

Abstract
Summary
Liquid chromatography mass spectrometry (LC-MS) is the favored method for untargeted metabolomic analysis of small molecules in biofluids. Here we present SimExTargId, an open-source R package for autonomous analysis of metabolomic data and real-time observation of experimental runs. This simultaneous, fully automated and multi-threaded (optional) package is a wrapper for vendor-independent format conversion (ProteoWizard), xcms- and CAMERA- based peak-picking, MetMSLine-based pre-processing and covariate-based statistical analysis. Users are notified of detrimental instrument drift or errors by email. Also included are two shiny applications, targetId for real-time MS2 target identification, and peakMonitor to monitor targeted metabolites.
Availability and implementation
SimExTargId is publicly available under GNU LGPL v3.0 license at https://github.com/JosieLHayes/simExTargId, which includes a vignette with example data. SimExTargId should be installed on a dedicated data-processing workstation or server that is networked to the LC-MS platform to facilitate MS1 profiling of metabolomic data.
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
Collection of untargeted metabolomic data, based on liquid chromatography-mass spectrometry (LC-MS) MS1-profiling, is subject to many potential pitfalls. For example, unobserved instrument failure can occur when an investigator is not present during experimental runs. Timely intervention after such a failure is crucial when processing precious samples. Minor leaks or partial blockages in LC systems can lead to retention time drift and loss of chromatographic resolution and column/ion-source degradation and mass-analyzer drift can lead to signal attenuation.
Here we present SimExTargId, an open source R package designed to approach autonomous and real-time analysis of metabolomic data. MetShot, a currently available R package, provides a framework to achieve nearly-online acquisition of spectra for features of statistical relevance (Neumann et al., 2013). In contrast to MetShot, SimExTargId provides an autonomous workflow that can also perform data preprocessing in real-time, thereby alerting the user to signal degradation or loss.
2 Worklow
SimExTargId is a wrapper function for peak peaking and normalization that exploits existing tools. This open-source software facilitates real-time monitoring of LC-MS data acquisition and processing via the Windows operating system. An overview of the SimExTargId autonomous workflow is shown in Figure 1, which addresses each of the following steps in the pipeline.
Fig. 1.
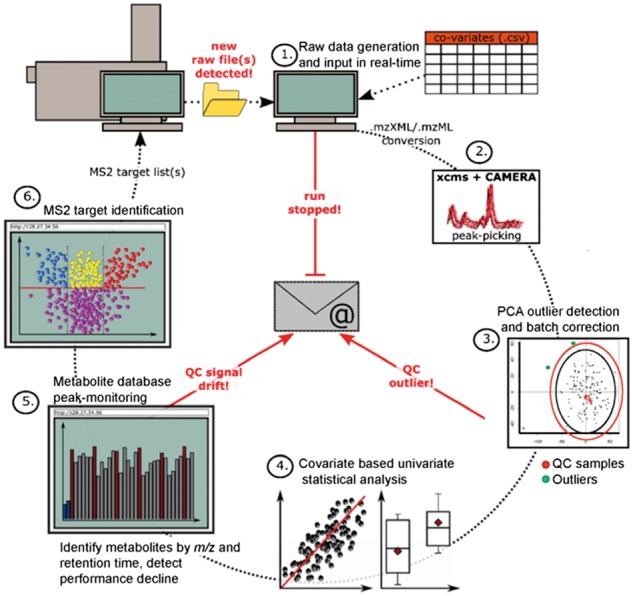
Overview of the autonomous simExTargId workflow and monitoring system. Each step is addressed in detail in the Workflow section
2.1 Initiation, raw file detection and conversion
Raw data from the MS are continuously monitored with a waiting-time counter, which determines whether the last data file exceeds a predetermined maximum time and then alerts the user by email. File sizes are also monitored, and an alert is sent if a file exceeds three absolute deviations from the total median file size, after a minimum of five files have been generated.
New raw data files are automatically converted to the mzXML open-file format using Proteowizard MSConvert (Chambers et al., 2012). A user-supplied worklist and table of covariates provides information for grouping xcms sub-directories, adjusting pooled-QC signals, filtering by coefficients of variation (CVs), and performing statistical analyses. This text file contains instructions for column conditioning, MS2 data collection and inclusion of negative controls & pooled-QC replicates.
2.2 xcms peak-picking and CAMERA MS1 deconvolution
Peak-picking (xcmsSet, Benton et al., 2010; Smith et al., 2006; Tautenhahn et al., 2008) is performed after a specified minimum number of samples has been collected, followed by retention-time correction and peak-grouping & filling. The CAMERA package is used to annotate isotopes, ESI adducts & in-source fragments and to identify pseudospectra (Kuhl et al., 2012).
2.3 MetMSLine preprocessing, PCA outlier detection and batch correction
Our previous R package MetMSLine is utilized for all pre-processing steps via the wrapper function preProc (Edmands et al., 2015). A principal components analysis (PCA) is performed and potential analytical outliers are detected (pcaOutId), with alerts if these are QC samples. PCA-based detection of batch effects is then performed (pcaClustId) by partitioning around medoids (PAM), clustering and regression of all covariates to any clusters detected. Batch effects are automatically adjusted with a linear model (batchAdj) and statistical analyses are performed on both batch-adjusted and unadjusted peak tables.
2.4 Covariate based automatic statistical analysis
Following pre-processing and outlier removal, all covariates are used to automatically select an appropriate univariate method for statistical analysis (coVarTypeStat). This function attempts to distinguish between continuous & categorical variables and then applies a suite of parametric or non-parametric statistical methods, including Wilcoxon-rank sums, Spearman correlation and ANOVA. Statistical analyses are performed on up to four peak tables (i.e. batch-adjusted & unadjusted tables and batch-adjusted and unadjusted weighted-mean mean tables).
2.5 Metabolite database peak-monitoring
peakMonitor can be used to monitor a list of previously known metabolites supplied as a .csv file. The function identifies peak groups (metabolites) in the xcms database file by m/z and retention time within user-defined parameters for mass accuracy and retention time deviation. Plots of median m/z & retention times, peak areas and PCAs are viewed using a shiny application (http://shiny.rstudio.com, Supplementary Fig. S1). The user is alerted if a user-defined percentage of attenuation for the monitored peak areas is observed.
2.6 MS2 target identification
targetId is a visualization tool for the statistical output from step 4 (Supplementary Fig. S2). This shiny application provides a volcano plot of both the raw P-values and multiple-testing adjusted P-values versus fold changes for all covariates. Peak areas are used to suggest the most suitable samples for obtaining particular MS2 spectra.
3 Conclusions and limitations
The SimExTargId R package is the first open-source package that provides comprehensive real-time automation and a standardized workflow for metabolomic profiling of MS1 data. The SimExTargId package has been tested primarily with data files from Agilent Q-TOF and Thermo FT-ICR mass spectrometers operating within a Windows environment, but can be readily extended to other platforms.
Supplementary Material
Acknowledgements
We gratefully acknowledge the assistance of Agilent Technologies (Santa Clara, CA, USA) for the loans of liquid-chromatography mass-spectrometry instruments used in the development of this package.
Funding
This research was supported by the National Institute of Environmental Health Sciences under grants P50ES018172, P01ES018172 and P42ES004705.
Conflict of Interest: none declared.
References
- Benton H.P. et al. (2010) Correction of mass calibration gaps in liquid chromatography-mass spectrometry metabolomics data. Bioinformatics, 26, 2488.. [DOI] [PubMed] [Google Scholar]
- Chambers M.C. et al. (2012) A cross-platform toolkit for mass spectrometry and proteomics. Nat. Biotechnol., 30, 918–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edmands W.M.B. et al. (2015) MetMSLine: an automated and fully integrated pipeline for rapid processing of high-resolution LC-MS metabolomic datasets. Bioinformatics, 31, 788–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhl C. et al. (2012) CAMERA: an integrated strategy for compound spectra extraction and annotation of liquid chromatography/mass spectrometry data sets. Anal. Chem., 84, 283–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann S. et al. (2013) Nearline acquisition and processing of liquid chromatography-tandem mass spectrometry data. Metabolomics, 9, 84–91. [Google Scholar]
- Smith C.A. et al. (2006) XCMS: processing mass spectrometry data for metabolite profiling using nonlinear peak alignment, matching and identification. Anal. Chem., 78, 779–787. [DOI] [PubMed] [Google Scholar]
- Tautenhahn R. et al. (2008) Highly sensitive feature detection for high resolution lc/ms. BMC Bioinformatics, 9, 504.. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.