

Abstract
Palatal tremor (PT) is an uncommon movement disorder that has been subdivided into essential and symptomatic forms. A distinct subgroup of the symptomatic form presents with progressive ataxia and PT. The histopathology of progressive ataxia and PT has not been previously determined. This study consisted of a clinical review, histopathology, and electron microscopy of the brain of a man with progressive ataxia and PT. The inferior olivary hypertrophy was symmetrical and homogenous, and no focal pathologic lesions could be identified in the brainstem. Insoluble tau deposits were found in neurons, exclusively infratentorially. We present the clinical and pathological evaluation of a case of progressive ataxia and PT that provide evidence for a unique form of 4R tauopathy.
Keywords: neurodegenerative disorders, neuropathology, ataxia, tremor
Symptomatic palatal tremor (SPT) may develop as a result of brainstem or cerebellar pathology involving the pathway between the dentate nucleus and inferior olive (IO), two sides of the “Guillain‐Mollaret triangle.”1, 2 The dentato‐(rubro)‐olivary pathway (DROP) originates from the dentate on one side, passes within the superior cerebellar peduncle, crosses over to the contralateral side, circles around the red nucleus (without rubral synaptic relay), and then joins the central tegmental tract to the IO. Associated with injuries in these pathways, inferior olivary hypertrophy (IOH) can develop as a result of trans‐synaptic degeneration.3, 4, 5, 6, 7 It takes approximately 3 weeks for IOH to appear after DROP lesions,8 with subsequent progression. If SPT were to develop, it presents several months later.9, 10 Etiology of SPT/IOH includes vascular, traumatic, and idiopathic entities.11, 12 One particular syndrome among the variety of etiologies is cerebellar ataxia with13 or without limb tremor, dysarthria, and, possibly, corticospinal tract involvement.11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24 This particular syndrome has been termed progressive ataxia and palatal tremor (PAPT).24 Etiology of PAPT has not been determined and no pathological evaluation of cases has been reported in the literature. We report here on the first case of PAPT with histologically proven, unique 4R tau pathology.
Patient and Methods
Case Report
A Caucasian right‐handed man first developed dysarthria and falls at the age of 63. In addition to the imbalance, his gait was noted to be wide based. He also had abnormal limb coordination and hand tremor. After years of relatively slow progression, his symptoms seemed to plateau. He had mouth angle twitching, which was later confirmed to correlate with his palatal tremor (PT). On specific request by physicians, the patient's wife observed that the facial twitching continued in sleep.
His first neurological exam, approximately 1 year after symptom onset, was conducted by a community neurologist and was incomplete, but was significant for dysarthric speech, postural hand tremor, titubation, PT without ear clicks, dyssynergia on finger‐to‐nose testing, and an ataxic respiratory pattern. There was neither nystagmus nor gaze palsy. His initial workup included normal brainstem auditory evoked potentials and vertebral and carotid circulation. The first MRI was noncontrast and showed ywo T2 hyperintense areas in the brainstem, later identified as bilateral hypertrophic IOs. A repeated MRI with gadolinium contrast provided no additional information. Because MRI was conducted at an outside institution, the images could not be retrieved, and our record only refers to the report in clinic notes. He was observed in our clinic for the first time 2 years after onset of symptoms. Additional findings on this exam included the presence of snout and palmomental reflexes, lack of extinction of glabello‐palpebral reflex, hypometric saccades, and irregular pursuit. PT was associated with rhythmic twitching of the mouth angles. The rate of PT was estimated at 3 Hz. He was started on buspirone and his ataxia dramatically improved. His follow‐up examination 4 years later in our clinic showed prominent primitive reflexes and hypometric saccades. PT frequency at this time was estimated at 1 to 2 Hz. Dysarthria was present. It was most consistent with a cerebellar pattern. He had bilateral postural tremor in the hands. There was significant dysmetria on finger‐to‐nose testing and a “no‐no” head tremor. On the most recent neurological exam that was available to us, with his neurologist 11 years after onset, there was scanning dysarthria and PT (without frequency estimate reported). On finger‐to‐nose testing, he had tremor and ataxia. His gait was wide based and moderately unsteady. He was hospitalized 12 years after onset and died from complications of a myocardial infarction.
Immunohistochemistry
The brain was cut coronally; 6‐μm sections from selected brain regions were stained with hematoxylin and eosin (H&E), Luxol fast blue, and Bielschowsky silver stain and processed for immunohistochemistry (IHC). The following panel of polyclonal and monoclonal (mAb) antibodies were employed: exon 10–specific polyclonal anti‐four repeat tau Ab (4R; dilution 1:1K); 17025 anti‐tau rabbit polyclonal Ab, raised against six recombinant human tau isoforms, (phosphorylation independent [pI]; highly phosphorylated anti‐neurofilament [NFH‐p+++]; dilution, 1:5); and LB509, anti‐α‐synuclein mouse mAb (pI; dilution, 1:1K) were provided by the Center for Neurodegenerative Disease Research at the University of Pennsylvania (Philadelphia, PA). Anti‐3R‐tau–mouse mAb was a gift from Dr. R. DeSilva (dilution, 1:1K); PHF1 (pD) mouse mAb (p‐ser262) was a gift from Dr. P. Davies (dilution, 1:1K); pD mouse mAb AT8 mouse mAb, tau antibody was from Innogenetics (pD; dilution, 1:1K; Ghent, Belgium); and 1510 anti‐ubiquitin, rabbit polyclonal Ab, dilution 1:1K (Chemicon, Temecula, CA).
Results
Gross Examination
Our examination revealed no evidence of swelling or herniation. There was no evidence of traumatic change. There were no pathologic changes in the cortical ribbon or the underlying white matter. Caudate nucleus, putamen, pallidum, claustrum, thalamus, mammillary bodies, and red nucleus were intact. The SN was well pigmented. The cerebellum appeared mildly atrophic. Serial sectioning of pons and medulla oblongata revealed hypertrophic inferior olivary nuclei, but no other pathologic changes.
Histopathology
IOs were markedly enlarged bilaterally, including the principal, medial, and dorsal accessory nuclei (Fig. 1A). On light microscopy, inferior olivary nuclei showed severe diffuse neuronal loss and gliosis, with a marked increase of activated microglial cells, which were rod‐like and devoid of branching processes. Hypertrophy of the inferior olivary nuclei was accompanied by atrophy of the amiculum. Unaffected neuronal cell bodies were enlarged and pleomorphic with distended processes and atypical cytoplasmic deposits. Granular basophilic cytoplasmic inclusions in the perikaryon and associated thickened processes were frequently present, whereas homogeneous, well‐demarcated cytoplasmic and filamentous inclusions, sometimes occurring in bundles in this region, were less abundant (Fig. 1B). Outside the IOs, cytoplasmic inclusions were observed in the locus coeruleus. In addition to changes in the neuronal perikaryon, there was marked hypertrophy of neuronal processes resulting in the formation of glomeruloid bodies throughout the IO (Fig. 1C). Silver stains revealed neuronal processes markedly thickened with apparently irregular orientation (Fig. 1C) in the absence of neuronal perikarya. Hypertrophic processes consistently showed prominent expression of neurofilament protein (Fig. 1D). Ubiquitin immunoreactivity in the medulla was confined to a small number of large “empty” neurons and was not colocalized with tau immunoreactivity (not shown). In the pons, 4R tau immunoreactivity was confined to granular intracellular inclusions and “classic” tangles (Fig. 1E,F). Note that 3R tau immunoreactivity was unremarkable and limited to a few neurons (Fig. 1G,H). Glomeruloid bodies consisting of enlarged perikarya and associated processes were frequently surrounded by markedly hypertrophic astrocytes, some of which showed atypical features, including irregular cytoplasmic filament distribution, cytoplasmic inclusions, and highly irregular nuclear contours. Different sections from both cerebellar hemispheres showed myelin loss around and within the dentate nucleus and focal and diffuse loss of myelin of other white matter regions toward the cortex. Purkinje and granular cells were markedly reduced in number, and preserved Purkinje cell (PC) bodies frequently were atrophic. Granular cytoplasmic inclusions were occasionally observed in degenerating PCs; however, the inclusions were smaller than those found in some of the IO neurons and were consistently silver stain negative. Neurons of the dentate nucleus were reduced in number, but devoid of significant cytological aberrations. The pontine tegmentum showed diffusely activated microglial cells, but there was no evidence of preferential involvement of the central tegmental tract. The SN was intact and well pigmented. No significant changes were observed in multiple sections taken from the thalamus, hypothalamus, and mammillary body. Insular cortex, claustrum, putamen, and pallidum, as well as random sections taken from frontal cortex, occipital cortex, and hippocampus, were structurally intact, and detailed structural and IHC search found no evidence of tau, other protein deposition, or other pathology. After a thorough examination (micro‐), infarcts were not found. IOs showed marked paired helical filament (PHF)1 immunoreactivity, confined to dense intracellular tangles and neutropil threads, indicating the presence of insoluble pathological tau (Supporting Fig. 1). Rabbit polyclonal α‐tau 17025 antibodies highlighted immunopositive tangles and threads (Supporting Fig. 2A). The staining pattern partially recapitulates PHF1 staining in the same region (Supporting Fig. 1). RMO24 (NHF‐p+++)‐positive dystrophic neurites were more abundant in the medulla of PAPT brain (lower left field) than in controls (data not shown), whereas no cellular staining was observed (lower right field). Prominent staining of two major types of 4R tau‐immunopositive intracellular inclusions were confined to granules and dense round structures. Granular structures were tightly “packed” in the cytoplasm of small neurons (Supporting Fig. 3A–D). Similar granules were not found in either Alzheimer's disease (AD) or PSP. Dense intracellular inclusions were scattered in a subset of neurons, and almost all were confined to “empty” (dead) large cells (Supporting Fig. 3E–J). Ultrastructural analysis using electron microscopy showed that the phosphorylation‐dependent tau mAb, PHF1, decorated the entire length of tau straight filaments (Fig. 2) in affected neurons. Locus coeruleus showed numerous PHF1‐ (Supporting Fig. 4A, B, D, and E) and AT8‐positive (Supporting Fig. 4F, G, H, I, K, and L) inclusions. Specific tau immunostaining was easily distinguishable from pigmented cells. No 3R tau (Supporting Fig. 4J and M) and ubiquitin (Supporting Fig. 4C) immunoreactivity was found in this region.
Figure 1.
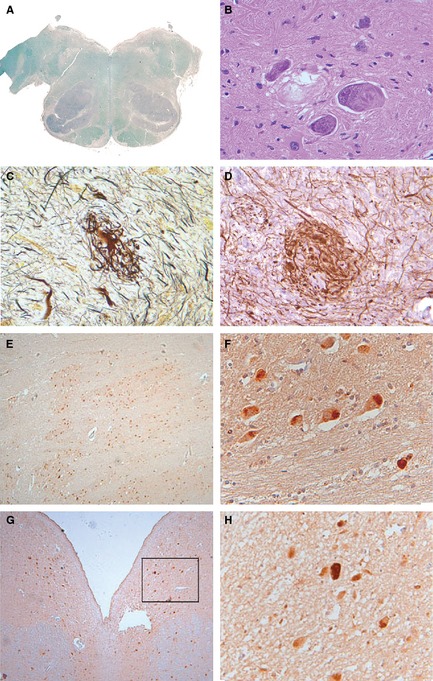
(A–D) Show staining of the IOs. (A) Luxol fast blue stain shows symmetrically enlarged IOs. (B) H&E stain shows filamentous bundled cytoplasmic inclusions in IO neurons. Many neuronal cells show prominent nuclei containing granular nucleoplasm and large nucleoli. (C) Glomeruloid bodies are found in IOs (Bielschowsky silver stain). (D) IHC with anti‐200‐kDa neurofilament protein in hypertrophic processes. (E–H) Pontine 4R tau and 3R tau immunoreactivity. (E and F) 4R tau immunoreactivity. (G and H) 3R tau immunoreactivity.
Figure 2.
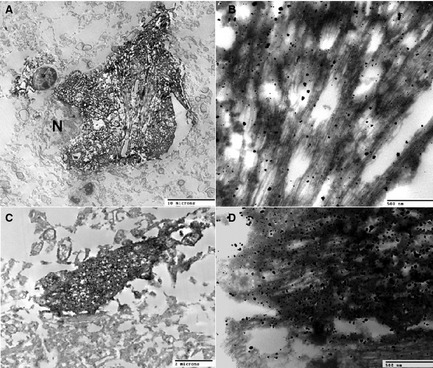
Immunolabeling of tau filaments using PHF1 antibody and ABC method plus silver enhancement (see an earlier report43 for the methodology). A neuron (A and B) and a neurite (C and D) granule are shown. N, nucleus.
All sections were negative for anti‐α‐synuclein (data not shown). No frozen tissue was available for biochemical analyses, thus the ratio of 4R:3R tau isoforms in this brain tissue could not be assessed.
Discussion
We present the first case of PAPT with tau pathology. It has previously been proposed that PT cases can be divided into essential PT and SPT.20 Idiopathic PAPT has been an established clinical and radiological entity with no obvious gross pathology to deafferent the IO.11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24 Our case is consistent with SPT, given the presence of IOH, lack of ear clicks, involvement of facial muscles, persistence of twitching during sleep, and presence of neurological symptoms in addition to PT. Cerebellar ataxia is part of this syndrome. PAPT therefore appears to be a distinct entity among degenerative diseases associated with SPT/IOH.
The course of our patient's PAPT syndrome is consistent with a slowly progressive neurodegenerative etiology. Clinically, this patient could be considered as having “possible MSA” based on the presence of gait ataxia with speech dysarthria and limb ataxia (“cerebellar criterion”), along with additional features, such as postural tremor and postural imbalance.25 However, the olivary pathology observed in MSA is very different from that observed in cases with IOH,26 and our case did not meet the pathological criteria for MSA.27 Importantly, this patient did not have parkinsonism or dysautonomia.
Our case did not meet the diagnostic criteria for PSP28 either. The main clinically distinguishing features were the lack of gaze palsy, pseudobulbar palsy, axial rigidity, and bradykinesia. The presence of palatal myoclonus is highly unusual in PSP and is not part of PSP syndrome. Also, long duration (more than 12 years) and slow progression of the disease were important features in this case. A summary distinguishing pathological features between PAPT and PSP, and between PAPT and IOH secondary to other causes (such as stroke affecting the central tegmental tract [CTT]), is available in Table 1. PAPT secondary to gluten sensitivity and celiac disease has been previously observed,29 but both were absent in our case. Small vascular lesions found in the CTT could also be causative, supporting observations by others that even very small lesions in the mid‐pontine region of the CTT can result in PT.30, 31 However, we believe that this is an unlikely possibility. First, our specimen was thoroughly observed for any brainstem pathology. Second, on thorough examination, there was virtually no evidence of cerebrovascular pathology in the rest of the brain. Third, the IOs were homogenously and symmetrically affected. This pattern of IOH would not be caused by such a small CTT lesion that could have been missed. Another unlikely possibility would be the presence of bilateral symmetrical small vascular lesions in the CTT, but very small vascular lesions would have been consistent with small‐vessel atherosclerotic disease, which this patient did not have. Fourth, the clinical course of gradual progression did not suggest a cerebrovascular event.
Table 1.
Comparative pathology of IOH and PSP
| IOH (Secondary, Other Than PAPT) | PSP | ||||
|---|---|---|---|---|---|
| Present | Absent | Present | Absent | ||
| PAPT (present case) | Present |
IOH; ↓ neurons in IO; NFH with ballooning neurons; dendritic enlargement with glomeruloid bodies; residual bodies; fibrillary astrocytosis with bizarre hypertrophic astrocytes; tegmental gliosis; IO hilar demyelination |
NFTs (tau positive); symmetric IOH; tightly packed tau‐positive granular intracellular lesions |
NFTs (tau positive); tegmental gliosis; IOH (cases with IOH); ↓ neurons in IO (cases with IOH); NFH with ballooning neurons (cases with IOH); dendritic enlargement with glomeruloid bodies; residual bodies (cases with IOH); fibrillary astrocytosis with bizarre hypertrophic astrocytes (cases with IOH) |
IOH (cases without IOH); ↓ neurons in IO (cases without IOH); NFH with ballooning neurons (cases without IOH); dendritic enlargement with glomeruloid bodies (cases without IOH); residual bodies (cases without IOH); fibrillary astrocytosis with bizarre hypertrophic astrocytes (cases without IOH); tightly packed tau positive granular intracellular lesions |
| Absent |
Vacuoles; asymmetric IOH; dentate/CTT lesion |
N/A |
Supratentorial involvement; nigral cell loss and gliosis; GVD |
N/A | |
NFH, high‐molecular‐weight neurofilament isoform; N/A, not applicable; GVD, granulovacuolar degeneration.
Although it has been proposed that PT might have been an unrelated finding in the pathologically proven cases of PSP,12, 32, 33 there is substantial overlap between PAPT and PSP pathology, with special emphasis on involvement of the olivo‐petal pathways. Virtually all PSP cases show some olivo‐petal histopathology,34 and OH is known to occur in PSP. Even in PSP cases without IOH, dentate pathology and olivary gliosis can be present.35 It is possible that the primary degenerative process affects neurons in the dentate and the olive, because two nuclei share anatomical and ontogenic features. The primary degenerative process affecting a portion of olivary neurons could trigger retrograde degeneration of the DROP fibers, which might cause secondary (deafferentation type) hypertrophic degeneration in other olivary neurons, perhaps through loss of axon collaterals. Partial deafferentation of the olive resulting from loss of dentate neurons could enhance this latter process. This could explain why we see the combination of primary (neurons with cytoplasmic tau accumulation) and secondary (glomeruloid bodies) degenerative changes in the olives. Alternatively, the primary neurodegenerative process in the olive could induce changes locally in other olivary neurons, which otherwise occur when the IO is deafferented. In the latter case, the fiber loss observed in the amiculum is suggestive of complete IO deafferentation and would be secondary to olivary degeneration. In either case, a primary neurodegenerative process is essential in the pathogenesis.
Although IOH is an almost obligate finding in patients with SPT, PT is observed only rarely in cases with IOH. In one report, only 2 of 29 IOH cases had PT.36 It appears that not only damage to the dentato‐olivary pathway is required for the PT to develop, but also the output from the inferior olivary nucleus (to the cerebellum) must be spared, probably along with some afferents of the IOs (other than DROP), such as those from the spinal cord. The pathophysiological importance of the spino‐olivary pathways in the generation of PT after DROP injury occurs has been discussed elsewhere.22 Dense pontine calcification, a lesion that could interrupt the dentate‐olivary pathway, though not observed in our case, has been recently reported in PAPT.37 An excellent model to explain PT has been proposed.38
Nearly all the unique pathological features consistent with transneuronal hypertrophic degeneration of the olives were observed in our case, with two important features: (1) numerous tau‐positive neuronal cytoplasmic inclusions and (2) symmetric bilateral involvement of the olives, without gross dentate, superior cerebellar peduncular, or CTT pathology. These are strongly suggestive of a primary degenerative etiology, together with lack of nondegenerative etiologies that can cause IOH (such as severe trauma, cerebrovascular disease, or tumor). PAPT has already been described, on clinical grounds, as a possible neurodegenerative entity.24
An unusual lack of intraneuronal vacuoles in the IO was found in our case, dissimilar to previous pathological reports on IOH.4, 36, 39, 40, 41 It is tempting to speculate that other pathological lesions (i.e., tau inclusions) could interfere with the process of vacuole formation. The presence of tau‐specific pathology, such as in PSP and our case, could play a protective role in explaining why neuronal vacuoles are not formed, even when other features of IOH are present. Alternatively, vacuole formation may be related to the rate of development of DROP lesions (see Supporting Information for additional discussion).
Analysis of tau isoforms and ultrastructural analyses of tau filaments have been widely used to characterize tau lesions in argyrophilic grain disease, Pick disease, frontotemporal dementia linked to chromosome 17, AD, PSP, and CBGD.42, 43, 44 Moreover, it has been noted that brain‐region–specific alterations in tau pathology can also distinguish between tauopathies.45 In our case, 4R tau pathology was almost selectively present in the brainstem, particularly in IOs, and was specifically absent supratentorially, making PAPT pathologically a unique form of 4R tauopathy.
PAPT may be a phenotypic variation of tauopathy with selective involvement of IO and DROP, whereas structures typically involved in other forms of tauopathies, such as basal ganglia in PSP or cortex in corticobasal degeneration, are spared. Tau‐positive neurofibrillary tangles (NFTs) observed in the IO in our case would support this notion. PSP neuropathology may vary,46 but supratentorial tau pathology is its characteristic, making PAPT uniquely different from PSP.
PAPT appears to be a neurodegenerative syndrome that can be pathologically defined by olivary hypertrophy, olivary neurofibrillary tangles/tau‐positive neuronal inclusions, marked atrophy of the amiculum, and sparing of basal ganglia and cortical structures. PAPT may represent an entity that can be placed on the spectrum of 4R tauopathies, distinguishing itself from other diseases on the spectrum, both clinically and pathologically, by the selective involvement of the dentato‐olivary fibers (especially in the amiculum) and the IOs.
Acknowledgments
The authors thank Ms. Magaly Rojas, Histopathology Laboratory, Dr. Mark Raffeld and Ms. Cynthia Harris, Immunohistochemistry Laboratory, Laboratory of Pathology, National Cancer Institute, Dr. Ranil DeSilva, Division of Intramural Research, National Heart, Lung, and Blood Institute, National Institutes of Health, and Teresa Schuck, Center for Neurodegenerative Disease Research, University of Pennsylvania, for their excellent histological and immunohistochemical preparations.
Author Roles
(1) Research Project: A. Conception, B. Organization, C. Execution; (2) Statistical Analysis: A. Design, B. Execution, C. Review and Critique; (3) Manuscript: A. Writing of the First Draft, B. Review and Critique.
Z.M.: 1A, 1B, 1C, 3A, 3B
A.J.M.H.: 3A, 3B
A.V.: 1B, 1C
V.Z.: 1C
K.U.: 1C
V.M.‐Y.L.: 1A, 1C
M.H.: 1A, 1B, 1C, 3A, 3B
Financial Disclosures
None.
Supporting information
SUPPORTING DATA 1. Supplementary discussion.
SUPPORTING FIG. 1. PHF1 (ser262; Pd) immunoreactivity in IOs. Three fields (1, 2, and 3) shown in 5× (first column) and 20× magnification (second column).
SUPPORTING FIG. 2. (A) Rabbit polyclonal α‐Tau 17025 and neurofilament specific RMO24 (NFH‐p+++) immunoreactivity in IOs. (B) Medullar 4R tau and 3R tau immunoreactivity.
SUPPORTING FIG. 3. Medullar 4R tau (E10, 4R specific mAb, 1:1,000) immunoreactivity. Five fields shown in 10× (first column: A,C, E, G, and I) and 40× magnification (second column: B, D, F, H, and J) from IO.
SUPPORTING FIG. 4. Locus ceruleus PHF1 (A, B, D, and E), AT8 (F, G, H, I, K, and L) tau, 3R tau (J and M), and ubiquitin (C) immunoreactivity (no other stains done for these sections). Magnification is 10× for A, D, F, H, and K, otherwise 40×.
References
- 1. Guillain G, Mollaret P. [Two cases of synchronous and rhythmic palato‐pharyngo‐laryngo‐oculo‐diaphragmatic myoclonus]. Rev Neurol (Paris) 1931;2:545–566. [Google Scholar]
- 2. Guillain G, Mollaret P, Bertrand I. Sur la lésion responsable du syndrome myoclonique de tronc cérébral [On the lesion responsible for the myoclonus syndrome of the brainstem]. Rev Neurol (Paris) 1933;3:666–674. [Google Scholar]
- 3. Foix C, Chavany J, Hillemand P. Le syndrome myoclonique de la calotte [The myoclonus syndrome]. Rev Neurol (Paris) 1926;33:942–956. [Google Scholar]
- 4. Horoupian DS, Wisniewski H. Neurofilamentous hyperplasia in inferior olivary hypertrophy. J Neuropathol Exp Neurol 1971;30:571–582. [DOI] [PubMed] [Google Scholar]
- 5. Lapresele J, Ben Hamida M. The dentato‐olivary pathway. Somatotopic relationship between the dentate nucleus and the contralateral inferior olive. Arch Neurol 1970;22:135–143. [DOI] [PubMed] [Google Scholar]
- 6. Cowan WM. Anterograde and retrograde transneuronal degeneration in the central and peripheral nervous system In: Nauta WJH, Ebbesson SOE, eds. Contemporary Research Methods in Neuroanatomy. Heidelberg: Springer‐Verlag; 1970:217–251. [Google Scholar]
- 7. Duchen LW. Transneuronal degeneration In: Adams JH, Duchen LW, eds. Greenfield's Neuropathology. 5th ed London: Edward Arnold; 1992:20–22. [Google Scholar]
- 8. Goto N, Kaneko M. Olivary enlargement: chronological and morphometric analyses. Acta Neuropathol (Berl) 1981;54:275–282. [DOI] [PubMed] [Google Scholar]
- 9. Matsuo F, Ajax ET. Palatal myoclonus and denervation supersensitivity in the central nervous system. Ann Neurol 1979;5:72–78. [DOI] [PubMed] [Google Scholar]
- 10. Goyal M, Versnick E, Tuite P, et al. Hypertrophic olivary degeneration: metaanalysis of the temporal evolution of MR findings. AJNR Am J Neuroradiol 2000;21:1073–1077. [PMC free article] [PubMed] [Google Scholar]
- 11. Deuschl G, Wilms H. Palatal tremor: the clinical spectrum and physiology of a rhythmic movement disorder. Adv Neurol 2002;89:115–130. [PubMed] [Google Scholar]
- 12. Deuschl G, Wilms H. Clinical spectrum and physiology of palatal tremor. Mov Disord 2002;17(suppl 2):S63–S66. [DOI] [PubMed] [Google Scholar]
- 13. Masucci E, Kurtzke J. Palatal myoclonus associated with extremity tremor. J Neurol 1989;236:474–477. [DOI] [PubMed] [Google Scholar]
- 14. Bender M, Nathanson M, Gordon GG. Myoclonus of muscles of eye, face and throat. Arch Neurol Psychiatry 1952;67:44–58. [DOI] [PubMed] [Google Scholar]
- 15. Nathanson M. Palatal myoclonus: further clinical and pathological observations. Arch Neurol Psychiatry 1956;75:285–296. [PubMed] [Google Scholar]
- 16. Herrmann C Jr, Brown JW. Palatal myoclonus: a reappraisal. J Neurol Sci 1967;5:473–492. [DOI] [PubMed] [Google Scholar]
- 17. Sperling MR, Herrmann C Jr. Syndrome of palatal myoclonus and progressive ataxia: two cases with magnetic resonance imaging. Neurology 1985;35:1212–1214. [DOI] [PubMed] [Google Scholar]
- 18. Leger JM, Duyckaerts C, Brunet P. Syndrome of palatal myoclonus and progressive ataxia: report of a case. Neurology 1986;36:1409–1410. [DOI] [PubMed] [Google Scholar]
- 19. Yokota T, Uchihara T, Ueki M, Tsukagoshi H. Spinocerebellar degeneration with oculopalatal myoclonus: a study on palato‐ocular synchrony. Rinsho Shinkeigaku 1988;28:382–387. [PubMed] [Google Scholar]
- 20. Deuschl G, Mischke G, Schenck E, Schulte‐Monting J, Lucking CH. Symptomatic and essential rhythmic palatal myoclonus. Brain 1990;113(Pt 6):1645–1672. [DOI] [PubMed] [Google Scholar]
- 21. Elble RJ. Inhibition of forearm EMG by palatal myoclonus. Mov Disord 1991;6:324–329. [DOI] [PubMed] [Google Scholar]
- 22. Deuschl G, Toro C, Valls‐Sole J, Zeffiro T, Zee DS, Hallett M. Symptomatic and essential palatal tremor. 1. Clinical, physiological and MRI analysis. Brain 1994;117(Pt 4):775–788. [DOI] [PubMed] [Google Scholar]
- 23. Phanthumchinda K. Syndrome of progressive ataxia and palatal myoclonus: a case report. J Med Assoc Thai 1999;82:1154–1157. [PubMed] [Google Scholar]
- 24. Samuel M, Torun N, Tuite PJ, Sharpe JA, Lang AE. Progressive ataxia and palatal tremor (PAPT): clinical and MRI assessment with review of palatal tremors. Brain 2004;127(Pt 6):1252–1268. [DOI] [PubMed] [Google Scholar]
- 25. Gilman S, Low PA, Quinn N, et al. Consensus statement on the diagnosis of multiple system atrophy. J Neurol Sci 1999;163:94–98. [DOI] [PubMed] [Google Scholar]
- 26. Sakurai A, Okamoto K, Yaguchi M, Fujita Y, Mizuno Y, Nakazato Y, Gonatas NK. Pathology of the inferior olivary nucleus in patients with multiple system atrophy. Acta Neuropathol (Berl) 2002;103:550–554. [DOI] [PubMed] [Google Scholar]
- 27. Osaki Y, Wenning GK, Daniel SE, et al. Do published criteria improve clinical diagnostic accuracy in multiple system atrophy? Neurology 2002;59:1486–1491. [DOI] [PubMed] [Google Scholar]
- 28. Litvan I, Agid Y, Calne D, et al. Clinical research criteria for the diagnosis of progressive supranuclear palsy (Steele‐Richardson‐Olszewski syndrome): report of the NINDS‐SPSP international workshop. Neurology 1996;47:1–9. [DOI] [PubMed] [Google Scholar]
- 29. Kheder A, Currie S, Romanowski C, Hadjivassiliou M. Progressive ataxia with palatal tremor due to gluten sensitivity. Mov Disord 2012;27:62–63. [DOI] [PubMed] [Google Scholar]
- 30. Lapresle J, Ben Hamida M. Correspondence somatotopique secteur par secteur des dégénérescences de l'olive bulbaire consécutive à de lésions limitées du noyau dentéle contralatéral [Somatotopic correspondence, region by region, of degeneration of the medullary olive following limited lesions of the contralateral dentate nucleus. Study of 4 anatomic case]. Rev Neurol (Paris) 1965;113:439–448. [PubMed] [Google Scholar]
- 31. Lapresle J, Ben Hamida M. Contribution à la connaissance de la voie dento‐olivaire [A contribution to the knowledge of the dento‐olivary pathway. Anatomical study of 2 cases of hypertrophic degeneration of the olivary nucleus following limited softening of the mesencephalic tegmentum]. Presse Méd 1968;76:1226–1230. [PubMed] [Google Scholar]
- 32. Suyama N, Kobayashi S, Isino H, Iijima M, Imaoka K. Progressive supranuclear palsy with palatal myoclonus. Acta Neuropathol 1997;94:290–293. [DOI] [PubMed] [Google Scholar]
- 33. Takeuchi M, Sasaki S, Ito A, Osawa M, Kobayashi I, Maruyama S. An autopsy case of progressive supranuclear palsy with olivary hypertrophy. No To Shinkei 1991;43:863–867. [PubMed] [Google Scholar]
- 34. Daniel SE, de Bruin VM, Lees AJ. The clinical and pathological spectrum of Steele‐Richardson‐Olszewski syndrome (progressive supranuclear palsy): a reappraisal. Brain 1995;118(Pt 3):759–770. [DOI] [PubMed] [Google Scholar]
- 35. Hanihara T, Amano N, Takahashi T, Itoh Y, Yagishita S. Hypertrophy of the inferior olivary nucleus in patients with progressive supranuclear palsy. Eur Neurol 1998;39:97–102. [DOI] [PubMed] [Google Scholar]
- 36. Jellinger K. Hypertrophy of the inferior olives. Report on 29 cases. Z Neurol 1973;205:153–174. [DOI] [PubMed] [Google Scholar]
- 37. Stamelou M, Adams M, Davagnanam I, Batla A, Sheerin U, Talbot K, Bhatia KP. Progressive ataxia and palatal tremor associated with dense pontine calcification: a unique case. Mov Disord 2013;28:1155–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Shaikh AG, Hong S, Liao K, et al. Oculopalatal tremor explained by a model of inferior olivary hypertrophy and cerebellar plasticity. Brain 2010;133(Pt 3):923–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Okamoto K, Hirai S, Iizuka T, Watanabe M. Fundamental morphological changes in human olivary hypertrophy. Acta Pathol Jpn 1992;42:408–413. [DOI] [PubMed] [Google Scholar]
- 40. Pierot L, Cervera‐Pierot P, Delattre JY, Duyckaerts C, Chiras J, Brunet P. Palatal myoclonus and inferior olivary lesions: MRI‐pathologic correlation. J Comput Assist Tomogr 1992;16:160–163. [PubMed] [Google Scholar]
- 41. Sohn D, Levine S. Hypertrophy of the olives: a report on 43 cases In: Zimmermann HM, ed. Prog Neuropathol 1971:202–213. [Google Scholar]
- 42. Zhukareva V, Shah K, Uryu K, et al. Biochemical analysis of tau proteins in argyrophilic grain disease, Alzheimer's disease, and Pick's disease: a comparative study. Am J Pathol 2002;161:1135–1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Buee L, Delacourte A. Comparative biochemistry of tau in progressive supranuclear palsy, corticobasal degeneration, FTDP‐17 and Pick's disease. Brain Pathol 1999;9:681–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Sergeant N, Wattez A, Delacourte A. Neurofibrillary degeneration in progressive supranuclear palsy and corticobasal degeneration: tau pathologies with exclusively “exon 10” isoforms. J Neurochem 1999;72:1243–1249. [DOI] [PubMed] [Google Scholar]
- 45. Tolnay M, Sergeant N, Ghestem A, et al. Argyrophilic grain disease and Alzheimer's disease are distinguished by their different distribution of tau protein isoforms. Acta Neuropathol (Berl) 2002;104:425–434. [DOI] [PubMed] [Google Scholar]
- 46. Dickson DW, Ahmed Z, Algom AA, Tsuboi Y, Josephs KA. Neuropathology of variants of progressive supranuclear palsy. Curr Opin Neurol 2010;23:394–400. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
SUPPORTING DATA 1. Supplementary discussion.
SUPPORTING FIG. 1. PHF1 (ser262; Pd) immunoreactivity in IOs. Three fields (1, 2, and 3) shown in 5× (first column) and 20× magnification (second column).
SUPPORTING FIG. 2. (A) Rabbit polyclonal α‐Tau 17025 and neurofilament specific RMO24 (NFH‐p+++) immunoreactivity in IOs. (B) Medullar 4R tau and 3R tau immunoreactivity.
SUPPORTING FIG. 3. Medullar 4R tau (E10, 4R specific mAb, 1:1,000) immunoreactivity. Five fields shown in 10× (first column: A,C, E, G, and I) and 40× magnification (second column: B, D, F, H, and J) from IO.
SUPPORTING FIG. 4. Locus ceruleus PHF1 (A, B, D, and E), AT8 (F, G, H, I, K, and L) tau, 3R tau (J and M), and ubiquitin (C) immunoreactivity (no other stains done for these sections). Magnification is 10× for A, D, F, H, and K, otherwise 40×.