

Abstract
The application of metabolic phenotyping to epidemiological studies involving thousands of biofluid samples presents a challenge for the selection of analytical platforms that meet the requirements of high-throughput precision analysis and cost-effectiveness. Here direct infusion–nanoelectrospray (DI–nESI) was compared with an ultra-performance liquid chromatography (UPLC)–high-resolution mass spectrometry (HRMS) method for metabolic profiling of an exemplary set of 132 human urine samples from a large epidemiological cohort. Both methods were developed and optimized to allow the simultaneous collection of high-resolution urinary metabolic profiles and quantitative data for a selected panel of 35 metabolites. The total run time for measuring the sample set in both polarities by UPLC–HRMS was 5 days compared with 9 h by DI–nESI–HRMS. To compare the classification ability of the two MS methods, we performed exploratory analysis of the full-scan HRMS profiles to detect sex-related differences in biochemical composition. Although metabolite identification is less specific in DI–nESI–HRMS, the significant features responsible for discrimination between sexes were mostly the same in both MS-based platforms. Using the quantitative data, we showed that 10 metabolites have strong correlation (Pearson’s r > 0.9 and Passing–Bablok regression slope of 0.8–1.3) and good agreement assessed by Bland–Altman plots between UPLC–HRMS and DI–nESI–HRMS and thus can be measured using a cheaper and less sample- and time-consuming method. A further twenty metabolites showed acceptable correlation between the two methods with only five metabolites showing weak correlation (Pearson’s r < 0.4) and poor agreement due to the overestimation of the results by DI–nESI–HRMS.
Keywords: ultra-performance liquid chromatography, direct infusion mass spectrometry, metabolic profiling, exploratory analysis, quantitative analysis, high-throughput analysis
1. Introduction
Metabolic profiling methods are now widely applied in molecular epidemiology studies and other large-scale phenotyping investigations of human biofluid and tissue collections, such as Biobanks, involving tens of thousands or more samples. The scale of analysis required is a strong driver for both the optimization of pre-existing standard analytical methods and the development of new techniques, with the aim of improving the measurement precision and the coverage of chemical species profiled but with high-throughput and economic feasibility in mind.1−5 The measurement of low-molecular-weight metabolites conducted on the population scale provides sufficient statistical power for the investigation of molecular mechanisms responsible for phenotypic variation in human populations and affords the potential for metabolome-wide association studies (MWAS) to elucidate metabolic signatures of disease and disease risk factors.6
Analytical methods for large-scale phenotyping are selected on the basis of the need to strike a balance between metabolite coverage, precision, and selectivity of the analysis with throughput and cost of analysis as well as the number of analytes and sample amount required. Various analytical platforms are typically used to carry out metabolic phenotyping, most notably, liquid and solid 1H NMR spectroscopy,2,7,8 LC–MS,9,1,10 and GC–MS (MS/MS).111H NMR spectroscopy is a robust and reliable technique that provides highly reproducible data and can achieve quantification of detectable metabolites in 4 to 5 min data acquisition time per sample.2 Under standard conditions the technique typically measures metabolite concentrations in the micromolar to millimolar concentration range, but this includes hundreds of biologically relevant compounds. Hyphenated ultraperformance liquid chromatography–mass spectrometry (UPLC–MS) has become a widely used tool in metabolic profiling because it offers an attractive combination of high sensitivity and selectivity of analysis, along with the possibility of structural elucidation for metabolite identification and quantification at low micromolar to nanomolar levels of concentration.1,9,12 The use of high-resolution mass spectrometry (HRMS) interfaced with LC can provide simultaneous global metabolic profiling in full-scan mode and quantitative analysis of a variety of analytes.13−19
Although UPLC–MS platforms, having an orthogonal dimension of chromatographic separation, provide more specific and comprehensive data, direct infusion mass spectrometry (DIMS) was shown to be useful for rapid diagnosis of biological samples.5,20−23 We have recently demonstrated the high-throughput capabilities of a direct infusion nanoelectrospray high-resolution mass spectrometry (DI–nESI–HRMS) method applied to urinary metabolic phenotyping in large-scale epidemiological studies.5,24 The method was developed using the TriVersa Advion NanoMate system in infusion mode that provides nESI of the sample coupled to HRMS QTOF Synapt G2-S (Waters, Manchester, U.K.) for direct measurement of m/z. The ratio of the intensities of each analyte and its corresponding isotopically labeled internal standard, added to all samples, was measured, and the quantification was performed using the slope of the calibration curve obtained for each metabolite by the method of standard additions in the pooled urine sample to account for any matrix effect. In addition to generating quantitative information for selected metabolites, the use of high-resolution full-scan data allowed further data mining in an untargeted manner and showed the potential for the discovery of metabolites responsible for the differences in metabolic phenotypes of two populations from the INTERMAP study.5
Studies comparing LC–MS and direct infusion mass spectrometry (DIMS) approaches for targeted MS/MS assays in proteomics and drug analysis,25,26 as well as in untargeted and targeted analysis of plant metabolites,27 metabonomic,28 and lipidomic29 studies are scarce. DIMS and LC–MS data from human serum samples have been reported, comparing kidney cancer patients and healthy individuals.28 This study showed that the DIMS and LC–MS methods offered comparable discrimination capabilities and that DIMS analysis was significantly faster. Quantification data for specific plant metabolites obtained by targeted direct infusion tandem mass spectrometry method (DIMSMS) using a linear ion trap mass spectrometer were compared to the results from targeted LC–MS/MS analysis, and it was shown that quantification by DIMSMS was reasonably accurate.27 In lipidomic analysis of biological samples, DIMS provided the most comprehensive lipid coverage due to the identification of some polar lipid classes not identified by the UPLC–MS method.29 Liquid chromatography–multiple reaction monitoring (LC–MRM) and direct infusion–multiple reaction monitoring (DI–MRM) assays have also been compared for quantification of heat shock proteins in cell lines,25 showing that the increased throughput of DI–MRM is useful for rapid parallel analysis of large sample batches. Other studies have combined DIMS and 1H NMR spectroscopic analysis of tissue extract samples30 and metabolic profiling of human neuroblastoma cells treated with neurotoxins.31
In the present work, we performed a practical comparison of DI–nESI–HRMS and reversed-phase UPLC–HRMS methods for metabolic profiling of an exemplar set of 132 samples obtained from the epidemiological cross-sectional study INTERMAP (INTERnational collaborative study of MAcronutrients, micronutrients and blood Pressure) investigating the relation between diet and blood pressure among 4680 men and women aged 40–59 in Japan, Peoples Republic of China, United Kingdom, and the United States.32 Both methods were optimized to enable the combined exploratory and targeted metabolic profiling. As an indirect way to assess the coverage provided by the two MS-based platforms and 1H NMR spectroscopy as well as their comparative usefulness for a typical untargeted metabolic profiling, we performed a multivariate analysis of the full-scan urinary profiles obtained and compared the predictive capability and discriminative features selected using sex as the basis for classification because we know a priori that there are differences attributable to sex in the urinary metabolic profiles. To assess the quantification capabilities of the two HRMS methods, we used the panel of metabolites (Table S-1, Supporting Information (SI)) with varying biological functions and related to blood pressure as in our previous publication using the DI–nESI–HRMS method for the analysis of urinary samples from the INTERMAP study.5 The quantitative results, sensitivity, and dynamic ranges for different metabolites as well as sample preparation, run time, and number of analytes amenable to quantification were assessed and compared between the two HRMS methods. For metabolites present at in microlar range of concentration, we validated results from the MS-based platforms using 1H NMR spectroscopy, which outperforms MS techniques in terms of linear dynamic range and analytical reproducibility.
To the best of our knowledge, this is the first comparative evaluation of DI–nESI–HRMS and UPLC–HRMS applied for simultaneous combined exploratory human urinary metabolic profiling and quantitative targeted analysis.
2. Experimental Section
2.1. Chemicals and Preparation of Standard Solutions
A description of the solvents and reference standards used for the MS-based analyses (Table S-1) as well as chemicals and materials used for the NMR measurements is provided in the SI. Buffer for the 1H NMR analysis of the urine specimens was prepared as previously reported.2
The labeled and nonlabeled standard stock solutions of chemicals were prepared at concentration of 1 mg/mL in methanol or methanol/water mixture. The multianalyte mixture of labeled internal standards and the multianalyte mixture of nonlabeled calibration standards were prepared at different levels of concentration (μg/mL) by mixing stock solutions in a total volume of 10 mL. The stock solutions were stored at 4 °C in tightly closed containers for the length of the analysis. The details of the preparation of different calibrator solutions are provided in the SI and Table S-2.
2.2. Sample Preparation
The urine samples were aliquots taken from the 24 h urine specimens collected in the INTERMAP study as a part of an epidemiological study investigating hypertension. Boric acid had been added as a preservative to the urine samples upon collection. The description of the collection of urinary specimens along with clinical and dietary data is provided elsewhere.32 Institutional ethics committee approval was obtained for each site, and all participants gave written informed consent. For this study, we used a test set of 132 randomly selected aliquots from the United States population, which included 22 pairs of blinded duplicate samples to assess the reproducibility of quantification.
The dilution factor used for the urine samples was optimized for UPLC–MS and DI–nESI–HRMS assays by assessing the effect of dilution on the intensities of endogenous metabolites and the added internal standards. All dilution factors have been taken into account for the quantification of metabolites and calculations of the limits of quantification (LOQ).
For DI–nESI–HRMS, 10 μL of thawed urine sample were pipetted in randomized order into deep-well plates and diluted 50-fold with ultrapure water. Aliquots of 50 μL of the diluted samples were transferred to the well plates, adding 25 μL of the multianalyte mixture of labeled internal standards prepared in methanol and making the total volume 150 μL by adding methanol (water–methanol final proportion 1:2). Aliquots of 10 μL of each thawed urine sample were mixed to obtain a pooled urine sample, which was used for the preparation of study reference (SR) sample, validation QC samples, and calibration series. The pooled urine samples were also diluted 50-fold.
For UPLC–HRMS, low- and high-concentration metabolites required different dilutions to accommodate the dynamic ranges of different urinary metabolites. The assay had to be performed twice with 20- and 3-fold diluted urine samples. To prepare two different dilutions, 20 or 50 μL of thawed urine samples were pipetted in randomized order into deep-well plates and diluted with water. Thereafter, aliquots of 50 μL of the diluted samples were transferred to the well plates, adding 25 μL of the multianalyte mixture of labeled internal standards prepared in methanol to each well. Aliquots of 10 μL of each thawed urine sample were mixed to obtain pooled urine samples used for the preparation of study reference (SR) sample and validation QC samples diluted by the corresponding factor.
For both DI–nESI–HRMS and UPLC–HRMS, the pooled SR samples were prepared identically to the study samples and measured throughout the analytical run to monitor system stability.
The protocol for 1H NMR spectroscopic sample preparation was described by Dona et al.2 In brief, 600 μL of thawed urine sample was transferred to an Eppendorf tube and centrifuged at 12 000g for 5 min at 4 °C. Following centrifugation, 540 μL was taken from the Eppendorf tube and transferred to a 5 mm SampleJet NMR tube, to which 60 μL of NMR sample buffer was also added (600 μL of volume per NMR tube) and mixed thoroughly.
Calibration series and validation QC samples for DI–nESI–HRMS were prepared by pipetting 50 μL of diluted pooled urine samples in a well plate, adding 25 μL of the multianalyte mixture of labeled internal standards at a fixed concentration and 25 μL of the corresponding calibrator solution (both prepared in methanol) and increasing the total volume to 150 μL by adding methanol. For UPLC–HRMS, the calibration series and validation QC samples were prepared by adding 25 μL of the multianalyte mixture of labeled internal standards at fixed concentration and 25 μL of the corresponding calibrator solution to 100 μL of ultrapure water. The sample plates were sealed and subjected to 1 min of ultrasonication, followed by 10 min of centrifugation at 1500g at 4 °C before MS analysis.
2.3. Chip-Based Nanoelectrospray MS (DI–nESI–HRMS) Analysis of Urine Samples
Chip-based nanoelectrospray infusion analysis was performed using the TriVersa NanoMate system (Advion BioSciences, Ithaca, NY) coupled to a Waters Synapt G2-Si (Waters MS Technologies, U.K.) operating in high-resolution continuum mode in negative and positive ion modes with automatic polarity switching. The MS experimental conditions are detailed in the SI.
2.4. Ultraperformance Liquid Chromatography–High-Resolution Mass Spectrometry (UPLC–HRMS) Analysis of Urine Samples
Chromatography was performed on a Waters Acquity UPLC system, equipped with a binary solvent manager, sample manager and column heater (Waters, Milford, MA). The chromatographic separations were performed on an HSS T3 (1.8 μm, 2.1 × 100 mm) column (Waters, Milford, MA) maintained at 40 °C. The mobile phase consisted of a linear gradient elution, previously described,9 and was performed at 0.4 mL/min using 0.1% formic acid in water and in acetonitrile. Mass spectrometry was performed on a Waters Synapt G2-S mass instrument (Waters MS Technologies, U.K.) equipped with an electrospray ion (ESI) source operated in high-resolution mode in positive (ESI+) and negative (ESI−) polarity. All mass spectral data were collected in centroid mode using the MSe mode of operation.33 The LC and MS experimental conditions are detailed in the SI.
2.5. 1H NMR Spectroscopic Analysis of Urine Samples
The analysis of urine biofluid samples by NMR spectroscopy was performed on a Bruker 600 MHz Advance III spectrometer, equipped with a BBI 600 MHz 5 mm Z gradient probe and automated tuning and matching unit, and a SampleJet autosampler, which retains the samples at 6 °C before spectral acquisition. For each urine sample, a standard 1D experiment using a pulse sequence with presaturation of the water resonance and a 2D J-resolved experiment were performed. All spectrometer tuning and configuration protocols and standard operating procedures, as well as the pulse sequence details, have been described by Dona et al.2
2.6. Quantification of Metabolites
The list of urinary metabolites quantified by the two HRMS methods is presented in Table S-1. The calibration curves in both methods were plotted as the intensity of metabolite divided by the intensity of its labeled internal standard against the added concentration of the calibration standard solution (μg/mL). In DI–nESI–HRMS, the standard addition method in pooled urine samples was used for quantification using the slope of the calibration curve for each metabolite, as previously described.5 In UPLC–HRMS, the study samples were quantified using a calibration curve obtained in water, as described above. The 1H NMR quantitative measurements on a total of 24 metabolites (SI) were provided by Bruker BioSpin (Section 2.5).
2.7. Validation of the UPLC–HRMS Method
Linear ranges and inter- and intra-day accuracy and precision were determined for the UPLC–HRMS method following the FDA guidelines for the validation of bioanalytical methods.34
The validation study was performed over separate days. On the first day, the well plate contained the calibration series consisting of six points plus zero point (spiked with the mixture of labeled internal standards only) and six replicates of validation QC samples prepared at low, medium, and high concentration levels for each metabolite (Table S-3),34 which were defined for each metabolite from the corresponding linear range (Table S-2). Sample blanks (water) were injected after the calibration series and each QC series to ascertain if there was carryover. On the second and third days, each plate contained a calibration series and one replicate of the QC series. The intra- and interday accuracy and precision were calculated as relative error (RE%) and coefficient of variance (CV%), respectively.
accuracy = mean (measured concentration)/theoretical concentration × 100
precision = mean SD/mean (measured concentration) × 100
The method LOQ was determined as 10 times the standard deviation of the y intercept divided by the slope of the calibration curves obtained in the analyte-free matrix.
2.8. Data Preprocessing and Analysis
MS data acquisition, subsequent visualization, and quality control were achieved using MassLynx 4.1 software (Waters, U.K.).
DI–nESI–HRMS raw data were converted to the mzML format with the ProteoWizard MSConvert software,35 followed by processing using in-house Python 2.7.4 scripts, as previously described.5 In brief, the processing included the sum of the scans acquired, interpolation of the data to a final resolution of 0.0028 m/z, local baseline estimation, and peak picking. Because the lock-mass function was turned off, the data were recalibrated post-acquisition by in-house scripts using the masses of the reference signals of labeled standards and endogenous metabolites present in all urine samples. After correction, the mass accuracy errors measured in the final data set were <10 ppm. For the untargeted analysis, the full-scan HRMS spectra were binned to 0.05 ppm, preprocessed, and normalized in MATLAB R2014a (Mathworks). Preprocessing consisted of removal of background signals by subtracting the spectrum of a blank sample from each sample profile and removal of the m/z features showing high variation in the pooled SR samples. After filtering, these data were normalized by probabilistic quotient normalization (PQN)36 to a median reference spectrum. Pareto and UV scaling were applied prior to multivariate analysis.
UPLC–HRMS profiling data (dilution 1/3) were preprocessed utilizing XCMS37 operating in an R statistical software environment.38 Centroid raw data files were converted to NetCDF open format using the DataBridge executable function (MassLynx software, Waters). Within XCMS, the centWave algorithm was used for peak detection, extraction, and integration using the following parameters: ppm = 25, prefilter = (8, 3000), peakwidth = (2, 8), snthresh = 10, mzCenterFun = “wMean”, integrate = 2, followed by peak grouping (using “density” method, bw = 1, mzwid = 0.01) and PQN normalization. Pareto and UV scaling were applied prior to multivariate analysis. The integration of chromatographic peaks for targeted analysis was performed using TargetLynx software (Waters, U.K.).
For untargeted analysis, the 1H NMR spectra were digitized (after the removal of water, urea, and TSP regions) to a resolution of 0.001 ppm, aligned, normalized by PQN, and mean-centered.
For both HRMS methods, calibration curves plotting, quantification, and corresponding calculations were performed in Microsoft Excel. Pearson’s correlation was used to calculate the association between the results of urinary metabolite quantification (μg/mL) obtained by the three techniques. DI–nESI–HRMS and UPLC–HRMS method comparison was performed by Passing–Bablok regression39 and Bland–Altman plots40 in R statistical software.38
Principal component analysis (PCA) and orthogonal projection to latent structures–discriminant analysis (OPLS-DA)41 were performed in SIMCA 14 (Umetrics, Sweden) to investigate the ability to identify sex-related metabolic differences in full-scan urine profiles obtained by the three techniques.
Putative metabolite annotation on the untargeted spectral profiles was performed using in-house and public databases (HMDB,42 Metlin,43 BioMagResBank44). For UPLC–HRMS feature annotation, accurate mass, isotopic pattern, retention time, and MS/MS data (obtained from the MSe mode high-collision energy function) were matched to the database information. The NMR J-Resolved data were used to verify peak multiplicities and J-coupling constants to confirm metabolite annotation in the 1H NMR analysis.
3. Results and Discussion
In this work, we compared two MS-based metabolic profiling platforms and assessed their capabilities for performing parallel exploratory metabolic profiling and targeted quantification of selected metabolites in urine samples.
3.1. Comparison of UPLC–HRMS and DI–nESI–HRMS
The global profiles of the pooled urine obtained by UPLC–HRMS (diluted 1/3) and DI–nESI–HRMS (diluted 1/50) in positive ionization mode are illustrated in Figure S-1. The total number of the UPLC–HRMS features (retention time-m/z) extracted by XCMS, as described in the Experimental Section, was 1945 for ESI+ and 3795 for ESI–. In DI–nESI–HRMS, after the interpolation of the full-scan data and binning to 0.05 ppm and before applying further preprocessing, as described above, the total number of features (m/z) was 11200 for both polarities.
To assess the performance of the two methods, for untargeted and quantitative urinary profiling, we used the widely accepted strategy of analyzing different types of QC samples. Pooled study reference urine samples (pooled SR) were periodically injected between the injections of the study samples to monitor system stability and feature variability throughout the runs. Dilution SR samples, prepared by diluting the pooled SR sample to known factors (100, 30, 10, and 5%), were analyzed to enable the selection of biologically relevant features in exploratory profiling.45
The LC–MS system stability can be estimated from the degree of similarity of the chromatograms obtained for pooled SR samples injected after the column conditioning (Figure S-2A). Similarly, the pooled SR samples were monitored throughout the DI–nESI–HRMS run. The overlay of high-resolution mass spectra obtained for five SR samples without any normalization (Figure S-2B–D) shows the assay’s robust performance at different time points throughout the run. For both methods, the variation of mass accuracy was <10 mDa (in ESI+ and ESI−), and the variation of retention time in UPLC–HRMS was <0.03 min throughout the run.
Assessing the CV% of the peak area (UPLC–HRMS) or height (DI–nESI–HRMS) across the pooled SR samples provided an indication of the repeatability as well as the reliability of the features as potential biomarkers. Here we applied the CV% filter approach (with a tolerance of 30%) to examine data acquired for the same sample set on the LC– and DI–nESI platforms. We observed that for the UPLC–HRMS method, 89 and 92% of the total features detected in ESI+ and ESI– modes, respectively, had a CV% < 30%, compared with 94 and 90% features generated by DI–nESI–HRMS. Taking a lower CV% cutoff value of CV% < 15%, 75% (ESI+) and 77% (ESI−) of features detected in UPLC–HRMS and 82% (ESI+) and 63% (ESI−) in DI–nESI–HRMS fell within the tolerance limit.
3.2. Untargeted Analysis by UPLC–HRMS, DI–nESI–HRMS, and 1H NMR Spectroscopy
The full-scan 1H NMR and MS data sets were analyzed by untargeted multivariate approaches to assess the differences and similarities captured by the three different analytical platforms as well as ascertaining the value of the complementary information obtained for the samples analyzed.
In the first instance, the whole data sets containing 132 samples and SR samples were modeled using PCA as an unsupervised approach, which allowed the detection of samples showing anomalous behavior. All three techniques identified the same samples to be outliers. The discriminant metabolites responsible for the outlying behavior in the three data sets were subsequently putatively annotated. There were two main groups of outliers, one due to high urinary glucose concentration and the other due to the presence of acetaminophen metabolites. The samples with high glucose resonance signals in representative 1H NMR spectra (Figure S-3A) were identified as the same samples from the HRMS spectra obtained by DI–nESI–HRMS in ESI+ (Figure S-3B), which show an intense signal at m/z = 203.06 corresponding to the Na+ glucose adduct. Similarly, the UPLC chromatograms for representative samples with high concentration of acetaminophen (m/z = 150.05, rt = 4.45 min) and its metabolites (acetaminophen glucuronide (m/z = 326.09, rt = 2.45 min), acetaminophen sulfate (m/z = 230.01, rt = 2.62 min)) (Figure S-3C) are identified as the same samples from the HRMS spectra obtained by DI–nESI–HRMS (Figure S-3D), showing matching m/z values.
To exemplify the classification capacity of the three techniques, after the first data assessment by PCA and removal of outliers, the remaining samples were subjected to supervised OPLS-DA approach using sex as the basis for discrimination. The cross-validated scores plots obtained for 1H NMR, UPLC–HRMS (ESI−), and DI–nESI–HRMS (ESI−) are shown in Figure 1. All models (Table S-4) found the explained sex-dependent variance values (R2X) to account for 10–40% of the variance in the data set, with the models for UPLC–HRMS data demonstrating a predictive value (Q2Y) of 20–30%. These model statistics are considered as acceptable thresholds for human populations.46 It is worth noting that the higher specificity and sensitivity of UPLC–HRMS metabolic profiling is reflected by better model statistics compared with the statistics of the models obtained for DI–nESI–HRMS data (Table S-4). We putatively annotated some discriminatory metabolites responsible for the separation according to sex in the data obtained by the three techniques (Table S-5). All methods identified creatine and citrate as being present at higher concentration in female urine samples, which is in agreement with previously reported data.47,48 Men were characterized by higher urinary concentrations of creatinine and ethanol, as determined by 1H NMR profiles, which could be related to dietary factors such as meat and alcohol consumption; the levels of creatinine also reflect greater muscle turnover.49 UPLC–HRMS in positive ion mode also indicated higher levels of caffeine metabolites, 1-methylxantine and 1-methylurate, in male urine samples, which is again indicative of sex differences in dietary intake. Both UPLC–HRMS and DI–nESI–HRMS showed higher levels of several acylcarnitines in positive ion mode and steroid metabolites in negative ion mode in urine samples obtained from men, indicating differences in lipid metabolism, which is consistent with previously reported data on sex-specific urine47 and serum metabolome.50 Although some information is lost, or the results are less specific in terms of metabolite identification for DI–nESI–HRMS compared with UPLC–HRMS, most of the significant features responsible for the discrimination of different sample groups (sex) in the exploratory multivariate analysis were detected by both HRMS methods. The improvement of metabolite identification capabilities of the direct infusion methods can be achieved with ultrahigh mass accuracy and high-resolution mass spectrometers such as Orbitrap51 and FT-ICR.23 The masses obtained during the measurement on these instruments are expected to be close to the theoretical mass of metabolites, which facilitates the prediction of ion elemental formulas and provides more confident metabolite identification.
Figure 1.
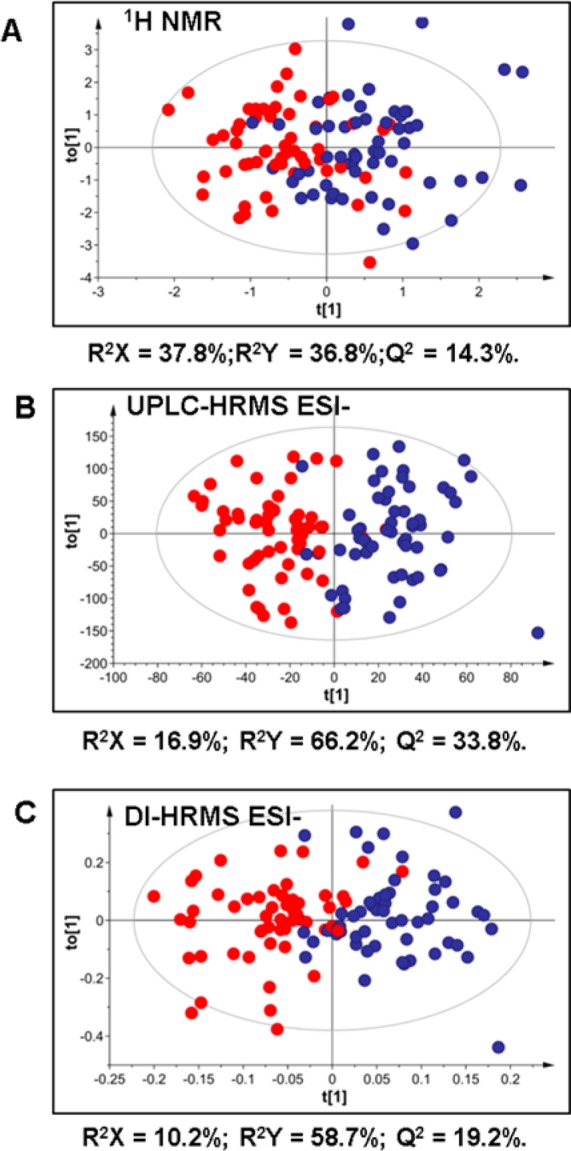
Cross-validated OPLS-DA scores plots showing the separation of profiles of urine samples from men (blue) and women (red) measured by 1H NMR (A), UPLC–HRMS (ESI−) (B), and DI-HRMS (ESI−) (C). UPLC–HRMS and DI-HRMS data were Pareto-scaled.
3.3. Targeted UPLC–HRMS
Modification of a conventional reversed-phase UPLC–MS method for global urinary metabolic profiling9 allowed simultaneous acquisition of both qualitative and quantitative HRMS information. As a model, we took the experimental design used in our DI–nESI–HRMS method described in the Introduction and elsewhere.5,24 Thus stable isotope-labeled standards (Table S-1) were added to each urine sample. Each analytical plate contained a calibration series and validation QC samples in addition to pooled SR samples traditionally used in UPLC–MS experiments to monitor system stability.9
In preliminary experiments aimed to optimize the UPLC–HRMS method for simultaneous quantitative analysis of the metabolites shown in Table 1, we observed that it was not possible to use the standard addition method as in DI–nESI–HRMS.5 In UPLC–HRMS, the additions of calibrator solutions (WS6-WS1) to the pooled urine samples were quickly saturating the detector. We therefore used water as a matrix for the calibration series in UPLC–HRMS and tested the parallel response of each metabolite added to the pooled urine sample and water by comparing the slopes of two calibration curves. In DI–nESI–HRMS, more severe matrix effect and ion suppression inherent of direct infusion MS (due to the fact that all ionizable components of a sample are infused simultaneously and compete for the charge) required using the pooled urine sample as a matrix for calibration and validation of QC samples by applying the method of standard additions for quantification, as previously described.5
Table 1. Correlation (Pearson’s r) between Metabolites Detected and Quantified in 132 Urine Samples by UPLC–HRMS and DI–nESI–HRMS and Passing–Bablok regression parameters.
| Passing–Bablok
regression |
|||
|---|---|---|---|
| metabolite | Pearson correlation r | slope | intercept |
| caffeic acid | 0.58 | 31.8 | 0.2 |
| carnitine | 0.96 | 1.2 | 16.6 |
| cholate | 0.71 | 19.8 | –0.4 |
| citrate | 0.9 | 1.04 | 49.6 |
| cotinineb | 0.97 | 1.6 | 0.2 |
| creatine | 0.98 | 1.2 | 73.2 |
| creatinine | 0.95 | 1.3 | –31.2 |
| glutamate | 0.68 | 0.3 | 18.0 |
| glycocholate | 0.7 | 24.6 | –0.8 |
| hippurate | 0.97 | 0.8 | –23.1 |
| homovanillatea | 0.56 | 22.4 | –17.4 |
| 3-hydroxycinnamatea | 0.39 | 56.3 | 0.1 |
| indoxyl sulfate | 0.98 | 0.6 | –0.5 |
| isovalerylglycine | 0.78 | 3.2 | 0.7 |
| kynureninea | 0.49 | 59.9 | 7.4 |
| leucine | 0.69 | 18.8 | 0.6 |
| N-acetylneuraminate | 0.88 | 1.2 | 1.1 |
| nicotineb | 0.35 | 1.4 | 1.9 |
| phenylacetylglutamine | 0.95 | 0.9 | –12.3 |
| phenylacetatea | 0.55 | 21.6 | –15.0 |
| proline betaine | 0.99 | 1.05 | 3.3 |
| propionylcarnitine | 0.89 | 1.03 | 0.2 |
| saccharin | 0.98 | 1.06 | –0.01 |
| succinate | 0.6 | 6.03 | 4.5 |
| tyraminea | 0.34 | 42.03 | 5.1 |
| vanillilmandelate | 0.58 | 1.97 | 5.0 |
Metabolites showing weak or moderately weak (r < 0.6) correlation between the two MS methods.
Passing–Bablok regression was done between the samples from smokers only.
In UPLC–HRMS, we observed that some high-concentration metabolites, namely, creatinine, creatine, citrate, phenylacetylglutamine, hippurate, carnitine, and proline betaine, required higher dilution of the urine sample for more precise quantification. Hence, for the simultaneous UPLC–HRMS analysis of all metabolites listed in Table S-1, we prepared two sets of samples diluted by two different factors of 3- and 20-fold. In comparison, the dilution factor used for urine samples analyzed by DI–nESI–HRMS was 50, which sufficed for the quantification of all selected metabolites.5 The UPLC–HRMS run time in two ionization modes for every sample dilution (1/3 or 1/20) was 5 days compared with the total analysis time by DI–nESI–HRMS of 9 h. Both targeted UPLC–HRMS and DI–nESI–HRMS are represented in Figure S-4 using the example of hippurate.
We validated the UPLC–HRMS method according to the reported guidelines for bioanalytical methods validation.34,52 Along with the validation QC samples prepared at three concentration levels (high, medium, and low; Table S-3) selected from the linear range for each metabolite (Table S-2), metabolites detected in the replicates of pooled SR urine samples analyzed in each validation plate were quantified. The results of validation as intra- and interday accuracy and precision as well as precision of metabolite quantification in pooled SR urine samples are shown in Table S-6. For UPLC–HRMS validation, we adapted the acceptance criteria for the reliability of quantitative analysis according to the FDA guidelines for biomarker and metabonomic studies, allowing a 30% limit of variability,34,52 as we have previously described for DI–nESI–HRMS.5
The validation results for the UPLC–HRMS show that only 5 out of 34 measured metabolites had errors >20% in the QC samples prepared at low concentration. However, the intra- and interday precision for these 5 metabolites measured in the pooled urine sample is <10%, which indicates that the method can be used for reliable quantification.
During the analysis of study samples in both UPLC–HRMS and DI–nESI–HRMS, the validation QC samples spiked with 35 reference standards at three levels of concentration (Table S-3) were measured periodically. The results of in-study validation expressed as accuracy (RE%) and precision (CV%) of quantification are shown in Table S-7. The set of 132 urine specimens analyzed in this study contained 22 pairs of blinded duplicate samples, which were also used to assess the reproducibility of quantification (CV%) achieved by the two HRMS methods (Table S-7). The comparison of duplicate samples showed an acceptable level of reproducibility for all metabolites measured by DI–nESI–HRMS. For UPLC–HRMS, three metabolites, expected to be present at low levels (caffeic acid, nicotine, and daidzein), were less precise. Note that acetylcarnitine could not be measured by UPLC–HRMS because it is highly polar and elutes early under the reversed-phase conditions, whereas using DI–nESI–HRMS, it can be measured with a high degree of precision (CV% < 20%).
3.4. Correlation and Agreement between UPLC–HRMS and DI–nESI–HRMS
To assess further the similarity of the results of targeted analysis performed by both HRMS methods, we initially calculated the Pearson’s correlation coefficient r for all metabolites that could be measured by both methods in 132 urine samples (Table 1). Table 1 also contains the parameters of the Passing–Bablok regression analysis. The quantification results obtained by MS-based methods for certain metabolites were also correlated to the 1H NMR spectroscopic results. The Pearson’s correlation coefficients r for UPLC–HRMS–1H NMR spectroscopic results were 0.99 for creatine, 0.54 for creatinine, 0.73 for citrate, 0.88 for hippurate, and 0.49 for succinate, whereas for DI–nESI–HRMS–1H NMR spectroscopic results they were 0.97, 0.54, 0.68, 0.84, and 0.24, respectively. Regarding the correlation between the quantification results obtained by UPLC–HRMS and DI–nESI–HRMS, we observed three scenarios: r values below 0.4 (weak correlation), r values higher than 0.4 but lower than 0.8 (moderate correlation), and r values higher than 0.9 (strong correlation).
We observed the agreement of the quantitative results from DI–nESI–HRMS and UPLC–HRMS using Bland–Altman plots and Passing–Bablok regression analysis. The former approach assesses the agreement between two different analytical methods by plotting the concentration differences for each specimen against the average of the two measured concentrations.40 In addition, mean difference and lower and upper limits of agreement are shown as horizontal lines with limits of agreement defined as mean difference ±1.96 times the standard deviation. The difference was calculated by subtracting the concentration values (μg/mL) obtained by UPLC–HRMS from those obtained by DI–nESI–HRMS. Passing–Bablok regression is a nonparametric statistical method used for method comparison studies.39 Passing–Bablok regression and Bland–Altman agreement plots for the three scenarios of different levels of correlation between two MS-based methods are shown in Figure 2. Proline betaine (Figure 2A) is, for instance, highly correlated, the Passing–Bablok regression having slope of 1.05 and Bland–Altman plot showing good agreement between the results obtained by the two MS-methods (mean difference centered at zero). Glutamate illustrates the case of moderate but significant correlation (Figure 2B), with the Passing–Bablok regression slope of 0.3 and mean difference in Bland–Altman plot centered at zero at low concentrations and becoming negative at higher concentrations, which means that the concentration values obtained by UPLC–HRMS were overestimated compared with DI–nESI–HRMS. Because glutamate is a highly polar metabolite, it elutes early under reversed-phase chromatographic conditions and is represented by poorly shaped chromatographic peak, which leads to less accurate results. 3-Hydroxycinnamate shows weak correlation (Figure 2C). The Bland–Altman plot presents the positive and increasing mean difference, and the slope of Passing–Bablok regression is 56.3, both facts pointing out that the concentration is overestimated by DI–nESI–HRMS. This metabolite has the same molecular formula and hence the same accurate mass as phenylpyruvate, which is present in urine, according to the reported data,42 at concentrations that are an order of magnitude higher than the concentration of 3-hydroxycinnamate. Thus the quantitative value in DI–nESI–HRMS is likely to be calculated from the combined sum of phenylpyruvate, 3-hydroxycinnamate, and its isomers. This type of overlap of isobaric species is common to all direct infusion MS methods due to the absence of chromatography, which precludes the separation of isomers and their reliable quantification.
Figure 2.

Passing–Bablok regression with Pearson’s r parameter and agreement (Bland–Altman) plots for selected metabolites measured by UPLC–HRMS and DI–nESI–HRMS showing different levels of correlation: strong correlation (proline betaine); moderate correlation (glutamate) (the results are underestimated in UPLC–HRMS data due to early elution); and weak correlation (3-hydroxycinnamate) due to the overestimation in the DI–nESI–HRMS data.
For two metabolites, nicotine and cotinine, we used reported smoking status of the participants from the metadata to obtain strong correlation and regression parameters (Table 1) only for the samples collected from the smokers (Figure S-5A,B). Using the data for all samples led to unacceptable results of Passing–Bablok regression presumably due to the lower specificity of the peak detection by DI–nESI–HRMS (Figure S-5C,D).
In summary, 5 of the total of 26 metabolites that could be detected and quantified by the two HRMS methods in the set of 132 urine samples showed weak or moderately weak (r < 0.6) correlation and agreement (Table 1). Ten metabolites, namely, carnitine, citrate, creatine, creatinine, hippurate, N-acetylneuraminate, phenylacetylglutamine, proline betaine, propionylcarnitine, and saccharin were strongly correlated and showed good agreement between both HRMS methods. Some polar metabolites (creatine, creatinine, proline betaine, citrate, glutamate, carnitine, acetylcarnitine) were quantified more accurately by DI–nESI–HRMS. These metabolites elute early under the reversed-phase conditions used in this work, giving irregular peak shape due to their weak retention on the column, and their analysis by UPLC–MS would require the use of a different stationary phase such as hydrophylic interaction liquid chromatography (HILIC).
4. Conclusions
We provided a practical comparison of two HRMS methods optimized for the simultaneous acquisition of high-resolution exploratory data and quantitative information for a selected panel of metabolites using the conventional settings for global metabolic profiling on the same mass spectrometer. DI–nESI–HRMS was able to detect a wide range of metabolites, and the information relating to all ionized components in a sample could be readily obtained.
We demonstrated advantages and limitations of both methods, assessing their classification and prediction capability in exploratory untargeted analysis of high-resolution spectra obtained for an exemplar epidemiological data set. We used the difference between genders as a basis for classification knowing a priori that there are differences in the urinary metabolic profiles. This allowed validation of the findings in each platform against a reasonable set of expected and previously validated biomarkers, making for a clearer comparison. For both HRMS methods the same discriminatory metabolites were found, with UPLC–HRMS providing more specific information in terms of metabolite identification.
The quantitative results demonstrated that carnitine, creatine, creatinine, citrate, glutamate, hippurate, N-acetylneuraminate, phenylacetylglutamine, proline betaine, propionylcarnitine, and saccharin showed good correlation and agreement between UPLC–HRMS and DI–nESI–HRMS measurements, and thus can be adequately quantified using a less time-consuming method and with a minimal amount of samples. However, some metabolites had weak correlation and agreement between the two MS methods, revealing the limitation of DI–nESI–HRMS, which lacks specificity for the detection of isobaric and isomeric metabolites in the present settings of MS1 high-resolution acquisition. It is also worth mentioning that despite high-resolution isolation the MS/MS spectra acquired in DIMS may contain the product ions not only from the ion of interest but also from unrelated ions with close masses.
UPLC–MS required variable dilution factors of the original samples for the adequate quantification of metabolites present in high versus low concentration, necessitating longer sample preparation and analysis times. The total run time of 132 samples in two polarities by UPLC–HRMS was on the order of 5 days, whereas the total analysis time using DI–nESI–HRMS in both ionization modes was achieved in 9 h; this illustrates the main advantages accrued from DI–nESI–HRMS, which are analysis time and associated costs. This suggests an important role for this method for ultrarapid screening of large sample sets such as those collected for Biobanks (hundreds of thousands of samples). However, this method is not meant to replace the hyphenated LC–MS techniques that provide higher specificity and, in many instances, higher sensitivity due to the reduced matrix effects and ion suppression. We recommend DI–nESI–HRMS as a fast screening method for large sample batches, whereas we recommend UPLC–HRMS for more comprehensive analysis of selected samples.
Acknowledgments
E.C., G.d.S.C., M.G.-R., and J.K.N. are supported by the National Institute for Health Research (NIHR) Imperial Biomedical Research Centre (BRC). The views expressed in this publication are those of the author(s) and not necessarily those of the NHS, the National Institute for Health Research, or the Department of Health. J.K.N. is director of the MRC-NIHR National Phenome Centre (MC-PC-12025). The INTERMAP Study is supported by grants R01-HL50490, R01-HL84228, and R01-HL135486 from the National Heart, Lung, and Blood Institute, National Institutes of Health (Bethesda, MD) and by national agencies in China, Japan, and the U.K. Q.C. and E.H. are investigators of the MRC-PHE Centre for Environment and Health. P.E. is Director of the MRC-PHE Centre for Environment and Health and acknowledges support from the Medical Research Council and Public Health England (MR/L01341X/1). P.E. acknowledges support from the NIHR Biomedical Research Centre at Imperial College Healthcare NHS Trust and Imperial College London and the NIHR Health Protection Research Unit in Health Impact of Environmental Hazards (HPRU-2012-10141).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jproteome.8b00413.
Experimental Section. 1. Chemicals and preparation of standard solutions; 2. Chip-based nanoelectrospray MS system; 3. UPLC–HRMS experimental conditions; 4. Quantification of the samples by 1H NMR spectroscopy. Table S-1. List of the measured metabolites and their labeled standards. Table S-2. Characteristics of the calibration curves plotted in DI–nESI–HRMS and UPLC–HRMS and LOQ values. Table S-3. Target concentrations (μg/mL) of the validation QC samples prepared at three concentration levels (high, medium, and low) and measured by UPLC–HRMS and DI–nESI–HRMS. Table S-4. Statistics of OPLS-DA models obtained for UPLC–HRMS data (UV- and Pareto-scaled) and DI–nESI–HRMS data (UV- and pareto-scaled). Table S-5. Putatively annotated metabolites that were discriminant between genders in the OPLS-DA analysis of the full-scan UPLC–HRMS, DI–nESI–HRMS, and 1H NMR spectral profiles. Table S-6. Validation of the UPLC–HRMS method (intra- and interday accuracy and precision) using validation QC samples spiked with nonlabeled standards and precision of the quantification of the study reference urine samples. Table S-7. In-study validation (accuracy and precision) of the UPLC–HRMS and DI–nESI–HRMS methods performed using the validation QC samples spiked with nonlabeled standards included in each sample plate and precision of the metabolite quantification in the duplicate samples (CV%). Figure S-1. Comparison of UPLC–HRMS global profile (ESI+) and DI–nESI–HRMS full-scan (ESI+) obtained for the pooled SR urine sample. Figure S-2. Overlay of the full-scan UPLC–HRMS profiles and DI–nESI–HRMS spectra at different m/z regions obtained for five pooled SR samples measured throughout the analytical run of one sample plate (96 injections). Figure S-3. Urine samples showing outlying behavior in PCA. Figure S-4. Schematic representation of the implementation of UPLC–HRMS and DI–nESI–HRMS for urinary targeted analysis of hippuric acid and comparison of calibration curves obtained in water in UPLC–HRMS and in pooled urine sample in DI–nESI–HRMS. Figure S-5. Passing–Bablok regression analysis of cotinine and nicotine including the data for reported smokers only and all samples measured by DI–nESI–HRMS and UPLC–HRMS. (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Lewis M. R.; Pearce J. T. M.; Spagou K.; Green M.; Dona A. C.; Yuen A. H. Y.; David M.; Berry D. J.; Chappell K.; Horneffer-van der Sluis V.; Shaw R.; Lovestone S.; Elliott P.; Shockcor J.; Lindon J. C.; Cloarec O.; Takats Z.; Holmes E.; Nicholson J. K. Development and Application of Ultra-Performance Liquid Chromatography-TOF MS for Precision Large Scale Urinary Metabolic Phenotyping. Anal. Chem. 2016, 88 (18), 9004–9013. 10.1021/acs.analchem.6b01481. [DOI] [PubMed] [Google Scholar]
- Dona A. C.; Jimenez B.; Schafer H.; Humpfer E.; Spraul M.; Lewis M. R.; Pearce J. T. M.; Holmes E.; Lindon J. C.; Nicholson J. K. Precision High-Throughput Proton NMR Spectroscopy of Human Urine, Serum, and Plasma for Large-Scale Metabolic Phenotyping. Anal. Chem. 2014, 86 (19), 9887–9894. 10.1021/ac5025039. [DOI] [PubMed] [Google Scholar]
- Zelena E.; Dunn W. B.; Broadhurst D.; Francis-McIntyre S.; Carroll K. M.; Begley P.; O’Hagan S.; Knowles J. D.; Halsall A.; Wilson I. D.; Kell D. B. Development of a Robust and Repeatable UPLC-MS Method for the Long-Term Metabolomic Study of Human Serum. Anal. Chem. 2009, 81 (4), 1357–1364. 10.1021/ac8019366. [DOI] [PubMed] [Google Scholar]
- Begley P.; Francis-McIntyre S.; Dunn W. B.; Broadhurst D. I.; Halsall A.; Tseng A.; Knowles J.; Goodacre R.; Kell D. B. Development and Performance of a Gas Chromatography-Time-of-Flight Mass Spectrometry Analysis for Large-Scale Nontargeted Metabolomic Studies of Human Serum. Anal. Chem. 2009, 81 (16), 7038–7046. 10.1021/ac9011599. [DOI] [PubMed] [Google Scholar]
- Chekmeneva E.; Dos Santos Correia G.; Chan Q.; Wijeyesekera A.; Tin A.; Young J. H.; Elliott P.; Nicholson J. K.; Holmes E. Optimization and Application of Direct Infusion Nanoelectrospray HRMS Method for Large-Scale Urinary Metabolic Phenotyping in Molecular Epidemiology. J. Proteome Res. 2017, 16 (4), 1646–1658. 10.1021/acs.jproteome.6b01003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes E.; Loo R. L.; Stamler J.; Bictash M.; Yap I. K. S.; Chan Q.; Ebbels T.; De Iorio M.; Brown I. J.; Veselkov K. A.; Daviglus M. L.; Kesteloot H.; Ueshima H.; Zhao L. C.; Nicholson J. K.; Elliott P. Human metabolic phenotype diversity and its association with diet and blood pressure. Nature 2008, 453 (7193), 396–U50. 10.1038/nature06882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckonert O.; Keun H. C.; Ebbels T. M. D.; Bundy J. G.; Holmes E.; Lindon J. C.; Nicholson J. K. Metabolic profiling, metabolomic and metabonomic procedures for NMR spectroscopy of urine, plasma, serum and tissue extracts. Nat. Protoc. 2007, 2 (11), 2692–2703. 10.1038/nprot.2007.376. [DOI] [PubMed] [Google Scholar]
- Beckonert O.; Coen M.; Keun H. C.; Wang Y. L.; Ebbels T. M. D.; Holmes E.; Lindon J. C.; Nicholson J. K. High-resolution magic-angle-spinning NMR spectroscopy for metabolic profiling of intact tissues. Nat. Protoc. 2010, 5 (6), 1019–1032. 10.1038/nprot.2010.45. [DOI] [PubMed] [Google Scholar]
- Want E. J.; Wilson I. D.; Gika H.; Theodoridis G.; Plumb R. S.; Shockcor J.; Holmes E.; Nicholson J. K. Global metabolic profiling procedures for urine using UPLC-MS. Nat. Protoc. 2010, 5 (6), 1005–1018. 10.1038/nprot.2010.50. [DOI] [PubMed] [Google Scholar]
- Roux A.; Xu Y.; Heilier J. F.; Olivier M. F.; Ezan E.; Tabet J. C.; Junot C. Annotation of the Human Adult Urinary Metabolome and Metabolite Identification Using Ultra High Performance Liquid Chromatography Coupled to a Linear Quadrupole Ion Trap-Orbitrap Mass Spectrometer. Anal. Chem. 2012, 84 (15), 6429–6437. 10.1021/ac300829f. [DOI] [PubMed] [Google Scholar]
- Chan E. C. Y.; Pasikanti K. K.; Nicholson J. K. Global urinary metabolic profiling procedures using gas chromatography-mass spectrometry. Nat. Protoc. 2011, 6 (10), 1483–1499. 10.1038/nprot.2011.375. [DOI] [PubMed] [Google Scholar]
- Wilson I. D.; Nicholson J. K.; Castro-Perez J.; Granger J. H.; Johnson K. A.; Smith B. W.; Plumb R. S. High resolution “Ultra performance” liquid chromatography coupled to oa-TOF mass spectrometry as a tool for differential metabolic pathway profiling in functional genomic studies. J. Proteome Res. 2005, 4 (2), 591–598. 10.1021/pr049769r. [DOI] [PubMed] [Google Scholar]
- Rochat B. From targeted quantification to untargeted metabolomics: Why LC-high-resolution-MS will become a key instrument in clinical labs. TrAC, Trends Anal. Chem. 2016, 84, 151–164. 10.1016/j.trac.2016.02.009. [DOI] [Google Scholar]
- Glauser G.; Grund B.; Gassner A. L.; Menin L.; Henry H.; Bromirski M.; Schutz F.; McMullen J.; Rochat B. Validation of the Mass-Extraction-Window for Quantitative Methods Using Liquid Chromatography High Resolution Mass Spectrometry. Anal. Chem. 2016, 88 (6), 3264–3271. 10.1021/acs.analchem.5b04689. [DOI] [PubMed] [Google Scholar]
- Krauss M.; Singer H.; Hollender J. LC-high resolution MS in environmental analysis: from target screening to the identification of unknowns. Anal. Bioanal. Chem. 2010, 397 (3), 943–951. 10.1007/s00216-010-3608-9. [DOI] [PubMed] [Google Scholar]
- Dahmane E.; Boccard J.; Csajka C.; Rudaz S.; Decosterd L.; Genin E.; Duretz B.; Bromirski M.; Zaman K.; Testa B.; Rochat B. Quantitative monitoring of tamoxifen in human plasma extended to 40 metabolites using liquid-chromatography high-resolution mass spectrometry: new investigation capabilities for clinical pharmacology. Anal. Bioanal. Chem. 2014, 406 (11), 2627–2640. 10.1007/s00216-014-7682-2. [DOI] [PubMed] [Google Scholar]
- Hamelin E. I.; Bragg W.; Shaner R. L.; Swaim L. L.; Johnson R. C. Comparison of high-resolution and tandem mass spectrometry for the analysis of nerve agent metabolites in urine. Rapid Commun. Mass Spectrom. 2013, 27 (15), 1697–1704. 10.1002/rcm.6621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufmann A.; Butcher P.; Maden K.; Walker S.; Widmer M. Quantitative and confirmative performance of liquid chromatography coupled to high-resolution mass spectrometry compared to tandem mass spectrometry. Rapid Commun. Mass Spectrom. 2011, 25 (7), 979–992. 10.1002/rcm.4952. [DOI] [PubMed] [Google Scholar]
- Zhang N. R.; Yu S.; Tiller P.; Yeh S.; Mahan E.; Emary W. B. Quantitation of small molecules using high-resolution accurate mass spectrometers - a different approach for analysis of biological samples. Rapid Commun. Mass Spectrom. 2009, 23 (7), 1085–1094. 10.1002/rcm.3975. [DOI] [PubMed] [Google Scholar]
- Kirwan J. A.; Broadhurst D. I.; Davidson R. L.; Viant M. R. Characterising and correcting batch variation in an automated direct infusion mass spectrometry (DIMS) metabolomics workflow. Anal. Bioanal. Chem. 2013, 405 (15), 5147–5157. 10.1007/s00216-013-6856-7. [DOI] [PubMed] [Google Scholar]
- Kirwan J. A.; Weber R. J.; Broadhurst D. I.; Viant M. R. Direct infusion mass spectrometry metabolomics dataset: a benchmark for data processing and quality control. Sci. Data 2014, 1, 140012. 10.1038/sdata.2014.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Draper J.; Lloyd A. J.; Goodacre R.; Beckmann M. Flow infusion electrospray ionisation mass spectrometry for high throughput, non-targeted metabolite fingerprinting: a review. Metabolomics 2013, 9 (1), 4–29. 10.1007/s11306-012-0449-x. [DOI] [Google Scholar]
- Southam A. D.; Weber R. J.; Engel J.; Jones M. R.; Viant M. R. A complete workflow for high-resolution spectral-stitching nanoelectrospray direct-infusion mass-spectrometry-based metabolomics and lipidomics. Nat. Protoc. 2017, 12 (2), 255–273. 10.1038/nprot.2016.156. [DOI] [PubMed] [Google Scholar]
- Chekmeneva E.; Correia G.; Denes J.; Gomez-Romero M.; Wijeyesekera A.; Perenyi D. R.; Koot Y.; Boomsma C.; Want E. J.; Dixon P. H.; Macklon N. S.; Chan Q.; Takats Z.; Nicholson J. K.; Holmes E. Development of nanoelectrospray high resolution isotope dilution mass spectrometry for targeted quantitative analysis of urinary metabolites: application to population profiling and clinical studies. Anal. Methods 2015, 7 (12), 5122–5133. 10.1039/C5AY00850F. [DOI] [Google Scholar]
- Xiang Y.; Koomen J. M. Evaluation of Direct Infusion-Multiple Reaction Monitoring Mass Spectrometry for Quantification of Heat Shock Proteins. Anal. Chem. 2012, 84 (4), 1981–1986. 10.1021/ac203011j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickremsinhe E. R.; Ackermann B. L.; Chaudhary A. K. Validating regulatory-compliant wide dynamic range bioanalytical assays using chip-based nanoelectrospray tandem mass spectrometry. Rapid Commun. Mass Spectrom. 2005, 19 (1), 47–56. 10.1002/rcm.1747. [DOI] [PubMed] [Google Scholar]
- Koulman A.; Cao M.; Faville M.; Lane G.; Mace W.; Rasmussen S. Semi-quantitative and structural metabolic phenotyping by direct infusion ion trap mass spectrometry and its application in genetical metabolomics. Rapid Commun. Mass Spectrom. 2009, 23 (15), 2253–2263. 10.1002/rcm.4142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin L.; Yu Q. A.; Yan X. M.; Hang W.; Zheng J. X.; Xing J. C.; Huang B. L. Direct infusion mass spectrometry or liquid chromatography mass spectrometry for human metabonomics? A serum metabonomic study of kidney cancer. Analyst 2010, 135 (11), 2970–2978. 10.1039/c0an00265h. [DOI] [PubMed] [Google Scholar]
- Lisa M.; Cifkova E.; Khalikova M.; Ovcacikova M.; Holcapek M. Lipidomic analysis of biological samples: Comparison of liquid chromatography, supercritical fluid chromatography and direct infusion mass spectrometry methods. J. Chromatogr A 2017, 1525, 96–108. 10.1016/j.chroma.2017.10.022. [DOI] [PubMed] [Google Scholar]
- Hao J.; Liebeke M.; Sommer U.; Viant M. R.; Bundy J. G.; Ebbels T. M. D. Statistical Correlations between NMR Spectroscopy and Direct Infusion FT-ICR Mass Spectrometry Aid Annotation of Unknowns in Metabolomics. Anal. Chem. 2016, 88 (5), 2583–2589. 10.1021/acs.analchem.5b02889. [DOI] [PubMed] [Google Scholar]
- Marshall D. D.; Lei S. L.; Worley B.; Huang Y. T.; Garcia-Garcia A.; Franco R.; Dodds E. D.; Powers R. Combining DI-ESI-MS and NMR datasets for metabolic profiling. Metabolomics 2015, 11 (2), 391–402. 10.1007/s11306-014-0704-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamler J.; Elliott P.; Dennis B.; Dyer A. R.; Kesteloot H.; Liu K.; Ueshima H.; Zhou B. F. INTERMAP: background, aims, design, methods, and descriptive statistics (nondietary). J. Hum. Hypertens. 2003, 17 (9), 591–608. 10.1038/sj.jhh.1001603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bateman R. H.; Carruthers R.; Hoyes J. B.; Jones C.; Langridge J. I.; Millar A.; Vissers J. P. C. A novel precursor ion discovery method on a hybrid quadrupole orthogonal acceleration time-of-flight (Q-TOF) mass spectrometer for studying protein phosphorylation. J. Am. Soc. Mass Spectrom. 2002, 13 (7), 792–803. 10.1016/S1044-0305(02)00420-8. [DOI] [PubMed] [Google Scholar]
- U.S. Department of Health and Human Services Food and Drug Administration. Bioanalytical Method Validation: Guidance for Industry. https://www.fda.gov/downloads/drugs/guidances/ucm070107.Pdf.
- Kessner D.; Chambers M.; Burke R.; Agus D.; Mallick P. ProteoWizard: open source software for rapid proteomics tools development. Bioinformatics 2008, 24 (21), 2534–2536. 10.1093/bioinformatics/btn323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dieterle F.; Ross A.; Schlotterbeck G.; Senn H. Probabilistic quotient normalization as robust method to account for dilution of complex biological mixtures. Application in H-1 NMR metabonomics. Anal. Chem. 2006, 78 (13), 4281–4290. 10.1021/ac051632c. [DOI] [PubMed] [Google Scholar]
- Smith C. A.; Want E. J.; O’Maille G.; Abagyan R.; Siuzdak G. XCMS: Processing mass spectrometry data for metabolite profiling using Nonlinear peak alignment, matching, and identification. Anal. Chem. 2006, 78 (3), 779–787. 10.1021/ac051437y. [DOI] [PubMed] [Google Scholar]
- R Development Core Team . R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2011. [Google Scholar]
- Passing H.; Bablok W. A New Biometrical Procedure for Testing the Equality of Measurements from Two Different Analytical Methods. Application of linear regression procedures for method comparison studies in Clinical Chemistry, Part I. Clin. Chem. Lab. Med. 1983, 21 (11), 709–720. 10.1515/cclm.1983.21.11.709. [DOI] [PubMed] [Google Scholar]
- Martin Bland J.; Altman D. G. Statistical Methods for Assessing Agreement between Two Methods of Clinical Measurement. Lancet 1986, 327 (8476), 307–310. 10.1016/S0140-6736(86)90837-8. [DOI] [PubMed] [Google Scholar]
- Bylesjo M.; Rantalainen M.; Cloarec O.; Nicholson J. K.; Holmes E.; Trygg J. OPLS discriminant analysis: combining the strengths of PLS-DA and SIMCA classification. J. Chemom. 2006, 20 (8–10), 341–351. 10.1002/cem.1006. [DOI] [Google Scholar]
- Bouatra S.; Aziat F.; Mandal R.; Guo A. C.; Wilson M. R.; Knox C.; Bjorndahl T. C.; Krishnamurthy R.; Saleem F.; Liu P.; Dame Z. T.; Poelzer J.; Huynh J.; Yallou F. S.; Psychogios N.; Dong E.; Bogumil R.; Roehring C.; Wishart D. S. The human urine metabolome. PLoS One 2013, 8 (9), e73076. 10.1371/journal.pone.0073076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith C. A.; O’Maille G.; Want E. J.; Qin C.; Trauger S. A.; Brandon T. R.; Custodio D. E.; Abagyan R.; Siuzdak G. METLIN - A metabolite mass spectral database. Ther. Drug Monit. 2005, 27 (6), 747–751. 10.1097/01.ftd.0000179845.53213.39. [DOI] [PubMed] [Google Scholar]
- Ulrich E. L.; Akutsu H.; Doreleijers J. F.; Harano Y.; Ioannidis Y. E.; Lin J.; Livny M.; Mading S.; Maziuk D.; Miller Z.; et al. BioMagResBank. Nucleic Acids Res. 2007, 36, D402–D408. 10.1093/nar/gkm957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croixmarie V.; Umbdenstock T.; Cloarec O.; Moreau A.; Pascussi J. M.; Boursier-Neyret C.; Walther B. Integrated Comparison of Drug-Related and Drug-Induced Ultra Performance Liquid Chromatography/Mass Spectrometry Metabonomic Profiles Using Human Hepatocyte Cultures. Anal. Chem. 2009, 81 (15), 6061–6069. 10.1021/ac900333e. [DOI] [PubMed] [Google Scholar]
- Swann J. R.; Spagou K.; Lewis M.; Nicholson J. K.; Glei D. A.; Seeman T. E.; Coe C. L.; Goldman N.; Ryff C. D.; Weinstein M.; Holmes E. Microbial-Mammalian Cometabolites Dominate the Age-associated Urinary Metabolic Phenotype in Taiwanese and American Populations. J. Proteome Res. 2013, 12 (7), 3166–3180. 10.1021/pr4000152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thevenot E. A.; Roux A.; Xu Y.; Ezan E.; Junot C. Analysis of the Human Adult Urinary Metabolome Variations with Age, Body Mass Index, and Gender by Implementing a Comprehensive Workflow for Univariate and OPLS Statistical Analyses. J. Proteome Res. 2015, 14 (8), 3322–3335. 10.1021/acs.jproteome.5b00354. [DOI] [PubMed] [Google Scholar]
- Rasmussen B.; Cloarec O.; Tang H. R.; Staerk D.; Jaroszewski J. W. Multivariate analysis of integrated and full-resolution H-1-NMR spectral data from complex pharmaceutical preparations: St. John’s wort. Planta Med. 2006, 72 (6), 556–563. 10.1055/s-2006-931567. [DOI] [PubMed] [Google Scholar]
- Heymsfield S. B.; Arteaga C.; Mcmanus C.; Smith J.; Moffitt S. Measurement of Muscle Mass in Humans - Validity of the 24-h Urinary Creatinine Method. Am. J. Clin. Nutr. 1983, 37 (3), 478–494. 10.1093/ajcn/37.3.478. [DOI] [PubMed] [Google Scholar]
- Mittelstrass K.; Ried J. S.; Yu Z. H.; Krumsiek J.; Gieger C.; Prehn C.; Roemisch-Margl W.; Polonikov A.; Peters A.; Theis F. J.; Meitinger T.; Kronenberg F.; Weidinger S.; Wichmann H. E.; Suhre K.; Wang-Sattler R.; Adamski J.; Illig T. Discovery of Sexual Dimorphisms in Metabolic and Genetic Biomarkers. PLoS Genet. 2011, 7 (8), e1002215. 10.1371/journal.pgen.1002215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erve J. C. L.; DeMaio W.; Talaat R. E. Rapid metabolite identification with sub parts-per-million mass accuracy from biological matrices by direct infusion nanoelectrospray ionization after clean-up on a ZipTip and LTQ/Orbitrap mass spectrometry. Rapid Commun. Mass Spectrom. 2008, 22 (19), 3015–3026. 10.1002/rcm.3702. [DOI] [PubMed] [Google Scholar]
- Dunn W. B.; Wilson I. D.; Nicholls A. W.; Broadhurst D. The importance of experimental design and QC samples in large-scale and MS-driven untargeted metabolomic studies of humans. Bioanalysis 2012, 4 (18), 2249–2264. 10.4155/bio.12.204. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.