

Abstract
Autosomal dominant cerebellar ataxias (ADCAs) are clinically heterogeneous disorders classified according to genetic subtype and collectively known as SCAs. In a few SCAs, movement disorders can be the most frequent extracerebellar sign. The aim of this article is to perform a systematic review of movement disorders frequency and characteristics in ADCAs. This work consisted of a structured search of electronic databases up to January 2013. Publications containing descriptions of ADCA clinical features written in several languages were selected initially based on title and abstract screening, followed by full‐text reading of potentially relevant publications. Clinical findings and demographic data on genetically confirmed patients were extracted. Analysis of individual patient data from subjects with movement disorders was performed using the chi‐square test and logistic regression. One thousand and sixty‐six publications reviewing 12,151 patients from 30 different SCAs were analyzed. Individual data were available from 755 patients with at least one type of movement disorder during overall disease course. Of 422 patients in whom onset symptom data were available, one third referred a movement disorder as the initial symptom. During overall disease course, parkinsonism was common in many SCA subtypes, frequently described in the absence of ataxia and characterized as responding to dopaminergic medications. Motor complications developed occasionally in some patients as did nigrostriatal imaging alterations. Other frequent features were dystonia, chorea, and myoclonus. Rare conditions, such as akathisia, paroxysmal nonkinesigenic dyskinesia, or stiff person‐like syndrome, were also reported. ADCA descriptions included a full range of movement disorders. Aside from postural or intention tremor, dopamine‐responsive parkinsonism and dystonia were the most common.
Keywords: autosomal dominant cerebellar ataxias, spinocerebellar ataxias, parkinsonism, dystonia, chorea, myoclonus
Autosomal dominant cerebellar ataxias (ADCAs), genetically defined as SCAs are clinically heterogeneous neurodegenerative disorders, characterized by progressive ataxia presenting alone or in combination with other neurological features. Only on rare occasions is ataxia absent. Genetic abnormalities are variable and include repeat expansions in coding and noncoding regions of genes, conventional mutations, or large gene rearrangements, explaining, in part, clinical manifestation heterogeneity as well as differences in age of onset, disease severity, and progression.1
Movement disorders are among the most common noncerebellar symptoms of ADCAs and include both hypo‐ and hyperkinetic manifestations, such as parkinsonism, dystonia, chorea, and myoclonus, among others.2 In some SCA subtypes, movement disorders are the predominant clinical expression, whereas in others they can be the sole manifestation or the only presenting feature. This makes presumptive ADCA diagnosis during initial stages a real challenge because it may resemble its idiopathic counterparts (e.g., Parkinson's disease [PD] or Huntington's disease [HD]).3, 4, 5 Diagnosis becomes even more difficult when patients show nigrostriatal pathway disruption on functional neuroimaging,6, 7 or partial or complete response to dopamine replacement therapy (DRT),5, 8or develop dopaminergic‐related motor complications, such as motor fluctuations and dyskinesias.9
A previous review described many types of movement disorders present in ADCAs, linking them to the most probable SCA subtype, in particular, cases when ataxia was associated with a given movement disorder.10 We aim to report the frequency and characteristics of movement disorders and analyze individual patient data to explore different characteristics of the most common movement disorders, such as parkinsonism and dystonia and their relationship to medication or ataxia. Also, to describe infrequent phenomenology or conditions, such as head tremor, myokymia, restless legs syndrome (RLS), tics, akathisia, paroxysmal nonkinesigenic dyskinesia (PNKD), stiff person‐like syndrome, stuttering, spasmodic‐like dysphonia, laryngeal stridor, and palatal tremor (myoclonus), albeit present in many types of ADCAs, which have received scarce attention in the medical literature.
Methods
Details of the search strategy, selection of studies, data extraction, and statistical analysis of the systematic review that served as the source of the present analysis were published elsewhere.11 In brief, a comprehensive systematic search of electronic databases up to January 2013 was undertaken to include original articles, clinical notes, case reports, letters, congress abstracts, or any other publications potentially containing phenomenological descriptions or proportions of ADCAs published in English, Spanish, French, German, Italian, or Portuguese. Some researchers were contacted to identify gray literature. Back‐searching of retrieved publication reference lists was conducted to scan for other relevant publications missed by the structured search. Selection of studies was based on a two‐step sequence: First, potentially relevant publications were identified after title and abstract screening. If no abstract was available, a publication could still be selected during the second step, in which full‐text reading was undertaken to identify clinical feature descriptions or rate reporting. Data were extracted only for patients with genetic SCA confirmation. Presence or absence of clinical features was recorded in a standardized data sheet only when explicitly mentioned in the text and extracted exactly as described (e.g., unspecific descriptions, such as extrapyramidal or dyskinesia). When possible, individual patient data were extracted, including demographics, clinical features, medication use, or complementary studies. Patients with drug‐induced movement disorders were excluded.
Descriptive data were presented as mean ± standard error of the mean or proportions. Categorical variables were analyzed by chi‐square test with Yates' correction and logistic regression. If overall P < 0.01, pair‐wise comparisons were done by chi‐square test. IBM SPSS software (version 20; SPSS, Inc., Chicago, IL) was used for statistical analysis.
Results
Eleven thousand one hundred and fifty‐two publications were screened during this literature search. A flow diagram shows the disposition of publications that the search yielded was reported elsewhere.11 Clinical features from 12,151 genetically confirmed patients among 30 SCA subtypes were extracted. Patients with movement disorders that were reported with individual data at disease onset and overall disease were available for 422 and 755 patients, respectively. Four patients with exposure to typical antipsychotic medication were excluded.
Movement Disorders at Disease Onset
Movement disorders found at disease onset of different genetically confirmed SCA subtypes are described in Table 1. Prevalence for all types of movement disorders at disease onset was inferior to 4%. Frequencies of movement disorders at disease onset based on individual data are shown in Table 2. In all, approximately 30% of patients began with a movement disorder, whether isolated or in combination with ataxia. Logistic regression, including age, gender, and main movement disorders as independent variables, disclosed that age at onset ≥34 years was, as an independent factor, related to the presence of parkinsonism (odds ratio [OR]: 2.14; 95% confidence interval [CI]: 1.23–3.72), chorea (OR, 2.15; 95% CI: 1.11–4.16), and myoclonus (OR, 0.35; 95% CI: 0.17–0.71).
Table 1.
Movement disorders found at disease onset of different SCA subtypes
| SCA Type | Intention or Postural Upper‐Limb Tremor | Head Tremor | Parkinsonism | Chorea | Myoclonus | Dystonia |
|---|---|---|---|---|---|---|
| SCA1 | 1/244 | |||||
| SCA2 | 8/1007 | 12/1007 | 53/1007 | 1/1007 | 7/1007 | |
| SCA3 | 3/603 | 28/606 | 8/603 | |||
| SCA4 | ||||||
| SCA5 | ||||||
| SCA6 | 1/544 | 9/545 | 6/544 | |||
| SCA7 | 4/286 | 1/286 | ||||
| SCA8 | 17/71 | 1/71 | ||||
| SCA10 | ||||||
| SCA11 | 1/24 | |||||
| SCA12 | 74/114 | 1/114 | ||||
| SCA13 | ||||||
| SCA14 | 8/100 | 8/100 | 1/100 | |||
| SCA15‐16 | 9/65 | 1/10 | ||||
| SCA17 | 2/126 | 15/126 | 7/126 | 6/126 | ||
| SCA18 | ||||||
| SCA19‐22 | ||||||
| SCA20 | 1/14 | |||||
| SCA21 | ||||||
| SCA23 | 1/10 | 1/10 | ||||
| SCA25 | ||||||
| SCA26 | ||||||
| SCA27 | 14/19 | |||||
| SCA28 | ||||||
| SCA29 | ||||||
| SCA30 | ||||||
| SCA31 | 2/258 | |||||
| SCA35 | ||||||
| SCA36 | ||||||
| DRPLA | 5/171 | 12/171 | 3/171 | 3/171 | ||
| Total (%) | 150/3945 (3.8) | 13/3945 (0.3) | 105/3945 (2.6) | 19/3945 (0.5) | 13/3945 (0.3) | 35/3945 (0.9) |
DRPLA, dentatorubro‐pallidoluysian atrophy; NOS, not otherwise specified.
Table 2.
Frequencies of movement disorders at disease onset based on patients with individual data
| Total No. of Patients | 422 (100%)a |
|---|---|
| Patients with ataxia as presenting symptom | 159 (38%) |
| Patients with a movement disorder as presenting symptom | 123 (29%) |
| Parkinsonism | 78 |
| Dystonia | 20 |
| Postural or intention upper‐limb tremor | 10 |
| Head tremor | 6 |
| Chorea | 5 |
| Myoclonus | 4 |
| Akathisia | 1 |
| Patients with ataxia plus any movement disorder at disease onset | 18 (4%) |
| Dystonia | 7 |
| Chorea | 5 |
| Head tremor | 2 |
| Parkinsonism | 1 |
| Myoclonus | 1 |
| Postural or intention upper‐limb tremor | 1 |
| Restless legs syndrome | 1 |
| Patients with ataxia plus any other neurological feature | 35 (8%) |
| Patients only with neurological features other than ataxia or movement disorders as initial symptom | 87 (21%) |
Total percentages are not always 100%, because some patients began with more than one type of movement disorder.
Movement Disorders During Overall Disease Course
The most common movement disorders found during overall disease course of different genetically confirmed SCA subtypes are described in Table 3. Uncommon movement disorders were described in rare cases, such as PNKD (SCA 27), tics (SCA17, SCA25, and DRPLA), stuttering and akathisia (SCA3), palatal tremor or myoclonus and spasmodic‐like dysphonia (SCA20), and stiff person‐like syndrome (SCA1 and SCA3).
Table 3.
Most common movement disorders rates of SCA subtypes during overall disease coursea
| SCA Type | Intention or Postural Upper‐Limb Tremor | Head Tremor | Parkinsonism | Chorea | Myoclonus | Dystonia | Myokymia | Restless Legs | Extrapyramidal Signs or Parkinsonism (NOS) | ||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Bradykinesia | Rigidity | Rest Tremor | |||||||||
| SCA1 | 123/277 | 1/1 | 11/78 | 15/233 | 12/205 | 24/345 | 15/256 | 47/394 | 8/62 | 4/23 | 47/330 |
| SCA2 | 322/646 | 76/177 | 100/258 | 97/408 | 85/356 | 52/568 | 84/451 | 127/862 | 9/317 | 9/43 | 34/233 |
| SCA3 | 82/280 | 2/4 | 179/737 | 176/1036 | 51/504 | 37/689 | 43/580 | 442/1825 | 95/252 | 108/672 | |
| SCA4 | |||||||||||
| SCA5 | 6/16 | 1/1 | 2/15 | 2/7 | 5/22 | 0/7 | |||||
| SCA6 | 52/259 | 1/1 | 9/227 | 23/444 | 7/173 | 5/341 | 4/363 | 19/452 | 4/15 | 9/177 | |
| SCA7 | 39/111 | 1/8 | 1/47 | 5/39 | 12/87 | 6/32 | 23/131 | 14/89 | 7/17 | 31/184 | |
| SCA8 | 32/52 | 3/21 | 2/16 | 1/15 | 0/1 | 8/37 | 3/3 | 0/21 | |||
| SCA10 | 9/16 | 2/2 | 1/1 | 0/27 | 0/27 | 1/28 | 0/76 | ||||
| SCA11 | 0/3 | 1/21 | |||||||||
| SCA12 | 77/115 | 2/4 | 9/10 | 2/16 | 2/6 | 0/1 | |||||
| SCA13 | 1/8 | 1/8 | |||||||||
| SCA14 | 6/10 | 7/33 | 1/19 | 3/42 | 0/13 | 2/14 | 13/87 | 10/49 | 6/30 | 1/8 | 0/6 |
| SCA15‐16 | 27/46 | 3/9 | 2/10 | 2/13 | 2/10 | 2/23 | |||||
| SCA17 | 11/25 | 1/4 | 17/22 | 54/68 | 4/20 | 40/83 | 4/15 | 58/110 | 24/60 | ||
| SCA18 | 2/7 | ||||||||||
| SCA19‐22 | 2/10 | 2/19 | 0/9 | ||||||||
| SCA20 | 2/14 | 7/13 | |||||||||
| SCA21 | 12/16 | 5/16 | 10/16 | ||||||||
| SCA23 | 3/4 | 2/4 | |||||||||
| SCA25 | 1/6 | 1/6 | 1/6 | ||||||||
| SCA26 | |||||||||||
| SCA27 | 14/18 | 7/16 | 10/17 | 0/1 | |||||||
| SCA28 | 1/11 | 1/24 | 3/24 | 3/24 | |||||||
| SCA29 | 1/20 | ||||||||||
| SCA30 | 0/6 | 1/6 | 0/6 | ||||||||
| SCA31 | 7/65 | 0/2 | 0/34 | 0/2 | 1/42 | 11/146 | |||||
| SCA35 | 5/12 | 4/11 | |||||||||
| SCA36 | 2/44 | 1/44 | 1/44 | 0/14 | |||||||
| DRPLA | 24/53 | 3/3 | 4/10 | 1/2 | 1/2 | 137/215 | 78/179 | 36/103 | 5/13 | ||
| Total (%) | 845/2100 (40) | 114/360 (32) | 356/1475 (24) | 364/2320 (16) | 165/1311 (13) | 321/2397 (13) | 260/2109 (12) | 780/4085 (19) | 131/558 (24) | 120/358 (33) | 276/2025 (14) |
Adapted from Rossi et al.11
DRPLA, dentatorubro‐pallidoluysian atrophy; NOS, not otherwise specified.
Figure 1 shows the relationship between the most frequent types of movement disorders present alone or in combination with other symptoms during overall disease course of patients with individual data. In all, 491 of 755 (65%) patients had a single movement disorder, whereas 264 (35%) showed two or more movement disorders combined. The most frequent combinations were parkinsonism plus dystonia (35%), chorea plus myoclonus (11%), chorea plus dystonia (9%), dystonia plus myoclonus (5%), and parkinsonism plus chorea (4%), followed by other less‐common combinations.
Figure 1.
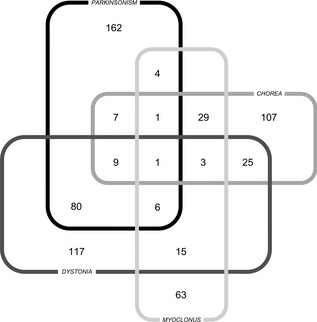
Number of patients with individual data of movement disorders during overall disease course.
Dystonia
Table 4 shows the frequencies of dystonic features of patients with individual data during overall disease course. In 9 of 140 patients (6%), botulinum toxin (BoT) use was reported, with a partial response observed in all. Dopamine replacement therapy (DRT) was also tried in 16 of 140 patients (11%), with partial response in 75% of the cases.
Table 4.
Frequencies of dystonic features of patients with individual data during overall disease course
| Total No. of Patients | 140 (%)a |
|---|---|
| Upper‐limb dystonia | 66/140 (47%) |
| Writer's cramp | 8/66 (12%) |
| Lower‐limb dystonia | 31/140 (22%) |
| Cervical dystonia | 42/140 (30%) |
| Blepharospasm | 10/140 (7%) |
| Craniofacial dystonia | 5/140 (4%) |
| Lingual dystonia | 3/140 (2%) |
| Dystonia NOS | 116 |
Percentages are not always 100%, because some patients had more than one type of movement disorder. NOS, not otherwise specified.
Parkinsonism
Parkinsonism was the movement disorder most frequently described in absence of ataxia during overall disease course (P = 0.01). However, of those “pure” parkinsonian patients, 30% exhibited cerebellar or brainstem atrophy on MRI. DRT use was reported in only half of the parkinsonian patients. When levodopa was used, mean daily dose was 572 ± 78 mg. In cases with parkinsonism, either alone or in combination with ataxia, response to DRT was inversely related to the presence of ataxia (P = 0.001; OR, 0.25; 95% CI: 0.07–0.72). Parkinsonism during overall disease duration responded to DRT in 107 of 138 (78%) patients described. Motor complications were described in a few of those patients (dyskinesias in 25 [23%] patients and motor fluctuations in 21 [20%]).
Functional Imaging of Parkinsonism
Dopaminergic imaging to evaluate nigrostriatal pre‐ and postsynaptic integrity was performed either with single‐photon emission CT (99mTc‐TRODAT‐1, [123]I‐FP‐CIT or [123]I‐IBZM) or PET ([18F]‐dopa or [11C]‐raclopride) in 52 of 271(19%) patients with parkinsonism, of which 47 (90%) showed alterations. Some patients underwent only presynaptic evaluation with radioligands, such as 99mTc‐TRODAT‐1, [123]I‐FP‐CIT, or PET [18F]‐dopa, whereas others included also postsynaptic functional imaging studies, such as [123]I‐IBZM or [11C]‐raclopride. Presynaptic involvement was found in 47 of 52 (90%) patients, whereas postsynaptic alterations were described in all 18 patients evaluated. In some cases, basal ganglia nuclei involvement was reported, corresponding to the putamen in 27 of 28 (96%) patients and to the caudate in 25 of 28 (89%). Asymmetrical alterations were present in 26 of 43 (60%) of the cases. Eight patients with ataxia without parkinsonism, 1 with myoclonus and 2 with dystonia, underwent dopaminergic imaging tests. Symmetric alterations were described in 6 (75%).
Discussion
This systematic review assessed the frequencies and characteristics of varied types of movement disorders in several genetic SCA subtypes. In almost one third of patients on whom individual data were available, movement disorder was the initial symptom. After intention or postural upper‐limb tremor, which can also form part of cerebellar syndrome, parkinsonism was the most frequent movement disorder at onset, followed by dystonia. Certain movement disorders were typically present at onset, in particular, SCA subtypes, as was the case of myoclonus in SCA14, parkinsonism in SCA17, and intention or postural upper‐limb tremor in SCA8, SCA12, SCA15‐16, and SCA27.
During overall disease, parkinsonism continued to be the most common movement disorder, followed in descending order by dystonia, chorea, myoclonus, myokymia, RLS, and head tremor. As had been observed for disease onset, during the course of the disease, some genetic subtypes had significantly higher proportion of a particular movement disorder, compared with the rest, such as parkinsonism, myokymia, intention or postural upper‐limb tremor and head tremor in SCA2, dystonia in SCA3, intention or postural upper‐limb tremor in SCA12, parkinsonism, chorea, and dystonia in SCA17, parkinsonism in SCA21, chorea in SCA27, and the combination of chorea, myoclonus, and dystonia in DRPLA11. A diagnostic scheme for movement disorders typically present at onset or during overall disease course in particular SCAs subtypes is depicted in Figure 2.
Figure 2.
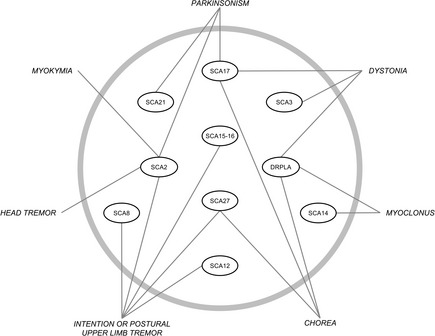
Diagnostic scheme for movement disorders typically present at onset or during overall disease course in particular SCA subtypes.
Intention or postural tremor was described in almost all SCA subtypes. Presence of l‐dopa‐responsive intention or postural tremor in young patients, associated with orthostatic tremor with a frequency range of 14 to 18 Hz, was described in SCA3.12
Rigidity and bradykinesia were the most frequent isolated parkinsonian signs, followed by rest tremor. Patients with isolated parkinsonism often developed ataxia during the course of disease. However, few presented parkinsonism without ataxia, even after 30 years of follow‐up.3, 8, 13 Many parkinsonian patients descriptions resembled those of idiopathic PD (iPD) patients, sharing clinical features, such as asymmetric onset, late age at onset, slow progression, and good response to DRT.3, 13, 14, 15, 16, 17, 18 However, one third of patients with isolated parkinsonism exhibited cerebellar or brainstem atrophy on MRI. Screening for SCA mutations in patients with parkinsonism without a cerebellar syndrome is not recommended, except for SCA2 (especially in Asian ethnic groups) and SCA17 in families with autosomal dominant parkinsonism.
DRT use was reported in only half the parkinsonian patients on whom individual data were available. Both complete and partial responses were observed of magnitude inversely related to the presence of ataxia. SCA patients with parkinsonism resembling iPD were commonly responsive to DRT.5, 8 Dopaminergic‐related motor complications, such as motor fluctuations and dyskinesias, were also described.5, 9, 13
Dopaminergic functional imaging with SPECT (99mTc‐TRODAT‐1, [123]I‐FP‐CIT, or [123]I‐IBZM) or PET ([18F]‐dopa or [11C]‐raclopride) was seldom performed in SCA patients with parkinsonism. Pre‐ and postsynaptic alterations of the nigrostriatal pathway were found. Asymmetric findings, such as those observed in iPD,18, 19 were common, though almost uniform affectation of putamen and caudate was found in most cases, distinguishing these patients from iPD cases, which tend to show predominant putamen involvement. Affectation of nigrostriatal pathway integrity might help explain the presence of dopamine‐responsive parkinsonism and occasional development of motor fluctuations or dyskinesias. However, the presence of presynaptic alteration on dopaminergic functional imaging and response to DRT are both findings for which further clarification is needed. Although rare, simultaneous iPD cannot be ruled out in the cases described. Recently, sequence variations of the glucocerebrosidase gene, which have been associated with iPD, were found in some SCA3 parkinsonian patients.20 Interestingly, symmetric nigrostriatal pathway disruption was also observed in some ataxic patients without parkinsonism.6, 7 Similarly, SN hyperechogenicity was frequently observed in ADCA patients, even those without movement disorders.21
Dystonia was the third‐most commonly described movement disorder during overall disease, whether in focal, segmental, or even generalized form.2, 10 Presence of dystonia strongly suggested diagnosis of SCA3, SCA17, and dentatorubral‐pallidoluysian atrophy (DRPLA).11 However, few SCA2 patients with cervical dystonia were also reported, which can be as frequent as 61% in some series.22 Upper‐limb involvement was present in almost half of the dystonic patients described, including writer's cramp. Other dystonic features reported were, in descending order, cervical dystonia, lower‐limb dystonia, blepharospasm, apraxia of eyelid opening, and craniofacial and lingual dystonia. In a few cases, DRT was tried, usually with partial response; seldom were responses marked.23, 24, 25 The most effective therapy was BoT, but, surprisingly, its use was rarely reported.26, 27
Chorea was frequently associated with SCA17, SCA27, and DRPLA, and, in several cases, phenotype was indistinguishable from HD.4, 11, 28 Myoclonus was observed often in SCA14 and DRPLA and rarely in other SCA subtypes.11 This could be of cortical, brainstem, or spinal origin and usually showed a poor response to different pharmacological treatments.29, 30 RLS was reported in six SCA subtypes with an overall frequency of 33%, which is superior to the 5% to 15% found in the general population.31 RLS rates increased with age and were mostly independent from peripheral neuropathy. This suggests a central origin, possibly resulting from dopaminergic dysfunction, also supported by the response to l‐dopa.32, 33
Less commonly reported were paroxysmal nonkinesigenic dyskinesia, stiff person‐like syndrome, akathisia, myokymia, stuttering, and tics.32, 34, 35, 36, 37 One SCA27 patient with PNKD differed clinically from patients with myofibrillogenesis regulator 1 gene mutations that frequently cause familial forms of this condition, by the development of mild mental retardation and lack of familial history.36, 38 One patient with SCA3 was described presenting with akathisia as the main neurological manifestation, which partially improved with clonazepan.37 A few patients with postural truncal dystonia, orofacial dyskinesia, and head tremor in SCA2 and gait apraxia in SCA10 were also reported.35
The main strengths of this systematic review were that the search was not restricted to a single database or English‐language publications, a large population of varied origin from multiple different publications was ascertained, and data extraction was limited to genetically confirmed ADCA patients. The study limitations include that statistical analysis may have been underpowered for rare SCA subtypes with few reported patients. Also, that conclusions on dopaminergic imaging or DRT response in patients with parkinsonism or dystonia, or BoT response in patients with dystonia, might be subject to reporting bias, because DRT use was reported in half of the patients with parkinsonism and only rarely was DRT or BoT treatment described in patients with dystonia.
In conclusion, movement disorders are frequently present in ADCAs. Sometimes, they can be the initial symptom, present even before development of ataxia, or sometimes even in the absence of ataxia for as long as for 30 years of disease duration. The most prominent is parkinsonism. In those cases, complementary studies are mandatory to rule out cerebellar atrophy. Alteration of the nigrostriatal pathway reported in some cases, as well as the response to DRT and development of motor fluctuations, are all findings requiring further clarification.
Author Roles
(1) Research Project: A. Conception, B. Organization, C. Execution; (2) Statistical Analysis: A. Design, B. Execution, C. Review and Critique; (3) Manuscript Preparation: A. Writing of the First Draft, B. Review and Critique.
M.R.: 1A, 1B, 1C, 2A, 2B, 2C, 3A, 3B
S.P.‐L.: 1A, 1B, 1C, 2A, 2B, 2C, 3A, 3B
D.C.: 1A, 1B, 1C, 2A, 2B, 2C, 3A, 3B
M.M.: 1A, 1B, 1C, 2A, 2B, 2C, 3A, 3B
Disclosures
Funding Sources and Conflicts of Interest: The authors report no sources of funding and no conflicts of interest.
Financial Disclosures for previous 12 months: Authors have no financial disclosures from previous 12 months to report.
Acknowledgments
The authors specially recognize the outstanding work done by their institutional medical librarians Floriana Colombo, Gabriela Tielas, and Lucina Freidemberg, to whom they are very grateful. The authors also thank Celia Podesta for language assistance.
Relevant disclosures and conflicts of interest are listed at the end of this article.
References
- 1. Durr A. Autosomal dominant cerebellar ataxias: polyglutamine expansions and beyond. Lancet Neurol 2010;9:885–894. [DOI] [PubMed] [Google Scholar]
- 2. Schols L, Peters S, Szymanski S, et al. Extrapyramidal motor signs in degenerative ataxias. Arch Neurol 2000;57:1495–1500. [DOI] [PubMed] [Google Scholar]
- 3. Furtado S, Farrer M, Tsuboi Y, et al. SCA‐2 presenting as parkinsonism in an Alberta family: clinical, genetic, and PET findings. Neurology 2002;59:1625–1627. [DOI] [PubMed] [Google Scholar]
- 4. Stevanin G, Fujigasaki H, Lebre AS, et al. Huntington's disease‐like phenotype due to trinucleotide repeat expansions in the TBP and JPH3 genes. Brain. 2003;126:1599–1603. [DOI] [PubMed] [Google Scholar]
- 5. Payami H, Nutt J, Gancher S, et al. SCA2 may present as levodopa‐responsive parkinsonism. Mov Disord 2003;18:425–429. [DOI] [PubMed] [Google Scholar]
- 6. Brockmann K, Reimold M, Globas C, et al. PET and MRI reveal early evidence of neurodegeneration in spinocerebellar ataxia type 17. J Nucl Med 2012;53:1074–1080. [DOI] [PubMed] [Google Scholar]
- 7. Yen TC, Lu CS, Tzen KY, et al. Decreased dopamine transporter binding in Machado‐Joseph disease. J Nucl Med 2000;41:994–998. [PubMed] [Google Scholar]
- 8. Lu CS, Wu Chou YH, Yen TC, Tsai CH, Chen RS, Chang HC. Dopa‐responsive parkinsonism phenotype of spinocerebellar ataxia type 2. Mov Disord. 2002;17:1046–1051. [DOI] [PubMed] [Google Scholar]
- 9. Ferrara JM, Adam OR, Ondo WG. Levodopa‐induced dyskinesias in spinocerebellar ataxia type 2. Arch Neurol 2010;67:114–115. [DOI] [PubMed] [Google Scholar]
- 10. van Gaalen J, Giunti P, van de Warrenburg BP. Movement disorders in spinocerebellar ataxias. Mov Disord 2011;26:792–800. [DOI] [PubMed] [Google Scholar]
- 11. Rossi M, Perez‐Lloret S, Doldan L, et al. Autosomal dominant cerebellar ataxias: a systematic review of clinical features. Eur J Neurol 2014;21:607–615. [DOI] [PubMed] [Google Scholar]
- 12. Bonnet C, Apartis E, Anheim M, et al. Tremor‐spectrum in spinocerebellar ataxia type 3. J Neurol 2012;259:2460–2470. [DOI] [PubMed] [Google Scholar]
- 13. Lu CS, Wu Chou YH, Kuo PC, Chang HC, Weng YH. The parkinsonian phenotype of spinocerebellar ataxia type 2. Arch Neurol 2004;61:35–38. [DOI] [PubMed] [Google Scholar]
- 14. Gwinn‐Hardy K, Chen JY, Liu HC, et al. Spinocerebellar ataxia type 2 with parkinsonism in ethnic Chinese. Neurology 2000;55:800–805. [DOI] [PubMed] [Google Scholar]
- 15. Khan NL, Giunti P, Sweeney MG, et al. Parkinsonism and nigrostriatal dysfunction are associated with spinocerebellar ataxia type 6 (SCA6). Mov Disord 2005;20:1115–1119. [DOI] [PubMed] [Google Scholar]
- 16. Kim JM, Hong S, Kim GP, et al. Importance of low‐range CAG expansion and CAA interruption in SCA2 Parkinsonism. Arch Neurol 2007;64:1510–1518. [DOI] [PubMed] [Google Scholar]
- 17. Shan DE, Liu RS, Sun CM, Lee SJ, Liao KK, Soong BW. Presence of spinocerebellar ataxia type 2 gene mutation in a patient with apparently sporadic Parkinson's disease: clinical implications. Mov Disord 2004;19:1357–1360. [DOI] [PubMed] [Google Scholar]
- 18. Yun JY, Kim JM, Kim HJ, Kim YE, Jeon BS. SCA6 presenting with young‐onset parkinsonism without ataxia. Mov Disord 2012;27:1067–1068. [DOI] [PubMed] [Google Scholar]
- 19. Marshall V, Grosset D. Role of dopamine transporter imaging in routine clinical practice. Mov Disord 2003;18:1415–1423. [DOI] [PubMed] [Google Scholar]
- 20. Siebert M, Donis KC, Socal M, et al. Glucocerebrosidase gene variants in parkinsonian patients with Machado Joseph/spinocerebellar ataxia 3. Parkinsonism Relat Disord 2012;18:185–190. [DOI] [PubMed] [Google Scholar]
- 21. Pedroso JL, Bor‐Seng‐Shu E, Felicio AC, Braga‐Neto P, Teixeira MJ, Barsottini OG. Transcranial sonography findings in spinocerebellar ataxia type 3 (Machado‐Joseph disease): a cross‐sectional study. Neurosci Lett 2011;504:98–101. [DOI] [PubMed] [Google Scholar]
- 22. Boesch SM, Muller J, Wenning GK, Poewe W. Cervical dystonia in spinocerebellar ataxia type 2: clinical and polymyographic findings. J Neurol Neurosurg Psychiatry 2007;78:520–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kitahara M, Shimohata T, Tokunaga J, Nishizawa M. Cervical dystonia associated with spinocerebellar ataxia type 2 successfully treated with levodopa: a case report. Mov Disord 2009;24:2163–2164. [DOI] [PubMed] [Google Scholar]
- 24. Nandagopal R, Moorthy SG. Dramatic levodopa responsiveness of dystonia in a sporadic case of spinocerebellar ataxia type 3. Postgrad Med J 2004;80:363–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wilder‐Smith E, Tan EK, Law HY, Zhao Y, Ng I, Wong MC. Spinocerebellar ataxia type 3 presenting as an L‐DOPA responsive dystonia phenotype in a Chinese family. J Neurol Sci 2003;213:25–28. [DOI] [PubMed] [Google Scholar]
- 26. Cardoso F, de Oliveira JT, Puccioni‐Sohler M, Fernandes AR, de Mattos JP, Lopes‐Cendes I. Eyelid dystonia in Machado‐Joseph disease. Mov Disord 2000;15:1028–1030. [DOI] [PubMed] [Google Scholar]
- 27. Zarubova K, Ruzicka E. Cervical dystonia in spinocerebellar ataxia type 2. Mov Disord 2006;21:1295–1296. [DOI] [PubMed] [Google Scholar]
- 28. Bauer P, Laccone F, Rolfs A, et al. Trinucleotide repeat expansion in SCA17/TBP in white patients with Huntington's disease‐like phenotype. J Med Genet 2004;41:230–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. De Rosa A, Striano P, Barbieri F, et al. Suppression of myoclonus in SCA2 by piracetam. Mov Disord 2006;21:116–118. [DOI] [PubMed] [Google Scholar]
- 30. Schelhaas HJ, Ippel PF, Beemer FA, Hageman G. Similarities and differences in the phenotype, genotype and pathogenesis of different spinocerebellar ataxias. Eur J Neurol 2000;7:309–314. [DOI] [PubMed] [Google Scholar]
- 31. Lavigne GJ, Montplaisir JY. Restless legs syndrome and sleep bruxism: prevalence and association among Canadians. Sleep 1994;17:739–743. [PubMed] [Google Scholar]
- 32. Abele M, Burk K, Laccone F, Dichgans J, Klockgether T. Restless legs syndrome in spinocerebellar ataxia types 1, 2, and 3. J Neurol 2001;248:311–314. [DOI] [PubMed] [Google Scholar]
- 33. Schols L, Haan J, Riess O, Amoiridis G, Przuntek H. Sleep disturbance in spinocerebellar ataxias: is the SCA3 mutation a cause of restless legs syndrome? Neurology 1998;51:1603–1607. [DOI] [PubMed] [Google Scholar]
- 34. Berciano J, Infante J, Garcia A, et al. Stiff man‐like syndrome and generalized myokymia in spinocerebellar ataxia type 3. Mov Disord 2006;21:1031–1035. [DOI] [PubMed] [Google Scholar]
- 35. Pedroso JL, Carvalho AA, Escorcio Bezerra ML, et al. Unusual movement disorders in spinocerebellar ataxias. Parkinsonism Relat Disord 2013;19:834–835. [DOI] [PubMed] [Google Scholar]
- 36. Shimojima K, Okumura A, Natsume J, et al. Spinocerebellar ataxias type 27 derived from a disruption of the fibroblast growth factor 14 gene with mimicking phenotype of paroxysmal non‐kinesigenic dyskinesia. Brain Dev 2012;34:230–233. [DOI] [PubMed] [Google Scholar]
- 37. Pedroso JL, Braga‐Neto P, Felicio AC, Barsottini OG, Jardim LB, Saraiva‐Pereira ML. Akathisia: an unusual movement disorder in Machado‐Joseph disease. Parkinsonism Relat Disord 2011;17:712–713. [DOI] [PubMed] [Google Scholar]
- 38. Bruno MK, Lee HY, Auburger GW, et al. Genotype‐phenotype correlation of paroxysmal nonkinesigenic dyskinesia. Neurology 2007;68:1782–1789. [DOI] [PubMed] [Google Scholar]