multiple system atrophy: a prospective study of self‐perceived impairment. Eur J Neurol 2007;14:66–72. [DOI] [PubMed] [Google Scholar]
- 134. Schrag A, Selai C, Mathias C, et al. Measuring health‐related quality of life in MSA: the MSA‐QoL. Mov Disord 2007;22:2332–2338. [DOI] [PubMed] [Google Scholar]
- 135. Olanow CW, Kieburtz K. Defining disease‐modifying therapies for PD–a road map for moving forward. Mov Disord 2010;25:1774–1779. [DOI] [PubMed] [Google Scholar]
- 136. Osaki Y, Ben‐Shlomo Y, Lees AJ, Wenning GK, Quinn NP. A validation exercise on the new consensus criteria for multiple system atrophy. Mov Disord 2009;24:2272–2276. [DOI] [PubMed] [Google Scholar]
- 137. Gaig C, Iranzo A, Tolosa E, Vilaseca I, Rey MJ, Santamaria J. Pathological description of a non‐motor variant of multiple system atrophy. J Neurol Neurosurg Psychiatry 2008;79:1399–1400. [DOI] [PubMed] [Google Scholar]
- 138. Schenck CH, Bundlie SR, Mahowald MW. Delayed emergence of a parkinsonian disorder in 38% of 29 older men initially diagnosed with idiopathic rapid eye movement sleep behaviour disorder. Neurology 1996;46:388–393. [DOI] [PubMed] [Google Scholar]
- 139. Brooks DJ, Seppi K. Proposed neuroimaging criteria for the diagnosis of multiple system atrophy. Mov Disord 2009;24:949–964. [DOI] [PubMed] [Google Scholar]
- 140. Seppi K, Peralta C, Diem‐Zangerl A, et al. Placebo‐controlled trial of riluzole in multiple system atrophy. Eur J Neurol 2006;13:1146–1148. [DOI] [PubMed] [Google Scholar]
- 141. Bensimon G, Ludolph A, Agid Y, Vidailhet M, Payan C, Leigh PN. Riluzole treatment, survival and diagnostic criteria in Parkinson plus disorders: the NNIPPS study. Brain 2009;132(Pt 1):156–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Dodel R, Spottke A, Gerhard A, et al. Minocycline 1‐year therapy in multiple‐system‐atrophy: effect on clinical symptoms and [(11)C] (R)‐PK11195 PET (MEMSA‐trial). Mov Disord 2010;25:97–107. [DOI] [PubMed] [Google Scholar]
- 143. Sacca F, Marsili A, Quarantelli M, et al. A randomized clinical trial of lithium in multiple system atrophy. J Neurol 2013;260:458–461. [DOI] [PubMed] [Google Scholar]
- 144. Poewe W, Barone P, Giladi N, et al. A randomized, placebo‐controlled clinical trial to assess the effects of rasagiline in patients with multiple system atrophy of the parkinsonian subtype. Mov Disord 2012;27(suppl. 1):1182.22744819 [Google Scholar]
- 145. Low PA, Robertson D, Gilman S, et al. Efficacy and safety of rifampicin for multiple system atrophy: a randomised, double‐blind, placebo‐controlled trial. Lancet Neurol 2014;13:268–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Holmberg B, Johansson JO, Poewe W, et al. Safety and tolerability of growth hormone therapy in multiple system atrophy: a double‐blind, placebo‐controlled study. Mov Disord 2007;22:1138–1144. [DOI] [PubMed] [Google Scholar]
- 147. Novak P, Williams A, Ravin P, Zurkiya O, Abduljalil A, Novak V. Treatment of multiple system atrophy using intravenous immunoglobulin. BMC Neurol 2012;12:131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Lee PH, Kim JW, Bang OY, Ahn YH, Joo IS, Huh K. Autologous mesenchymal stem cell therapy delays the progression of neurological deficits in patients with multiple system atrophy. Clin Pharmacol Ther 2008;83:723–730. [DOI] [PubMed] [Google Scholar]
- 149. Lee PH, Lee JE, Kim HS, et al. A randomized trial of mesenchymal stem cells in multiple system atrophy. Ann Neurol 2012;72:32–40. [DOI] [PubMed] [Google Scholar]
- 150. Pezzoli G, Tesei S, Canesi M, et al. The effect of repeated administrations of granulocyte colony stimulating factor for blood stem cells mobilization in patients with progressive supranuclear palsy, corticobasal degeneration and multiple system atrophy. Clin Neurol Neurosurg 2010;112:65–67. [DOI] [PubMed] [Google Scholar]