

Abstract
The continuity of life depends on mechanisms in the germline that ensure the integrity of the genome. The DNA damage response/checkpoint kinases ATM and ATR are essential signaling factors in the germline. However, it remains unknown how a downstream transducer, Checkpoint Kinase 1 (CHEK1 or CHK1), mediates signaling in the male germline. Here, we show that CHEK1 has distinct functions in both the mitotic and meiotic phases of the male germline in mice. In the mitotic phase, CHEK1 is required for the resumption of prospermatogonia proliferation after birth and the maintenance of spermatogonia. In the meiotic phase, we uncovered two functions for CHEK1: one is the stage-specific attenuation of DNA damage signaling on autosomes, and the other is coordination of meiotic stage progression. On autosomes, the loss of CHEK1 delays the removal of DNA damage signaling that manifests as phosphorylation of histone variant H2AX at serine 139 (γH2AX). Importantly, CHEK1 does not have a direct function in meiotic sex chromosome inactivation (MSCI), an essential event in male meiosis, in which ATR is a key regulator. Thus, the functions of ATR and CHEK1 are uncoupled in MSCI, in contrast to their roles in DNA damage signaling in somatic cells. Our study reveals stage-specific functions for CHEK1 that ensure the integrity of the male germline.
Introduction
The continuity of life depends on the germline, which is protected and maintained through numerous mechanisms that monitor the integrity of the genome. In the mammalian germline, such mechanisms are present in both the mitotic and subsequent meiotic phases of germ cells. During the mitotic phase, germ cells are highly susceptible to DNA damage and abnormal germ cells are readily eliminated to maintain the integrity of germline, thereby ensuring a lower mutation rate than somatic cells (1,2). During meiosis, genomic fidelity is also strictly monitored. These processes in the mitotic and meiotic phases are ensured by DNA damage response (DDR)/checkpoint proteins (3). In somatic cells, these proteins recognize DNA damage and, in response, mediate cell cycle checkpoint and DNA repair mechanisms (4,5). However, the specific functions of cell cycle checkpoint machinery and DNA repair mechanisms in the germline remain less understood.
Importantly, DDR/checkpoint proteins regulate normal developmental processes such as meiotic recombination. Meiotic recombination is catalyzed by SPO11, which induces programmed double-stranded DNA breaks beginning in the leptotene stage of meiotic prophase (6–8). In response to this, the DDR/checkpoint kinase Ataxia Telangiectasia Mutated (ATM) phosphorylates Serine 139 of the histone variant H2AX (γH2AX), a post-translational modification that mediates DNA damage signaling (9,10). Another DDR/checkpoint kinase, Ataxia Telangiectasia and Rad3-Related (ATR), catalyzes a subsequent wave of γH2AX formation in response to unsynapsed chromatin in the zygotene stage (11). Following the completion of synapsis in the pachytene stage, ATR-dependent γH2AX is established on the unsynapsed sex chromosomes in males, leading to the transcriptional silencing of the sex chromosomes, a process termed meiotic sex chromosome inactivation (MSCI) (12–14). MSCI is essential for the subsequent progression of male germ cells through meiosis, as its failure leads to complete germ cell elimination (15,16). Although DDR/checkpoint proteins are important for cell cycle regulation in somatic cells, it remains unknown whether DDR/checkpoint proteins regulate meiotic stage progression.
In somatic cells, a downstream transducer kinase propagates ATR-mediated DNA damage signaling. This transducer, Checkpoint Kinase 1 (CHEK1), plays an essential role in the S and G2 cell cycle checkpoints, and is activated by ATR-mediated phosphorylation at Serine 345 (17,18). CHEK1 also has key functions in unperturbed somatic cells, and global deletion of Chek1 in mice leads to apoptosis, which results in embryonic lethality (18–20). In mouse oocytes, CHEK1 is essential for meiotic cell cycle regulation (21). However, despite its general importance in cell cycle regulation, the functions of CHEK1 remain unknown in the male germline. Notably, CHEK2, another checkpoint kinase, is important for the DNA damage checkpoint pathway in oocytes (22,23) and for a pachytene checkpoint in mouse spermatocytes (24), emphasizing the question of CHEK1’s role in the male germline.
To determine the functions of CHEK1 in the male germline, we generated conditional-deletion mouse models for Chek1. Our results unravel previously unknown functions of CHEK1 during spermatogenesis, both in its mitotic and meiotic phases. In the mitotic phase of germ cells, CHEK1 is required for the resumption of proliferation of prospermatogonia [also known as gonocytes (25,26)] after birth, as well as the subsequent maintenance of spermatogonia. In meiosis, we uncovered two closely related Chek1-mutant phenotypes: first, with respect to autosomes, we observed a stark delay in the removal of γH2AX signals that are initiated at the onset of meiotic prophase; second, we observed an abnormal pattern of meiotic stage progression in which the leptotene and zygotene stages of prophase are prolonged, while the duration of the mid-pachytene stage is significantly shortened. Importantly, CHEK1 does not have a direct function in the initiation of MSCI, indicating that ATR and CHEK1 are uncoupled in MSCI. In summary, our study reveals stage-specific functions of CHEK1 that ensure the integrity of the male germline.
Results
CHEK1 is abundantly expressed during spermatogenesis but is not detectable on meiotic chromosome axes
To probe the possible involvement of CHEK1 in spermatogenesis, we examined the expression of CHEK1 protein in mouse testes. By western blotting, we detected a consistently abundant level of CHEK1 protein in testicular extracts from juvenile mice at 8–17 days postpartum (dpp), a time period in which the first wave of spermatogenesis undergoes semisynchronous progression from mitotic spermatogonia to meiotic primary spermatocytes (Fig. 1A). Although the CHEK1 antibody recognized multiple nonspecific bands in testicular extracts, we confirmed that the specific band for CHEK1 (54 kDa) was absent in testicular extracts of Chek1 conditional knockout mice (Ddx4-Cre-ERT2: Chek1cKO; described below). Thus, CHEK1 is expressed in spermatogonia through to spermatocytes in meiotic prophase.
Figure 1.

CHEK1 is required for the proliferation of prospermatogonia. (A) Western blotting of testicular lysate obtained from wild-type mice at 8, 11, 14 and 17 dpp, and from Chek1ctrl and Ddx4-Cre-ERT2: Chek1cKO mice at 31 dpp (tamoxifen injections were at 3 weeks of age, or 21 dpp, and testes were harvested 10 days later). Asterisks indicate the positions of non-specific bands. Anti-CHEK1 antibody (Sigma #C9358) was used. (B) Schematic for Chek1 deletion by Ddx4-Cre. (C) Picture of Chek1ctrl and Chek1cKO testes at 54 dpp. Scale bar: 1 cm. (D) Immunostaining of WT1 and PLZF on testicular sections at 24 dpp. Scale bars: 100 μm. (E) Immunostaining of SCML2 on testicular sections at 3 and 5 dpp. White arrowheads indicate SCML2-positive cells in the Chek1cKO. Nuclei were counterstained with DAPI. Scale bars: 100 μm.
Given its abundant expression in testes, we sought to determine the localization of CHEK1 during spermatogenesis. Previous studies reported that immunofluorescent signals from an anti-CHEK1 antibody localized on chromosome axes during meiosis (27), while signals from another anti-CHEK1 antibody and an anti-phospho-CHEK1 on Serine 345 (pCHEK1Ser345) antibody—the latter of which detects the activated form of CHEK1—localized on the unsynapsed axes of the sex chromosomes in pachytene spermatocytes (28). To confirm these results, we tested CHEK1 immunolocalization using four independent anti-CHEK1 antibodies on meiotic chromosome spreads. Surprisingly, we detected no fluorescent signals in our analyses of chromosome spreads, neither on chromatin nor on chromatin axes (Supplementary Material, Fig. S1A–D). Additionally, we tested two independent anti-pCHEK1Ser345 antibodies. Immunostaining with one of the anti-pCHEK1Ser345 antibodies resulted in no detectable fluorescent signals (Supplementary Material, Fig. S1E). Using the second anti-pCHEK1Ser345 antibody, we observed immunofluorescent signals on the sex chromosome axes (Supplementary Material, Fig. S1F), but we concluded that these signals comprised a false positive as they were routinely present in pachytene spermatocytes of the Chek1cKO (Supplementary Material, Fig. S1G). This result indicates that the second anti-pCHEK1Ser345 antibody cross-reacts with targets other than CHEK1. Indeed, we used this antibody for the western blotting of testicular crude extracts and found that, while pCHEK1Ser345 was present on the blotted membrane, the antibody cross-reacted with many different targets in the testicular extract (Supplementary Material, Fig. S1H). It is possible that this is due to the abundance of phosphorylated proteins in male germ cells; of note, a large number of phosphoproteins are present on the sex chromosomes during meiosis. We further tested these antibodies using slides prepared through an independent method that preserves the relative 3D chromatin structure of germ cells (3D slides) (29,30), and we confirmed that these antibodies do not show any specific signals (Supplementary Material, Fig. S1I–N). Taken together, our data—which include the use of ideal negative controls (i.e. CHEK1-null tissues)—strongly suggest that CHEK1 does not localize on chromosome axes during meiosis. These results may be explained by the previous observation that CHEK1 dissociates from chromatin after it is phosphorylated, thereby mediating DNA damage signaling at specific target sites (31,32). As a consequence, non-chromatin-bound CHEK1 may be washed away in the permeabilization step of chromosome spread preparation. To test this possibility, we performed immunohistochemistry with cryosections that preserve the integrity of cells. However, these CHEK1 antibodies did not show any specific signals in the cryosections (Supplementary Material, Fig. S2).
CHEK1 is essential for the resumption of prospermatogonia proliferation after birth
To address the function of CHEK1 in germ cells, we generated a germline-specific Chek1cKO mouse model because the constitutive deletion of Chek1 is embryonic lethal (18,19). We crossed mice harboring floxed Chek1 alleles (18) with mice harboring a Ddx4-Cre transgene, which catalyzes germline-specific Cre recombination on embryonic day 15 (E15) (Fig. 1B) (33). In Ddx4-Cre: Chek1cKO mice (Ddx4-Cre: Chek1F/-), testes were much smaller than that of control littermates (either Chek1F/+ or Chek1F/-: Chek1ctrl) (Fig. 1C). In testicular sections of the Chek1cKO at 24 dpp, all testicular cells were positive for the Sertoli cell-marker WT1, while no cells were positive for PLZF, a marker for a population of spermatogonia that includes undifferentiated spermatogonia (Fig. 1D). These results indicate that CHEK1 is essential for the survival of germ cells.
In the male germline, quiescent prospermatogonia resume proliferation during the time period from 1 to 4 dpp (34). Because CHEK1 is activated during normal S-phase progression in unperturbed cells (35), we hypothesized that CHEK1 is required for the proliferation of prospermatogonia after birth. To test this hypothesis, we examined testicular sections at 3 and 5 dpp for germ cell survival by immunostaining against SCML2, a germ cell marker (36). Although SCML2-positive germ cells were observed in both Chek1ctrl and Ddx4-Cre: Chek1cKO mice at 3 dpp, SCML2-positive germ cells were rarely observed in the Ddx4-Cre: Chek1cKO at 5 dpp (Fig. 1E). These data indicate an elimination of germ cells in the period between 3 and 5 dpp. To determine whether proliferation defects in the Ddx4-Cre: Chek1cKO lead to germ cell elimination, we performed immunostaining with an antibody against Ki67, a marker of cell proliferation. While most of the TRA98-positive germ cells of Chek1ctrl were positive for Ki67 at 5 dpp, the rarely present TRA98-positive germ cells of the Ddx4-Cre: Chek1cKO were devoid of Ki67 at 5 dpp (Supplementary Material, Fig. S3). Thus, we confirmed a function for CHEK1 in the resumption of prospermatogonia proliferation after birth. Because CHEK1 plays essential roles in cell proliferation (18,19), defective proliferation likely underlies germ cell depletion in Chek1 mutants.
CHEK1 is required for maintenance of spermatogonia
Since the Chek1cKO model driven by Ddx4-Cre showed germ cell loss soon after birth—before the initiation of first-wave spermatogenesis—questions as to the function of CHEK1 in spermatogenesis remained unanswered. Thus, we sought to understand the function of CHEK1 in spermatogenesis using a mouse model with a Cre-mediated Chek1 deletion that can be induced after initiation of the first wave of spermatogenesis. To that end, we generated a Chek1cKO model harboring the Rosa26-Cre-ERT2 transgene, which enables inducible, global deletion of Chek1 via treatment with tamoxifen (37). However, Chek1 deletion with Rosa26-Cre-ERT2 in mice aged 8–9 weeks resulted in lethality within 10 days of the first tamoxifen injection (Supplementary Material, Fig. S4). Thus, to circumvent lethality, we generated a conditional deletion model with the Ddx4-Cre-ERT2 transgene, which enables the induction of germ cell-specific Cre recombination upon treatment with tamoxifen (38). Widespread Cre expression of the Ddx4-Cre-ERT2transgene in testicular germ cells was previously demonstrated using β-galactosidase reporter mice (38). We initially tried to inject juvenile mice with tamoxifen, but this turned out to be lethal regardless of the genotype. In turn, we induced Chek1 deletion through tamoxifen injections of mice at 3 weeks of age (21 dpp), and testes were harvested 10 days later (31 dpp; 10 days after tamoxifen injection into 3-week-old mice, termed the ‘10-days-3-weeks’ condition: Fig. 2A). To precisely evaluate the phenotype after tamoxifen injections, we treated mutant mice and control littermates (Chek1ctrl: Chek1F/F) with equal doses of tamoxifen. Testis sizes between Ddx4-Cre-ERT2: Chek1cKO and control littermates at 31 dpp were comparable (Fig. 2B), and CHEK1 depletion in Ddx4-Cre-ERT2: Chek1cKO testes at 31 dpp was confirmed by western blotting analysis (Fig. 1A). Since Ddx4-Cre-ERT2 induces germ cell-specific Chek1 deletion, and germ cells comprise the vast majority of cells in the testes, the western blot data suggest that the absence of CHEK1 expression in testes results from efficient targeted deletion in germ cells. However, due to the lack of an anti-CHEK1 antibody suitable for immunostaining, we were unable to determine the deletion efficiency on a cell-by-cell basis.
Figure 2.

CHEK1 is required for the maintenance of spermatogonia. (A) Schematic of the experimental design of the 10-day-3-weeks method. (B) Picture of Chek1ctrl and Chek1cKO testes at 31 dpp. Scale bar: 1 cm. Testis weights are shown as mean ± SEM for three independent pairs of Chek1ctrl and Chek1cKO mice. n.s.: not significant. Significance was determined utilizing an unpaired t-test. (C) Sections stained with H & E at 31 dpp. Scale bars: 100 μm. (D) Immunostaining of H1t and γH2AX on testicular sections at 31 dpp. Asterisks indicate seminiferous tubules at stages IX–XI. Dashed squares are magnified in the right-hand panels. Scale bars: 100 μm. (E) Immunostaining of WT1 and PLZF on testicular sections at 31 dpp. Scale bars: 100 μm. Numbers of PLZF positive cells per seminiferous tubule are shown as mean ± SEM for three independent pairs of Chek1ctrl and Chek1cKO mice. *P < 0.05 (unpaired t-test). (F) Immunostaining of PLZF and Ki-67 on testicular sections at 31 dpp. Arrowheads indicate cells that are double positive for PLZF and Ki-67. Scale bars: 100 μm. Percentages of PLZF-positive cells that are also positive for Ki-67 are shown as the mean ± SEM for three independent pairs Chek1ctrl and Chek1cKO mice. n.s.: not significant. Unpaired t-test. (G) Immunostaining of SYCP3 and γH2AX on meiotic chromosome spreads at 31 dpp. Scale bars: 10 μm. Stage populations during meiotic prophase are shown as the mean ± SEM for five independent pairs of Chek1ctrl and Chek1cKO mice. Total numbers of analyzed nuclei are indicated in the panels. Scale bars: 10 μm. *P < 0.05, **P< 0.01, ***P < 0.001 (unpaired t-test).
To determine the effect of CHEK1 depletion, we sought to define the phenotype of Ddx4-Cre-ERT2: Chek1cKO testes obtained under the 10-days-3-weeks condition. Spermatogenesis is a continuous, cyclic process that can be divided into 12 broad, semisynchronous stages (39). This enabled us to distinguish and judge the status of spermatogenic development in both the presence and absence of CHEK1. Although the overall sizes of testicular tubules, judged by staining testicular sections with hematoxylin and eosin (H & E), appeared to be comparable between Ddx4-Cre-ERT2: Chek1cKO and control littermates at 31 dpp (Fig. 2C), we found a clear mutant phenotype through immunofluorescence analyses with specific stage markers. One such marker is H1t, a histone variant specific to the male germline that accumulates in nuclei during and after the mid-pachytene stage of prophase (i.e. H1t accumulates to a high degree in spermatocytes after stage VIII). Thus, stage IX–XI tubules were judged by the presence of H1t-positive late-pachytene or diplotene spermatocytes that comprise the middle layer of the tubules; these nuclei evinced focused γH2AX staining on the sex chromosomes (Fig. 2D and D′; staging criteria for stage IX–XI tubules are described in Supplementary Material, Fig. S5A). In stage IX–XI tubules of Chek1 control littermates, we observed the normal presence of spermatocytes in the leptotene and zygotene stages of prophase that comprise a peripheral layer of the tubules; this was determined by the presence of nucleus-wide γH2AX immunofluorescent signals and the concomitant absence of H1t signals (Fig. 2D′). By contrast, we observed the broad depletion of leptotene and zygotene spermatocytes in a peripheral layer of stage IX-XI tubules; staging was judged by nucleus-wide H1t signals and concomitant γH2AX staining limited to the sex chromosomes in late-pachytene and/or diplotene spermatocytes of the Ddx4-Cre-ERT2: Chek1cKO (Fig. 2D″). Together, these results indicate that the depletion of CHEK1 caused a reduction in the numbers of leptotene and zygotene spermatocytes.
CHEK1 is important for mitotic proliferation (18–20), so we suspected that a decrease in mitotically proliferating spermatogonia, resulting from depletion of CHEK1, led to a reduction in the numbers of leptotene and zygotene spermatocytes. Thus, to test whether the absence of CHEK1 resulted in a reduction of spermatogonia, we immunostained testicular sections with PLZF, which marks a spermatogonia population, and WT1, a marker of Sertoli cells (Fig. 2E). The numbers of PLZF-positive spermatogonia per seminiferous tubule were significantly decreased in the mutants as compared to littermate controls under the 10-days-3-weeks condition (Fig. 2E), indicating that CHEK1 is required for the maintenance of spermatogonia. Next, we examined whether the cell cycle was altered in mutant spermatogonia under the 10-days-3-weeks condition. By immunostaining with an antibody against Ki67, we observed that populations of Ki67-positive undifferentiated spermatogonia in mutant testes were normal (Fig. 2F). These data exclude the possibility that the depletion of spermatogonia in Chek1cKO is due to cell cycle arrest at a particular point in the cell cycle.
Next, we sought to determine the function of CHEK1 during meiosis under the 10-days-3-weeks condition. Consistent with the immunostaining of testicular sections, we observed a reduction in leptotene and zygotene spermatocytes in Chek1cKO versus littermate controls (Fig. 2G). However, we observed an increase in the relative proportions of spermatocytes in the mid-pachytene stage and subsequent stages of prophase. We interpreted this as an indirect consequence of depletion of spermatocytes prior to the mid-pachytene stage in the mutants: one cycle of the seminiferous epithelial cycle lasts for 8.6 days and it takes four cycles—or approximately 35 days—for spermatogonial stem cells to differentiate and mature into spermatozoa (40). Thus, the 10-day interim after tamoxifen injection is not sufficient for CHEK1 depletion to penetrate all stages of meiotic prophase. For example, if Chek1 deletion occurs in Type B spermatogonia, 10 days of spermatogenic development places cells descended from these Type B spermatogonia in the mid-pachytene stage (Supplementary Material, Fig. S5B). Therefore, through analyses of mutants and controls under the 10-days-3-weeks condition, we conclude that CHEK1 is required for the maintenance of spermatogonia; however, for a thorough characterization of meiosis, we recognized a need to wait a longer amount of time after treatment with tamoxifen.
CHEK1 regulates meiotic progression and the removal of autosomal γH2AX
To address the function of CHEK1 during meiosis, we examined the phenotype at 20 days after tamoxifen treatment of 3-week-old Ddx4-Cre-ERT2: Chek1cKO mice (41 dpp; termed the “20-days-3-weeks” condition: Fig. 3A). This condition covers all stages of meiotic prophase starting from Type B spermatogonia (Supplementary Material, Fig. S5C). Under the 20-days-3-weeks condition, testis sizes were comparable between Chek1cKO and control littermates that received equal treatments of tamoxifen (Fig. 3B), although the seminiferous tubules of the Chek1cKO were grossly disorganized as judged by H & E staining (Fig. 3C). Many of the mutant tubules were devoid of γH2AX and/or H1t positive cells (Fig. 3D: a), indicating that depletion of germ cells occurred in the mutants. In addition, some seminiferous tubules contained leptotene and zygotene spermatocytes but were devoid of H1t-positive germ cells (Fig. 3D: b), suggesting abnormal meiotic progression. Interestingly, seminiferous tubules at stages I–V, as judged by the presence of H1t-positive round spermatids, showed an ectopic retention of leptotene and zygotene spermatocytes (Fig. 3D: c, and Supplementary Material, Fig. S5A). In normal spermatogenesis, stage I–V tubules contained early pachytene spermatocytes without leptotene and zygotene spermatocytes, which populate stage IX–XII tubules (39). Thus, these data indicate that the absence of CHEK1 disrupted the coordination of seminiferous cycles, suggesting a longer duration for the leptotene and zygotene stages compared to controls.
Figure 3.

CHEK1 depletion disrupts the seminiferous cycle independent of apoptosis. (A) Schematic of the experimental design for the 20-day-3-weeks method. (B) Picture of Chek1ctrl and Chek1cKO testes at 41 dpp. Scale bar: 1 cm. Testis weights are shown as the mean ± SEM for three independent pairs of Chek1ctrl and Chek1cKO mice. n.s.: not significant (unpaired t-test). (C) Sections stained with H & E at 41 dpp. Scale bars: 100 μm. (D) Immunostaining of H1t and γH2AX on testicular sections at 41 dpp. A seminiferous tubule at stages I–V was judged by the presence of H1t positive round spermatids. The dashed circle labeled with ‘a’” indicates seminiferous tubules with an absence of germ cells. ‘b’ indicates seminiferous tubules including only leptotene and zygotene spermatocytes. ‘c’ indicates a seminiferous tubule at stages I–V. Dashed squares are magnified in the right-hand panels. Scale bars: 100 μm. (E) Immunostaining of H1t and Cleaved Caspase-3 on testicular sections at 41 dpp. Dashed squares are magnified in the right-hand panels. White arrowheads show Cleaved Caspase-3 positive cells. Nuclei were counterstained with DAPI. Scale bars: 100 μm. Population of H1t-positive seminiferous tubules containing Cleaved Caspase-3 positive cells are shown as the mean ± SEM for three independent pairs of Chek1ctrl and Chek1cKO mice. Total numbers of analyzed seminiferous tubules are indicated in the panels. n.s.: not significant (Chi-squared test).
To determine whether abnormal meiotic progression in the mutants results from an increase of apoptotic cells, we examined apoptotic cell death. We evaluated H1t-positive tubules by immunostaining against Cleaved Caspase-3, which detects cells undergoing apoptosis. To our surprise, the frequency of Cleaved Caspase-3-positive tubules did not increase among H1t-positive mutant tubules that undergo spermatogenic differentiation (Fig. 3E). This result suggests that abnormal meiotic progression in the mutants cannot be attributed to increased levels of apoptotic cell death.
To further elucidate the meiotic phenotype that results from Chek1 deficiency, we immunostained chromosome spreads from Ddx4-Cre-ERT2: Chek1cKO testes under the 20-days-3-weeks condition. We used an anti-SYCP3 antibody to detail the substages of meiotic prophase, as these can be precisely distinguished based on the status of chromosome synapsis (41). Consistent with the results of testicular sections, the relative populations of leptotene and early zygotene spermatocytes increased in Chek1cKO testes versus control testes, while the relative populations of mid- and late-pachytene spermatocytes decreased (Fig. 4A). Further, we observed that the removal of γH2AX from autosomes was delayed relative to SYCP3-based meiotic staging in the Chek1cKO as compared to controls. In normal meiosis, γH2AX accumulated throughout the whole nuclei of leptotene and early zygotene spermatocytes, during the initial formation of recombination-associated DSBs (Fig. 4B). Then, as spermatocytes progressed into the late-zygotene stage, γH2AX accumulation transitioned from a nucleus-wide, diffuse signal to a concentrated accumulation on the chromatin of unsynapsed chromosome axes (Fig. 4B). At the onset of the early pachytene stage, autosome synapsis was completed and γH2AX accumulation was largely confined to the unsynapsed sex chromosomes. γH2AX accumulation concentrated on the sex chromatin during the progression from the pachytene stages to the diplotene stages. Interestingly, the Chek1-deficient phenotype deviates from this regimen in surprising ways: from a broad period of spermatogenic development beginning in the late-zygotene stage and persisting to the mid-pachytene stage, γH2AX remained on autosomes in the Chek1cKO far longer than in Chek1 controls (Fig. 4B).
Figure 4.

CHEK1 coordinates meiotic progression and the removal of γH2AX from autosomes. (A) Stage populations during prophase are shown as the mean ± SEM for five independent pairs of Chek1ctrl and Chek1cKO mice at 41 dpp. *P < 0.05, **P < 0.01, n.s.: not significant (unpaired t-test). (B) Immunostaining of SYCP3 and γH2AX on meiotic chromosome spreads at 41 dpp. Distribution patterns of γH2AX were classified into three types by criteria described in the figure. Scale bars: 10 μm. (C) Population of each γH2AX pattern in each stage of meiotic prophase for three independent pairs of Chek1ctrl and Chek1cKO mice. **P < 0.01, ***P < 0.001. n.s.: not significant (Chi-squared test). (D) Integrative analyses of spermatocytes classified by the distribution of γH2AX on autosomes in different stages of prophase from three independent pairs of Chek1ctrl and Chek1cKO mice. Total numbers of analyzed nuclei are indicated in the panels.
To understand the significance of persistent γH2AX accumulation on the autosomes of the Chek1cKO, we pursued an experimental strategy to quantify γH2AX accumulation in the context of prophase development. To that end, we judged the stages of prophase by chromosome synapsis status via anti-SYCP3 immunostaining, as we performed previously (41), and we organized the accumulation patterns of γH2AX on autosomes into three categories, termed patterns I, II and III (Fig. 4B). We define pattern I as broad, uninterrupted γH2AX accumulation throughout the nucleus (Fig. 4B: early zygotene in the Chek1ctrl). Pattern II is defined as the partial accumulation of γH2AX, in which large disconnected regions of accumulation coincide with accumulation on autosome axes (Fig. 4B: late zygotene and early pachytene in the Chek1ctrl). Pattern III is defined by little or no γH2AX accumulation on autosomes (Fig. 4B: mid pachytene and late pachytene in the Chek1ctrl; the dynamics of γH2AX accumulation on the sex chromatin is described separately below). In the Chek1cKO, we observed a significant number of spermatocytes with delayed patterns of γH2AX removal relative to SYCP3 patterns in the late-zygotene to mid-pachytene stages (Fig. 4B and C). For example, although mutant spermatocytes entered the late-zygotene stage, as defined by the status of chromosome axes, we observed persistent nucleus-wide γH2AX accumulation similar to the preceding leptotene and early zygotene stages (Fig. 4B and C). Further, we observed broad, ectopic γH2AX accumulation in early- and mid-pachytene spermatocytes of the Chek1cKO, in contrast to the slight accumulation or general absence of γH2AX on autosomal chromatin in the Chek1ctrl (Fig. 4B and C). These data uncover a form of discoordination in the early stages of prophase: The accumulation status of autosomal γH2AX is uncoupled from the synapsis status of chromosome axes. Thus, these results suggest that CHEK1 plays a role in the timely cessation of autosomal DDR signaling during meiotic prophase.
Because we observed a mutant phenotype in which the synapsis status of chromosome axes is uncoupled from the termination of autosomal γH2AX signaling (Fig. 4B and C), we plotted the distribution of autosomal γH2AX accumulation with respect to prophase stage distributions to illuminate the overall phenotype throughout prophase (Fig. 4D). These data suggest a longer duration for leptotene and zygotene spermatocytes in mutants versus controls, while the duration of mid-pachytene spermatocytes was shortened in the mutants. Notably, Chek1cKO and control spermatocytes were able to proceed into the diplotene stage at comparable frequencies (Fig. 4A). Together, these data suggest that the reduction in pachytene spermatocyte populations in the Chek1cKO is not from cell death, since apoptotic cell death did not increase in the mutants (Fig. 3E), but instead results from a decrease in the duration of the mid- and late-pachytene stages. This raises two intriguing possibilities: (1) CHEK1 functions in a checkpoint to attenuate γH2AX signals from autosomes to ensure the duration of the pachytene stage, and (2) there are separate mechanisms at work in spermatocytes that can support the progression through prophase in the absence of CHEK1.
Notably, in spite of a general discoordination of prophase, we observed comparable numbers of MLH1 foci, which represent sites of crossover recombination, between Chek1cKO and control spermatocytes at the mid- and late-pachytene stages (Fig. 5A). Therefore, we excluded the possibility that CHEK1 regulates meiotic recombination, indicating that abnormal meiotic progression in the mutants is not due to defects in meiotic recombination. To determine whether delayed retention of γH2AX signals on autosomes in the mutants is associated with persistent DSBs in the mutants, we immunostained against RAD51, a recombinase that accumulates at sites of persistent DSBs. We found that the number of RAD51 foci on autosomes and sex chromosomes was comparable at the mid-pachytene stage of mutants and controls, and retention of γH2AX signals occurred independent of RAD51 foci (Fig. 5B). These results further suggest that CHEK1 does not function in meiotic recombination, and that CHEK1 functions specifically in the removal of γH2AX from autosomes independent of persistent DSBs in pachytene spermatocytes.
Figure 5.
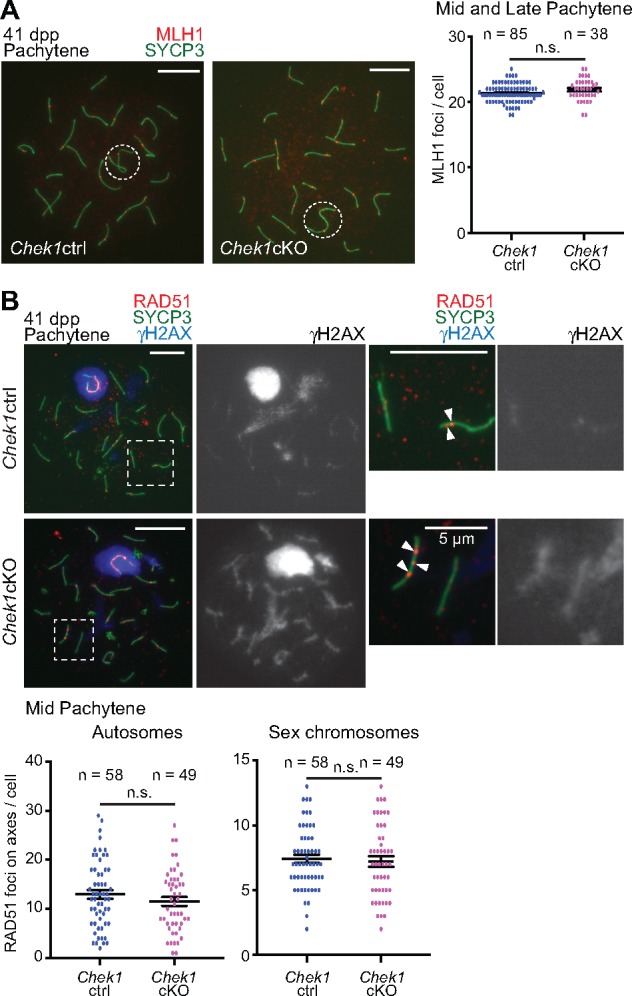
CHEK1 function is independent of meiotic recombination. (A) Immunostaining of SYCP3 with MLH1 on meiotic chromosome spreads at 41 dpp. Dashed circles indicate the XY body. Scale bars: 10 μm. Distributions of data from three independent pairs of Chek1ctrl and Chek1cKO mice are shown as dots. Mean ± SEM. Total numbers of analyzed nuclei are indicated in the panels. n.s.: not significant (unpaired t-test). (B) Immunostaining of SYCP3 with RAD51 and γH2AX on meiotic chromosome spreads at 41 dpp, and the number of RAD51 foci on axes. Dashed squares in left panels are magnified in the two right panels. Arrowheads indicate RAD51 foci. Scale bars: 10 μm unless shown otherwise in the panel. Distributions of data from three independent pairs of Chek1ctrl and Chek1cKO mice are shown as dots. Mean ± SEM. Total numbers of analyzed nuclei are indicated in the panels. n.s.: not significant (unpaired t-test).
CHEK1 does not directly regulate meiotic sex chromosome inactivation
Transcriptional silencing of the male sex chromosomes takes place through a process known as MSCI and is essential for meiotic progression (15,16). In cytological observations, the initiation of MSCI is tightly coupled with the accumulation of γH2AX on the chromosome-wide domain of the sex chromosomes in early pachytene spermatocytes. Although the domains of the sex chromosomes are indefinite at the early pachytene stage, sex chromosomes form a distinct, round-shaped domain termed the XY body (or sex body) that is protruded from the rest of the nucleus in the mid-pachytene stage. The eventual establishment of an XY body in Chek1 mutants (Fig. 4B: late pachytene in Chek1cKO) suggests that CHEK1 is not required for the initiation of MSCI. This is intriguing because ATR catalyzes the broad accumulation of γH2AX on the sex chromatin, an essential event in the initiation of MSCI (11). Additionally, in somatic cells, ATR and CHEK1 are functionally linked (42). Therefore, in contrast to the somatic DDR, the functions of ATR and CHEK1 seem to be uncoupled in the initiation of MSCI. However, since DDR signaling takes place through a feedforward protein network, it remains possible that CHEK1 directs separate events in ATR-dependent DDR signaling on the sex chromosomes. Thus, to clarify the role of CHEK1 in the regulation of the sex chromosomes, we examined the sex chromosome phenotype in the Chek1cKO.
In normal meiosis, γH2AX spreads through the chromosome-wide domain of the sex chromosomes as the late-zygotene stage transitions into the early pachytene stage. In the early pachytene stage, γH2AX spreading throughout the chromosome-wide domain of the sex chromosomes was completed, although the shape of the sex chromosome domain is indefinite, having not yet formed a distinct, round-shaped domain. We term this γH2AX accumulation pattern on the sex chromosomes ‘semi-ordered’ (SO: Fig. 6). After the establishment of the XY body at the mid-pachytene stage, the accumulation of γH2AX appeared to have a distinct boundary, and we designate this as ‘ordered’ γH2AX accumulation (O: Fig. 6). In Chek1cKO spermatocytes, the semiordered γH2AX accumulation pattern persisted into the mid-pachytene stage at a significantly higher proportion than in littermate controls (Fig. 6A and B). Intriguingly, upon entry into the late-pachytene stage, we observed that the majority of Chek1cKO spermatocytes had an ordered γH2AX domain, suggesting that the ordered XY body is formed by this point in Chek1-deficient prophase. Because of the shift in prophase population distribution in the mutants (Fig. 4A), we plotted the distribution of γH2AX accumulation patterns on sex chromosomes in the context of our population distribution data (Fig. 6C). The delayed acquisition of an ordered γH2AX domain in mutant mid-pachytene spermatocytes correlates with a relative reduction in the population of mid-pachytene spermatocytes. Thus, we infer that a checkpoint is lost or weakened in Chek1-deficient spermatocytes, and the decoupling of an ordered γH2AX domain from the mid-pachytene stage is a manifestation of this. Because there is a delay in the removal of autosomal γH2AX in the mid-pachytene stage, the delay in the efficient formation of ATR-dependent γH2AX on the sex chromosomes could be an indirect effect. It is possible that the coordinated removal of γH2AX from autosomes ensures the timely establishment of an ordered γH2AX domain on the sex chromosomes in the early to mid-pachytene stage transition.
Figure 6.
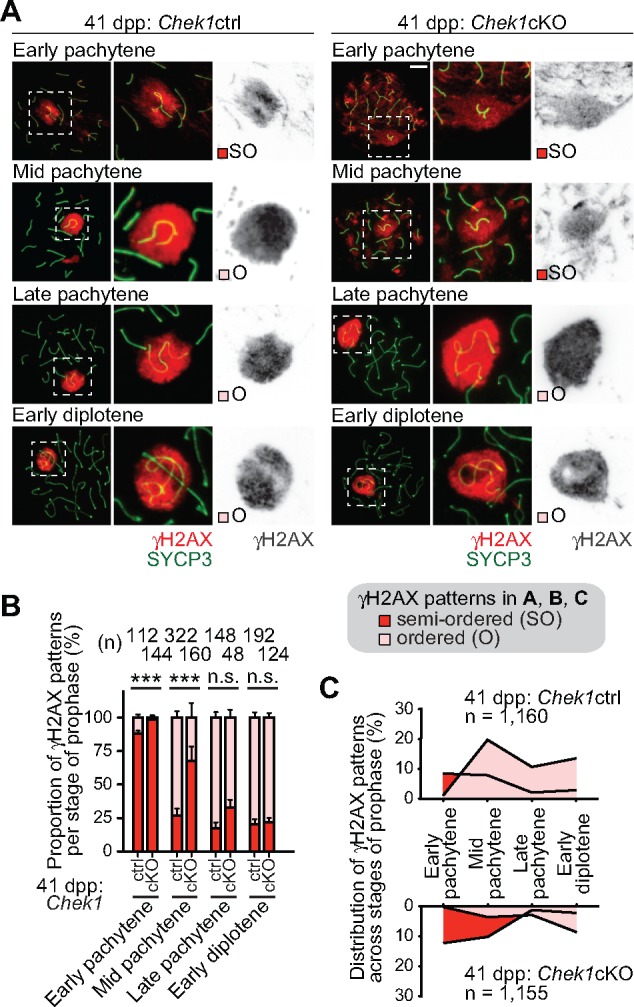
Loss of Chek1 leads to a delay in the establishment of ATR-dependent γH2AX in MSCI. (A) Immunostaining of SYCP3 and γH2AX on meiotic chromosome spreads. The γH2AX domain gradually becomes confined to sex chromatin; the boundary of the γH2AX domain is initially indeterminate (semiordered: SO) and, later, becomes tightly bounded (ordered: O). Scale bars: 10 μm. (B) Population of each γH2AX pattern in each substage for three independent pairs of Chek1ctrl and Chek1cKO mice. **P < 0.01, ***P < 0.001. n.s.: not significant (Chi-squared test). (C) Integrative analyses of spermatocytes classified by the distribution of γH2AX on the sex chromatin in stages of prophase from three independent pairs of Chek1ctrl and Chek1cKO mice. Total numbers of analyzed nuclei are indicated in the panels. ***P < 0.001. n.s.: not significant (Chi-squared test).
To test whether CHEK1 directly regulates the ATR-dependent DDR pathway in MSCI, we examined the localization of key DDR factors on the sex chromosomes. BRCA1, which regulates the localization of ATR (43) and amplifies ATR signaling along the unsynapsed sex chromosome axes (44), evinced normal localization on the unsynapsed sex chromosome axes of the Chek1cKO (Fig. 7A). Likewise, ATR and its co-activator TOPBP1 accumulated normally on both the unsynapsed axes and XY chromatin in the Chek1cKO, consistent with normal meiosis (Fig. 7A). MDC1, a binding partner of γH2AX that functions to initiate MSCI by spreading ATR-dependent γH2AX through the sex chromatin (16), also accumulated normally on sex chromatin in the Chek1cKO (Fig. 7A). The establishment of ATR, TOPBP1 and MDC1 on the XY body suggests that CHEK1 does not directly regulate ATR-mediated signaling on the sex chromosomes. So while CHEK1 interacts with BRCA1, ATR and TOPBP1 in the somatic DDR (18,45–47), the depletion of CHEK1 did not disturb MSCI and, therefore, the function of CHEK1 in MSCI is distinct from that in the somatic DDR.
Figure 7.

CHEK1 is dispensable for the regulation of DDR factors that establish MSCI, and a model of CHEK1 action during meiosis. (A) Immunostaining of SYCP3 with the indicated DDR factors, which are involved in the establishment of MSCI, on meiotic chromosome spreads at 41 dpp. Dashed circles indicate the XY body. Scale bars: 10 μm. (B) Model of action for CHEK1 during meiosis. CHEK1 functions in the context of the ATM-mediated DDR, and phospho-CHEK1 negatively regulates ATM-dependent γH2AX.
Since CHEK1 indirectly regulates DDR signaling in MSCI, we anticipated that CHEK1 deficiency does not disturb downstream epigenetic changes that are regulated by DDR signaling (41,48). To test this prediction, we performed immunostaining of repressive and active histone modifications in chromosome spreads obtained from Chek1cKO and littermate controls under the 20-days-3-weeks condition. On the XY body, the repressive histone modification of dimethylation of histone H3 at Lysine 9 (H3K9me2) is regulated by the Fanconi anemia protein network (41,49), while another repressive modification, ubiquitination of histone H2A at Lysine 119 (H2AK119ub), is suppressed by the germline-specific Polycomb protein SCML2 (36,50). Confirming our hypothesis, CHEK1 deficiency did not affect the distribution of H3K9me2 and H2AK119ub on the sex chromosomes (Supplementary Material, Fig. S6A and B), suggesting that the function of CHEK1 in MSCI is separated from the functions of the Fanconi anemia network and SCML2. Next, we examined the accumulation of an active modification, dimethylation of histone H3 at Lysine 4 (H3K4me2), which is dependent on the DDR protein RNF8 in a mechanism that later gives rise to essential gene activation in postmeiotic spermatids (48). In the Chek1cKO, the establishment of H3K4me2 was comparable to that of controls (Supplementary Material, Fig. S6C), further confirming our hypothesis. In somatic cells, CHEK1 represses transcription through the suppression of H3K9 acetylation (H3K9ac) (51). However, CHEK1 depletion did not disturb the distribution of H3K9ac during meiosis (Supplementary Material, Fig. S6D). Therefore, we conclude that CHEK1 does not regulate the deposition of these histone modifications, lending further evidence that CHEK1 indirectly regulates the DDR signaling involved in MSCI.
Taken together, we conclude that CHEK1 plays important and revelatory roles in the regulation of meiotic progression but is not essential for MSCI. Our study reveals distinct roles for CHEK1 in both the mitotic and meiotic phases of male germ cell development, and these findings unravel how a shifting, dynamic DDR/checkpoint network ensures the integrity of the male germline.
Discussion
CHEK1 is required for germ cell proliferation
In this study, we demonstrate that CHEK1 has two critical functions in the male germline: one is in promoting mitotic proliferation and the other is in the regulation of stage progression in meiotic prophase. Ddx4-Cre-mediated Chek1 deletion causes near-complete loss of germ cells when prospermatogonia resume proliferation. Cre- induction by Ddx4-Cre-ERT2 further demonstrates that CHEK1 is important for the maintenance of spermatogonia. Since CHEK1 monitors DNA synthesis in S-phase and functions in the G2/M checkpoint in mitosis, and since CHEK1 deficiency induces apoptosis (18,19), it is reasonable to assume that the essential function of CHEK1 is in the S-phase and G2/M checkpoint of proliferating germ cells. While near-complete loss of germ cells occurs when prospermatogonia resume proliferation in Ddx4-Cre mutants, the inducible deletion of CHEK1 by Ddx4-Cre-ERT2 in spermatogonia leads to a reduction in the numbers of spermatogonia while allowing differentiating spermatocytes to enter meiotic stages. These results emphasize that mitotically proliferating prospermatogonia and spermatogonia may have a differential susceptibility to CHEK1 deficiency. Because the germline has a lower mutation rate than that of somatic cells (1,2), it is possible that the mitotic phase of germ cells is highly susceptible to DDR mutations due to the need to maintain the integrity of the germline. Therefore, one possible interpretation is that, amidst the stages of mitotic proliferation, the proliferation of prospermatogonia is a critical window for monitoring the integrity of the germline.
Notably, a specific function of the DDR factor ATM has been described in undifferentiated spermatogonia (52). ATM deficiency causes an accumulation of DDR signaling, leading to cell cycle arrest at the G1 phase and an increase in apoptosis in undifferentiated spermatogonia (52). Since we did not observe cell cycle arrest in Chek1 mutant spermatogonia, the function of CHEK1 could be distinct from that of ATM in undifferentiated spermatogonia. Indeed, CHEK1 is regulated by ATR rather than ATM in somatic cells (53).
CHEK1 controls meiotic progression and the duration of meiotic prophase
Since, in the somatic DDR, CHEK1 is a downstream factor in the ATR pathway, we initially anticipated that CHEK1 would have a specific function in MSCI, which is regulated by an ATR-dependent DDR pathway. To our surprise, depletion of CHEK1 did not directly disturb MSCI. Thus, the functions of ATR and CHEK1 are uncoupled in MSCI. Because the initiation of MSCI is essential for meiotic progression (15,16), the normal initiation of MSCI in Chek1 mutants is one possible reason why mutant cells eventually persist through meiosis.
However, conditional deletion of Chek1 with the Ddx4-Cre-ERT2 transgene disrupted the seminiferous cycle and induced abnormal meiotic progression. This phenotype differed between the 10-days-3-weeks and 20-days-3-weeks conditions. Under the 10-days-3-weeks condition, mid-to-late-pachytene spermatocytes, along with subsequent germ cells, were apparently normal due to incomplete deletion of CHEK1, as described in Supplementary Material, Fig. S5B. Therefore, earlier spermatocytes, in which CHEK1 was depleted, were relatively decreased. Under the 20-days-3-weeks condition, CHEK1 depletion had sufficient time to penetrate all stages of meiotic prophase, as described in Supplementary Material, Fig. S5C. Thus, under this condition, we were able to observe a clear meiotic phenotype. The loss of CHEK1, under the 20-days-3-weeks condition, led to a delayed progression through the leptotene and zygotene stages, followed by rapid progression through the mid-pachytene stage. In addition, γH2AX persisted longer on autosomes in Chek1 mutants.
Although the function of ATR on autosomes has not been determined, we infer that CHEK1 is activated downstream of ATR on autosomes (Fig. 7B). In the leptotene stage, ATM mediates γH2AX in response to SPO11-dependent DSBs (9,10) and, subsequently, ATR mediates γH2AX on the unsynapsed chromatin of autosomes in the zygotene stage (11). At this stage, we speculate that CHEK1 is activated through phosphorylation by ATR, and activated CHEK1 has either a direct or indirect role in the attenuation of γH2AX signaling on autosomes (Fig. 7B).
Because depletion of CHEK1 delays both the removal of γH2AX from autosomes and general meiotic progression through prophase, as judged by the synapsis status of chromosomes, we propose that autosomal DDR signaling is a key determinant of the duration of each stage of meiotic prophase independent of meiotic recombination. Delayed meiotic progression through the leptotene and zygotene stages could be caused directly by abnormal γH2AX signals on autosomes. This could be through a CHEK1-dependent checkpoint, as discussed below. An alternative, though not mutually exclusive, possibility is that the delayed establishment of γH2AX on the sex chromosomes may regulate the duration of the pachytene stage. However, since abnormal accumulation of autosomal γH2AX disrupts γH2AX signaling on the sex chromosomes (54), the delayed establishment of γH2AX on the sex chromosome in Chek1 mutants may be an indirect consequence of prolonged γH2AX accumulation on autosomes.
It is an intriguing possibility that CHEK1 may have a role in checkpoint regulation in meiosis, since CHEK1 functions in the G2/M DNA damage checkpoint of somatic cells (55). Of course, any checkpoint-related functions of CHEK1 in male meiosis are likely to be tailored to the requirements of this specific process. Chek1 mutants are notable for a delay in the removal of γH2AX from autosomes in the pachytene stage, concomitant with prolonged leptotene and zygotene stages and a more rapid pachytene stage. These phenotypes could all be due to the abrogation of a CHEK1-dependent checkpoint. First, CHEK1 could serve to delay progression from zygotene into early pachytene until γH2AX is largely removed from the autosomes. Thus, the effect of a defect in this checkpoint would be to delay the leptotene and zygotene stages since there is insufficient removal of γH2AX from autosomes. Indeed, we observe that there is recombination-independent retention of γH2AX on autosomes in spermatocytes of Chek1 mutants in which autosome synapsis is complete, as indicated by SYCP3 immunostaining. Why the recombination-independent removal of γH2AX from autosomes is coordinated with entry into the pachytene stage is an important topic for further investigation. One possibility, though, is that the proper organization of γH2AX on the XY body in the mid-pachytene stage is a consequence of a CHEK1-dependent checkpoint at the zygotene-pachytene transition. Additionally, it should be noted that, despite the delayed removal of γH2AX from Chek1 mutant autosomes, the numbers of RAD51 foci and MLH1 foci were consistent between mutants and controls. These results suggest that progression from the pachytene stage into subsequent stages occurs independent of crossover formation in meiotic recombination.
Consistent with the original definition established in somatic cells (56), cell cycle checkpoints, such as our proposed CHEK1-dependent checkpoint in meiosis, serve to coordinate cell cycle events so that they occur in the proper order. The fact that CHEK1 is not required for the progression of male meiosis is consistent with it playing a role in the coordinated timing of events, rather than having a direct role in events such as the removal of γH2AX from autosomes and the completion of synapsis. It should be pointed out that there could be other events during meiosis that are regulated by a CHEK1-dependent checkpoint which remain to be discovered. Elucidating these events should eventually illuminate how the DDR ensures the successful completion of meiosis.
Together, our results unravel functions of CHEK1 during spermatogenesis, both in mitotically cycling and meiotic phases. In particular, the meiotic function of CHEK1 is distinct from the role of ATR. A promising future direction is to elucidate the distinct pathways regulated by CHEK1 in both the mitotically cycling and meiotic phases. Because meiotic progression eventually occurs in the absence of CHEK1, another important future direction is to determine the identity of putative mechanisms that facilitate meiotic progression in the absence of CHEK1. Elucidation of these processes is likely to illuminate how DDR/checkpoint pathways act to maintain germline integrity.
Materials and Methods
Mice
Chek1 flox/flox mice (Chek1F/F), in which exon 2 was targeted as the locus of deletion (18), were obtained (Jackson Laboratory; #019520). We used two separate Ddx4-Cre transgenic mouse models to induce Chek1 deletion. The first was Ddx4-Cre mice, in which germ cell-specific Cre is active in the embryo at embryonic days 15–18 (33) (Jackson Laboratory; #006954). To obtain the conditional knockout Ddx4-Cre: Chek1F/- (Chek1cKO) mice, males with the genotype of Ddx4-Cre: Chek1+/- were crossed with females with the genotype of Chek1F/F. This is because the Ddx4-Cre allele must be transmitted from the paternal allele to generate mice with a germ cell-specific conditional deletion. Male littermates with Chek1F/- or Chek1F/+ were used as controls (Chek1ctrl). As described in a previous report (20), the Chek1 null allele was confirmed by using the following primer set: F1 (5′-ACC TGC CCG CAA CTC CCT TTC-3′) and R2 (5′-TGC AAC AGC TTC AGT TAT TC-3′).
The second Ddx4-Cre transgenic mouse model took advantage of the Ddx4-Cre-ERT2 construct, which enabled us to control germ cell-specific Cre activation via treatment with tamoxifen (38) (Jackson Laboratory; #024760). To induce Chek1 deletion, all males with Ddx4-Cre-ERT2: Chek1F/F at 21 dpp were treated with 2 mg of tamoxifen dissolved in 100 μl of corn oil for three consecutive days by intraperitoneal injection. Control males with the genotype of Chek1F/F were also treated with the same tamoxifen regimen. Mice treated with tamoxifen were harvested at either day 10 or day 20 after the first tamoxifen injection. Highly efficient Cre-mediated excision of Chek1 floxed alleles was confirmed by PCR for all of the mutant mice that we examined. All experimental work was approved by the Institutional Animal Care and Use Committee protocol no. IACUC2015–0032.
Antibodies
All antibodies used in this study are described in Supplementary Material, Table S1.
Western blotting
Testis pieces were homogenized in RIPA buffer (50 mm Tris–HCl, pH 7.5; 150 mm NaCl; 0.1% SDS; 1% Triton -100; 1% sodium deoxycholate) containing a protease inhibitor cocktail (Roche; 11697498001) and a phosphatase inhibitor cocktail (Sigma; P0044). Then, for CHEK1 detection, 20 or 40 μg of proteins were separated by electrophoresis with a 10% SDS-PAGE gel. The proteins were transferred onto a PVDF membrane (EMD Millipore; IPVH00010) and western blotting was performed. The membranes were blocked with StartingBlock™ T20 (TBS) Blocking Buffer (ThermoFisher Scientific; 37543) for 30 min at room temperature and then incubated with primary antibodies at 4°C overnight. Membranes were then incubated with secondary antibodies conjugated to HRP (Abcam; ab131366 or ab131368) for 1 h at room temperature, and bands were visualized using an ECL kit (EMD Millipore; WBKLS0500).
Immunofluorescence of meiotic chromosome spreads
Meiotic chromosome spreads were prepared as described previously (41,57). The slides that preserves relative 3D chromatin structure of germ cells (3D slides) were prepared as described previously (29,30). For immunostaining experiments, surface spreads were washed in PBST for 30 min and blocked with antibody dilution buffer (0.15% BSA, 0.1% Tween 20 in PBS) for 30 min. Primary antibodies were added to surface spreads and incubated overnight in humid chambers at room temperature. The slides were incubated with secondary antibodies conjugated to fluorophores for 1 h at room temperature in humid chambers in darkness. Slides were mounted with #1.5 thickness coverslips (ThermoFisher Scientific; 12–544G) using ProLong Gold (ThermoFisher Scientific; P36930) after incubation in PBS containing DAPI (1 µg/ml) for 5 min in darkness. Images were obtained with an ECLIPSE Ti-E microscope (Nikon) equipped with a Zyla 5.5 sCMOS camera (Andor Technology) and an 100× CFI Apochromat TIRF oil immersion objective NA 1.4 (Nikon), and were processed with NIS-Elements (Nikon), Photoshop (Adobe) and Illustrator (Adobe).
Analyses of meiotic staging based on anti-SYCP3 and anti-γH2AX immunostaining
Sample images of spermatocytes stained with anti-SYCP3 and anti-γH2AX antibodies were blinded and manually scored with the ImageJ (National Institutes of Health) processing package Fiji (58). Sample images were blinded, scored, unblinded and sorted through the workflow described in (41). Data were imported to Prism 6 (GraphPad) for statistical analyses. Graphs of staging and accumulation patterns were composed with Excel (Microsoft), Prism 6 and Illustrator (Adobe).
Hematoxylin and eosin staining and immunohistochemistry
For the preparation of testicular paraffin blocks, testes were fixed with 4% paraformaldehyde (PFA) at 4°C overnight. Testes were dehydrated and embedded in paraffin. For histological analyses, paraffin sections at 6 μm thick were deparaffinized. H & E staining was performed as described previously (36). For immunostaining, sections were autoclaved in Target Retrieval Solution (DAKO; S-1700) at 121°C for 20 min. The sections were blocked with Blocking One Histo (Nacalai USA; 06349–64) for 10 min at room temperature, and then incubated with primary antibodies at 4°C overnight. The resulting signals were detected by incubation with secondary antibodies conjugated to fluorophores. Sections were counterstained with DAPI (1 μg/ml). Images were obtained by confocal laser scanning microscope (A1R, Nikon) and processed with NIS-Elements (Nikon), Image J (National Institutes of Health) and Photoshop (Adobe).
Immunofluorescence microscopy of cryosections
Testes were fixed in 4% PFA overnight at 4°C prior to processing for cryosections. For histological analyses, samples were washed several times with PBTx (PBS containing 0.1% Triton X-100) before incubation in PBTx for 10 min at room temperature. Then, sections at 20 μm thick were blocked with Blocking One Histo (Nacalai USA; 06349–64) for 10 min at room temperature before incubation with primary antibodies at 4°C overnight. For negative controls, sections were incubated in PBS without primary antibodies at 4°C overnight. The resulting signals were detected by incubation with secondary antibodies conjugated to fluorophores. Sections were counterstained with DAPI (1 μg/ml). Images were obtained by confocal laser scanning microscope (A1R, Nikon) and processed with NIS-Elements (Nikon), Image J (National Institutes of Health) and Photoshop (Adobe).
Statistical analyses
Means were compared using unpaired two-tailed Student’s t tests for experiments involving two groups. Pearson’s chi-square test was used to identify categorical differences in the accumulation of γH2AX on autosomes (Fig. 4D) and on the sex chromatin (Fig. 6C). Pearson’s chi-square test was also used to identify categorical differences in the analyses of seminiferous tubes, including the frequency of Cleaved Caspase-3 positive tubules, between groups. Excel (Microsoft) and Prism 6 (GraphPad) were used for these analyses.
Supplementary Material
Supplementary Material is available at HMG online.
Supplementary Material
Acknowledgements
We thank members of the Namekawa laboratory for discussion and helpful comments, and we thank Mary Ann Handel for providing the anti-H1t antibody and Junjie Chen for providing the anti-TOPBP1 antibody.
Conflict of Interest statement. None declared.
Funding
This work was supported by the Research Grant (FY13–510) from the March of Dimes Foundation to S.H.N., NIH Grants R01HD089932 to Q.P., and R01GM098605 to S.H.N.
References
- 1. Drost J.B., Lee W.R. (1995) Biological basis of germline mutation: comparisons of spontaneous germline mutation rates among drosophila, mouse, and human. Environ. Mol. Mutagen., 25, 48–64. [DOI] [PubMed] [Google Scholar]
- 2. Lynch M. (2010) Rate, molecular spectrum, and consequences of human mutation. Proc. Natl. Acad. Sci. U. S. A., 107, 961–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gunes S., Al-Sadaan M., Agarwal A. (2015) Spermatogenesis, DNA damage and DNA repair mechanisms in male infertility. Reprod. Biomed. Online, 31, 309–319. [DOI] [PubMed] [Google Scholar]
- 4. Ciccia A., Elledge S.J. (2010) The DNA damage response: making it safe to play with knives. Mol. Cell, 40, 179–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Polo S.E., Jackson S.P. (2011) Dynamics of DNA damage response proteins at DNA breaks: a focus on protein modifications. Genes. Dev., 25, 409–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kim S., Peterson S.E., Jasin M., Keeney S. (2016) Mechanisms of germ line genome instability. Semin. Cell. Dev. Biol., 54, 177–187. [DOI] [PubMed] [Google Scholar]
- 7. Hunter N. (2015) Meiotic recombination: the essence of heredity. Cold. Spring. Harb. Perspect. Biol., 7, a016618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gray S., Cohen P.E. (2016) Control of meiotic crossovers: from double-strand break formation to designation. Annu. Rev. Genet., 50, 175–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Barchi M., Mahadevaiah S., Di Giacomo M., Baudat F., de Rooij D.G., Burgoyne P.S., Jasin M., Keeney S. (2005) Surveillance of different recombination defects in mouse spermatocytes yields distinct responses despite elimination at an identical developmental stage. Mol. Cell. Biol., 25, 7203–7215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bellani M.A., Romanienko P.J., Cairatti D.A., Camerini-Otero R.D. (2005) SPO11 is required for sex-body formation, and Spo11 heterozygosity rescues the prophase arrest of Atm-/- spermatocytes. J. Cell. Sci., 118, 3233–3245. [DOI] [PubMed] [Google Scholar]
- 11. Royo H., Prosser H., Ruzankina Y., Mahadevaiah S.K., Cloutier J.M., Baumann M., Fukuda T., Hoog C., Toth A., de Rooij D.G.. et al. (2013) ATR acts stage specifically to regulate multiple aspects of mammalian meiotic silencing. Genes. Dev., 27, 1484–1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Turner J.M.A. (2007) Meiotic sex chromosome inactivation. Development, 134, 1823–1831. [DOI] [PubMed] [Google Scholar]
- 13. Ichijima Y., Sin H.S., Namekawa S.H. (2012) Sex chromosome inactivation in germ cells: emerging roles of DNA damage response pathways. Cell. Mol. Life. Sci., 69, 2559–2572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Inagaki A., Schoenmakers S., Baarends W.M. (2010) DNA double strand break repair, chromosome synapsis and transcriptional silencing in meiosis. Epigenetics, 5, 255–266. [DOI] [PubMed] [Google Scholar]
- 15. Fernandez-Capetillo O., Mahadevaiah S.K., Celeste A., Romanienko P.J., Camerini-Otero R.D., Bonner W.M., Manova K., Burgoyne P., Nussenzweig A. (2003) H2AX is required for chromatin remodeling and inactivation of sex chromosomes in male mouse meiosis. Dev. Cell, 4, 497–508. [DOI] [PubMed] [Google Scholar]
- 16. Ichijima Y., Ichijima M., Lou Z., Nussenzweig A., Camerini-Otero R.D., Chen J., Andreassen P.R., Namekawa S.H. (2011) MDC1 directs chromosome-wide silencing of the sex chromosomes in male germ cells. Genes. Dev., 25, 959–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sanchez Y., Wong C., Thoma R.S., Richman R., Wu Z., Piwnica-Worms H., Elledge S.J. (1997) Conservation of the Chk1 checkpoint pathway in mammals: linkage of DNA damage to Cdk regulation through Cdc25. Science (New York, N.Y.), 277, 1497–1501. [DOI] [PubMed] [Google Scholar]
- 18. Liu Q., Guntuku S., Cui X.S., Matsuoka S., Cortez D., Tamai K., Luo G., Carattini-Rivera S., DeMayo F., Bradley A.. et al. (2000) Chk1 is an essential kinase that is regulated by Atr and required for the G(2)/M DNA damage checkpoint. Genes. Dev., 14, 1448–1459. [PMC free article] [PubMed] [Google Scholar]
- 19. Takai H., Tominaga K., Motoyama N., Minamishima Y.A., Nagahama H., Tsukiyama T., Ikeda K., Nakayama K., Nakanishi M., Nakayama K. (2000) Aberrant cell cycle checkpoint function and early embryonic death in Chk1(-/-) mice. Genes. Dev., 14, 1439–1447. [PMC free article] [PubMed] [Google Scholar]
- 20. Lam M.H., Liu Q.H., Elledge S.J., Rosen J.M. (2004) Chk1 is haploinsufficient for multiple functions critical to tumor suppression. Cancer Cell, 6, 45–59. [DOI] [PubMed] [Google Scholar]
- 21. Chen L., Chao S.B., Wang Z.B., Qi S.T., Zhu X.L., Yang S.W., Yang C.R., Zhang Q.H., Ouyang Y.C., Hou Y.. et al. (2012) Checkpoint kinase 1 is essential for meiotic cell cycle regulation in mouse oocytes. Cell Cycle (Georgetown, Tex.), 11, 1948–1955. [DOI] [PubMed] [Google Scholar]
- 22. Bolcun-Filas E., Rinaldi V.D., White M.E., Schimenti J.C. (2014) Reversal of female infertility by Chk2 ablation reveals the oocyte DNA damage checkpoint pathway. Science (New York, N.Y, ), 343, 533–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Rinaldi V.D., Bolcun-Filas E., Kogo H., Kurahashi H., Schimenti J.C. (2017) The DNA damage checkpoint eliminates mouse oocytes with chromosome synapsis failure. Mol. Cell, 67, 1026–1036.e1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Pacheco S., Marcet-Ortega M., Lange J., Jasin M., Keeney S., Roig I., Lichten M. (2015) The ATM signaling cascade promotes recombination-dependent pachytene arrest in mouse spermatocytes. PLoS Genet, 11, e1005017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. McCarrey J.R. (2013) Toward a more precise and informative nomenclature describing fetal and neonatal male germ cells in rodents. Biol. Reprod., 89, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Culty M. (2013) Gonocytes, from the fifties to the present: is there a reason to change the name? Biol. Reprod., 89, 1–6. [DOI] [PubMed] [Google Scholar]
- 27. Flaggs G., Plug A.W., Dunks K.M., Mundt K.E., Ford J.C., Quiggle M.R.E., Taylor E.M., Westphal C.H., Ashley T., Hoekstra M.F., Carr A.M. (1997) Atm-dependent interactions of a mammalian chk1 homolog with meiotic chromosomes. Curr. Biol., 7, 977–986. [DOI] [PubMed] [Google Scholar]
- 28. Fedoriw A.M., Menon D., Kim Y., Mu W., Magnuson T. (2015) Key mediators of somatic ATR signaling localize to unpaired chromosomes in spermatocytes. Development, 142, 2972–2980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Namekawa S.H., Lee J.T. (2011) Detection of nascent RNA, single-copy DNA and protein localization by immunoFISH in mouse germ cells and preimplantation embryos. Nat. Protoc., 6, 270–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Namekawa S.H. (2014) Slide preparation method to preserve three-dimensional chromatin architecture of testicular germ cells. J. Vis. Exp., 83, e50819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Smits V.A., Reaper P.M., Jackson S.P. (2006) Rapid PIKK-dependent release of Chk1 from chromatin promotes the DNA-damage checkpoint response. Curr. Biol., 16, 150–159. [DOI] [PubMed] [Google Scholar]
- 32. Zhang Y.W., Otterness D.M., Chiang G.G., Xie W., Liu Y.C., Mercurio F., Abraham R.T. (2005) Genotoxic stress targets human Chk1 for degradation by the ubiquitin-proteasome pathway. Mol. Cell, 19, 607–618. [DOI] [PubMed] [Google Scholar]
- 33. Gallardo T., Shirley L., John G.B., Castrillon D.H. (2007) Generation of a germ cell-specific mouse transgenic Cre line, Vasa-Cre. Genesis, 45, 413–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Culty M. (2009) Gonocytes, the forgotten cells of the germ cell lineage. Birth. Defects. Res. C. Embryo. Today, 87, 1–26. [DOI] [PubMed] [Google Scholar]
- 35. Sorensen C.S., Syljuasen R.G. (2012) Safeguarding genome integrity: the checkpoint kinases ATR, CHK1 and WEE1 restrain CDK activity during normal DNA replication. Nucleic Acids Res., 40, 477–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hasegawa K., Sin H.-S., Maezawa S., Broering T.J., Kartashov A.V., Alavattam K.G., Ichijima Y., Zhang F., Bacon W.C., Greis K.D.. et al. (2015) SCML2 establishes the male germline epigenome through regulation of histone H2A ubiquitination. Dev. Cell, 32, 574–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ventura A., Kirsch D.G., McLaughlin M.E., Tuveson D.A., Grimm J., Lintault L., Newman J., Reczek E.E., Weissleder R., Jacks T. (2007) Restoration of p53 function leads to tumour regression in vivo. Nature, 445, 661–665. [DOI] [PubMed] [Google Scholar]
- 38. John G.B., Gallardo T.D., Shirley L.J., Castrillon D.H. (2008) Foxo3 is a PI3K-dependent molecular switch controlling the initiation of oocyte growth. Dev. Biol., 321, 197–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Russell L.D., Ettlin R.A., SinhaHikim A.P., Clegg E.D. (1990) Histological and Histopathological Evaluation of the Testis. Cache River Press, St Louis, MO, USA. [Google Scholar]
- 40. Oakberg E.F. (1956) Duration of spermatogenesis in the mouse and timing of stages of the cycle of the seminiferous epithelium. Am. J. Anat., 99, 507–516. [DOI] [PubMed] [Google Scholar]
- 41. Alavattam K.G., Kato Y., Sin H.S., Maezawa S., Kowalski I.J., Zhang F., Pang Q., Andreassen P.R., Namekawa S.H. (2016) Elucidation of the Fanconi anemia protein network in meiosis and its function in the regulation of histone modifications. Cell Rep., 17, 1141–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Smits V.A., Gillespie D.A. (2015) DNA damage control: regulation and functions of checkpoint kinase 1. FEBS J., 282, 3681–3692. [DOI] [PubMed] [Google Scholar]
- 43. Turner J.M., Aprelikova O., Xu X., Wang R., Kim S., Chandramouli G.V., Barrett J.C., Burgoyne P.S., Deng C.X. (2004) BRCA1, histone H2AX phosphorylation, and male meiotic sex chromosome inactivation. Curr. Biol., 14, 2135–2142. [DOI] [PubMed] [Google Scholar]
- 44. Broering T.J., Alavattam K.G., Sadreyev R.I., Ichijima Y., Kato Y., Hasegawa K., Camerini-Otero R.D., Lee J.T., Andreassen P.R., Namekawa S.H. (2014) BRCA1 establishes DNA damage signaling and pericentric heterochromatin of the X chromosome in male meiosis. J. Cell. Biol., 205, 663–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Yarden R.I., Pardo-Reoyo S., Sgagias M., Cowan K.H., Brody L.C. (2002) BRCA1 regulates the G2/M checkpoint by activating Chk1 kinase upon DNA damage. Nat. Genet., 30, 285–289. [DOI] [PubMed] [Google Scholar]
- 46. Furuya K., Poitelea M., Guo L., Caspari T., Carr A.M. (2004) Chk1 activation requires Rad9 S/TQ-site phosphorylation to promote association with C-terminal BRCT domains of Rad4TOPBP1. Genes. Dev., 18, 1154–1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Liu S., Bekker-Jensen S., Mailand N., Lukas C., Bartek J., Lukas J. (2006) Claspin operates downstream of TopBP1 to direct ATR signaling towards Chk1 activation. Mol. Cell. Biol., 26, 6056–6064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Sin H.S., Barski A., Zhang F., Kartashov A.V., Nussenzweig A., Chen J., Andreassen P.R., Namekawa S.H. (2012) RNF8 regulates active epigenetic modifications and escape gene activation from inactive sex chromosomes in post-meiotic spermatids. Genes. Dev., 26, 2737–2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kato Y., Alavattam K.G., Sin H.S., Meetei A.R., Pang Q., Andreassen P.R., Namekawa S.H. (2015) FANCB is essential in the male germline and regulates H3K9 methylation on the sex chromosomes during meiosis. Hum. Mol. Genet., 24, 5234–5249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Luo M., Zhou J., Leu N.A., Abreu C.M., Wang J., Anguera M.C., de Rooij D.G., Jasin M., Wang P.J., Schimenti J.C. (2015) Polycomb protein SCML2 associates with USP7 and counteracts histone H2A ubiquitination in the XY chromatin during male meiosis. PLoS Genet., 11, e1004954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Shimada M., Niida H., Zineldeen D.H., Tagami H., Tanaka M., Saito H., Nakanishi M. (2008) Chk1 is a histone H3 threonine 11 kinase that regulates DNA damage-induced transcriptional repression. Cell, 132, 221–232. [DOI] [PubMed] [Google Scholar]
- 52. Takubo K., Ohmura M., Azuma M., Nagamatsu G., Yamada W., Arai F., Hirao A., Suda T. (2008) Stem cell defects in ATM-deficient undifferentiated spermatogonia through DNA damage-induced cell-cycle arrest. Cell Stem Cell, 2, 170–182. [DOI] [PubMed] [Google Scholar]
- 53. Marechal A., Zou L. (2013) DNA damage sensing by the ATM and ATR kinases. Cold Spring Harb. Perspect. Biol., 5, a012716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Mahadevaiah S.K., Bourc'his D., de Rooij D.G., Bestor T.H., Turner J.M., Burgoyne P.S. (2008) Extensive meiotic asynapsis in mice antagonises meiotic silencing of unsynapsed chromatin and consequently disrupts meiotic sex chromosome inactivation. J. Cell. Biol., 182, 263–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Furnari B., Rhind N., Russell P. (1997) Cdc25 mitotic inducer targeted by chk1 DNA damage checkpoint kinase. Science (New York, N.Y.), 277, 1495–1497. [DOI] [PubMed] [Google Scholar]
- 56. Hartwell L.H., Weinert T.A. (1989) Checkpoints: controls that ensure the order of cell cycle events. Science, 246, 629–634. [DOI] [PubMed] [Google Scholar]
- 57. Peters A.H., Plug A.W., van Vugt M.J., de Boer P. (1997) A drying-down technique for the spreading of mammalian meiocytes from the male and female germline. Chromosome Res., 5, 66–68. [DOI] [PubMed] [Google Scholar]
- 58. Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C, Saalfeld S., Schmid B.. et al. (2012) Fiji: an open-source platform for biological-image analysis. Nat. Methods, 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.