

Abstract
Mitochondrial NAD kinase deficiency (NADK2D, OMIM #615787) is a rare autosomal recessive disorder of NADPH biosynthesis that can cause hyper-lysinemia and dienoyl-CoA reductase deficiency (DECRD, OMIM #616034). NADK2 deficiency has been reported in only three unrelated patients. Two had severe, unremitting disease; one died at 4 months and the other at 5 years of age. The third was a 10 year old female with CNS anomalies, ataxia, and incoordination. In two cases mutations in NADK2 have been demonstrated. Here, we report the fourth known case, a 15 year old female with normal intelligence and a mild clinical and biochemical phenotype presumably without DECRD. Her clinical symptoms, which are now stable, became evident at the age of 9 with the onset of decreased visual acuity, bilateral optic atrophy, nystagmus, episodic lower extremity weakness, peripheral neuropathy, and gait abnormalities. Plasma amino acid levels were within normal limits except for mean lysine and proline levels that were 3.7 and 2.5 times the upper limits of normal. Whole exome sequencing (WES) revealed homozygosity for a g.36241900 A>G p. Met1Val start loss mutation in the primary NADK2 transcript (NM_001085411.1) encoding the 442 amino acid isoform. This presumed hypomorphic mutation has not been previously reported and is absent from the v1000GP, EVS, and ExAC databases. Our patient’s normal intelligence and stable disease expands the clinical heterogeneity and the prognosis associated with NADK2 deficiency. Our findings also clarify the mechanism underlying NADK2 deficiency and suggest that this disease should be ruled out in cases of hyperlysinemia, especially those with visual loss, and neurological phenotypes.
Keywords: 2,4 dienoyl-CoA reductase deficiency; hyperlysinemia; NADK2; NADPH; optic atrophy; start-loss codon
1 |. INTRODUCTION
Recent studies have shown that NADK2 produces NADPH which is an essential co-substrate for and also acts as a molecular chaperone that activates and stabilizes alpha-aminoadipic semialdehyde synthase (AASS), the first enzyme in the lysine degradation pathway, and dienoyl-CoA reductase (DECR) (Houten et al., 2014, 2013). NADK2 deficiency (NADK2D, OMIM #615787) results in the accumulation of lysine and C10:2-carnitine (Houten et al., 2014). Previous cases of NADK2 deficiency presented with hyperlysinemia, dienoyl-CoA reductase deficiency (DECRD), and a severe phenotype (see Table 1) (Houten et al., 2014; Roe et al., 1990; Tort et al., 2016). The first patient had failure to thrive, hypotonia, microcephaly, and delayed milestones. She had elevated plasma levels of C10:2-carnitine (2-trans, 4-cisd ecadienoylcarnitine) and died due to respiratory acidosis at age 4 months. DNA analysis was not reported (Roe et al., 1990). The second patient developed metabolic acidosis, elevated plasma levels of C10:2-carnitine, severe encephalopathy, and leukodystrophy. Whole Exome Sequencing (WES) and follow-up in vitro experiments revealed homozygosity for a p.Arg340* mutation in the NADK2 gene on chromosome 5p13.2 which encodes mitochondrial NAD kinase, an essential enzyme for NADP biosynthesis. He ultimately died due to aspiration pneumonia at the age of 5. Both patients had hyperlysinemia and DECR activities were 17% in muscle and 10% in fibroblasts, respectively (Houten et al., 2014). The third patient presented with prenatal central nervous system abnormalities including hypoplasia of the corpus callosum, and colpocephaly (Tort et al., 2016). Post-natal findings included ataxia, microcephaly, intractable seizures, and generalized cerebellar atrophy. Metabolic workup showed elevated lysine levels in blood and cerebrospinal fluid as well as high levels of C10:2-carnitine. WES detected a homozygous splice site mutation in NADK2 (c.956 + 6A>G; p.Trp319Cysfs*21) that causes exon skipping, and is associated with virtual absence of NADK2 mRNA possibly due to nonsense mediated decay (Tort et al., 2016).
TABLE 1.
Characteristics and treatments of NADK2D cases
| Characteristic | Patient 1 | Patient 2 | Patient 3 | Our patient |
|---|---|---|---|---|
| Report | Roe et al. (1990) | Houten et al. (2014) | Tort et al. (2016) | |
| Gender | Female | Male | Female | Female |
| Onset | 2 days of age | 8 weeks of age | Birth | 9 years of age |
| Age/cause of death | 4 months: respiratory acidosis | 5 years | Alive at 10 years | Alive at 16 years |
| Developmental delay | Severe | Severe | Moderate | Normal |
| Neurological Findings | Severe encephalopathy, hypotonia, feeding difficulties & microcephaly | Severe encephalopathy, dystonia, cortical blindness, central apnea & intractable epilepsy | Axial hypotonia, uncoordinated movements, microcephaly, astatic myoclonic epilepsy | Optic atrophy, reduced vision, peripheral neuropathy & intermittent gait abnormalities |
| C10:2 carnitine | “Positive” on mass spectrometry | Positive (1.3umol/L | Positive (>0.11 umol/L) | Negative (<0.11 umoL/L) |
| Brain ultrasound/MRI | Bilateral ventriculomegaly | Progressive leukodystrophy, generalized cerebral atrophy, ventriculomegaly & bilateral basal ganglia T2 abnormalities | Ventriculomegaly, colpocephaly, and hypoplasia of the corpus callosum, generalized cerebellar atrophy | Age 9: Normal. Age 11: Optic pathway atrophy, parenchymal volume loss & abnormal morphology of the lateral ventricles with multiple septations within. |
| Plasma Lysine Levels | Elevated 5.0 times “upper limit of normal” 1052 nmol/L (normal 49–204 nmol/L) | Elevated 4.0 times “upper limit normal” 831 mol/L (normal 49–204 nmol/L) | “Elevated” (normal 49–204 nmol/L) | Elevated 3.7 times upper limit of normal- 890 nmol/L (103–255 mol/L) |
| Plasma proline levels | Normal (normal 85–303 nmol/L) | Normal (normal 85–303 nmol/L) | Normal (normal 85–303 nmol/L) | Elevated 2.5 times normal-920 nmol/L (normal 97–368 nmol/L) |
| NADK2 mutation | Not studied | Homozygous p.Arg340* c.1018C. T (NM_001085411.1) | Homozygous splice site (c.956 +6T>C; p.Trp319Cysfs*21) | Homozygous start loss g.36241900 T>C p. Met1Val |
| Therapy | Low-lysine formula (portagen®) & carnitine | Low-lysine diet, carnitine & medium chain fatty acid supplements | Low lysine diet, pyridoxal phosphate, biotin, thiamine, carnitine, creatine, vitamin E, idebenone & ubidecarenone supplements | NADH supplement |
In this report of the fourth documented case of NADK2 deficiency with hyperlysinemia, we highlight the clinical and molecular heterogeneity that can be associated with hypomorphic mutations.
2 |. CASE REPORT
Our patient is a 15-year-old female with a history of episodic weakness; intermittently elevated creatine phosphokinase (CPK); and hepatic enzyme levels, and bilateral optic atrophy was referred to the UDN (Table 1). She was the first of two children of non-consanginous parents and there are no similarly affected relatives. Developmental age is on target for chronologic age.
She initially presented with sudden onset of partial vision loss at the age of 8. She was evaluated by an ophthalmologist who diagnosed her as having optic atrophy, likely secondary to an autosomal dominant form of optic atrophy. At the age of 10, after a diagnosis of mononucleosis and pneumonia, she developed severe lower extremity weakness and walking difficulties that lasted several days. These symptoms resolved but recurred months later with associated weakness, numbness, paresthesias, and difficulty walking and climbing stairs. She has subsequently had multiple, similar episodes, most recently at 13 years of age. At that time, she had functional weakness in her hands and feet as well as difficulty walking. Episodes were associated with elevated CPK levels (up to 18,000 IU/L) and liver enzyme elevations. On physical examination a short philtrum, mild bilateral nystagmus, reduced deep tendon reflexes and vibratory sensation of her lower extremities bilaterally and an abnormal Romberg and Barre were noted without microcephaly or other dysmorphic features.
Metabolic studies include plasma amino acids that showed elevated lysine levels (890 nmol/L [normal 103–255 mol/L]) and proline levels (920 nmol/L [normal 97–368 nmol/L]), without elevations of citrulline or alanine (Table 1). The acylcarnitine profile (ACP) performed during an episode showed elevations of C10 to C18 suggestive of mild Multiple Acyl-CoA-Dehydrogenase Deficiency (MADD). But other ACP values have been within normal limits, including, most notably, C10:2 (normal <0.11 umol/L). Though we did not measure DECR activity in this patient, we infer that she does not have clinically significant DECRD because her C10:2 Level is normal. Urine organic acids during an episode showed severe lactic aciduria, but no elevation in glutaric acid, making MADD unlikely. Free and total carnitine levels were indicative of mild secondary carnitine deficiency, and vitamin B12 Levels were decreased, prompting daily carnitine, and monthly B12 supplementation. Additional tests have included chromosomal microarray, and Nuclear Mitochondrial Genome panel (nucSEEK) which identified several variants of unknown significance that were not consistent with the patient’s phenotype. This panel also included genes that are associated with MADD. A comprehensive DMD gene analysis was also normal.
Brain MRI at age 9 that was normal. Repeat brain MRI at age 11 showed optic pathway atrophy, possible parenchymal volume loss, and abnormal morphology of the lateral ventricles. Spine MRI showed minimal left inferior cerebellar tonsillar ectopia, likely of little clinical significance.
EMG showed “electrophysiological evidence of impaired conduction of the left anterior visual pathway” and “severe chronic active primary axonal sensory neuromotor peripheral neuropathy and mild proximal myopathy.” Muscle biopsy showed denervation atrophy and slight lipid excess in type one fibers of unknown significance.
The patient applied to the Undiagnosed Diseases Network (UDN) and was enrolled at the Vanderbilt clinical site. Family history did not reveal other affected family members and parental consanguinity was denied. Her UDN evaluation included WES, which revealed homozygosity for a g.36241900 A>G(p.Met1Val) start loss mutation in her NADK2 gene (Figure 1 top). Both parental samples were heterozygous for this p.Met1Val variant and their only other child, an unaffected son, was homozygous normal. No large shared parental haplotype containing the NADK2 gene was detected. No other viable candidate genes were identified. The primary NADK2 transcript encodes a 442 amino acid isoform (Figure 1 bottom). Use of the next in frame Met codon which is embedded in a weak Kozak sequence would result in the synthesis of a 279 amino acid isoform that is predicted to be nonfunctional because it has the mitochondrial target signal deleted (Ohashi, Kawai, & Murata, 2012). Immunoblot studies with antibodies against NADK2 showed a protein that is clearly reduced in amount and which may also be smaller in size (Figure 2a). Also the band for the patient appears in the cytosolic fraction rather than, as expected, in the mitochondrial fraction, in contrast to controls (Figure 2b).
FIGURE 1.
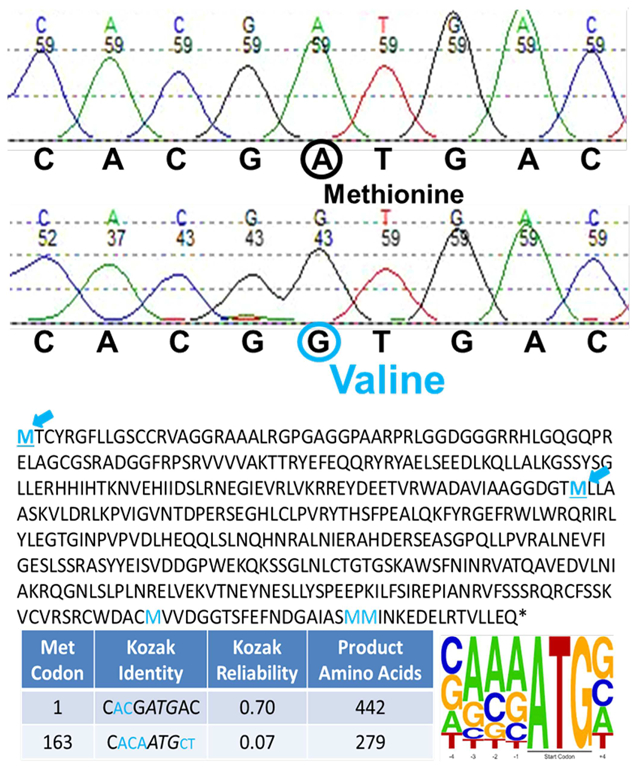
Sanger confirmation of p.Met1Val (top), and protein products and Kozak scores corresponding to alternative NADK2 methionines (bottom). Color figure can be viewed at wileyonlinelibrary.com
FIGURE 2.
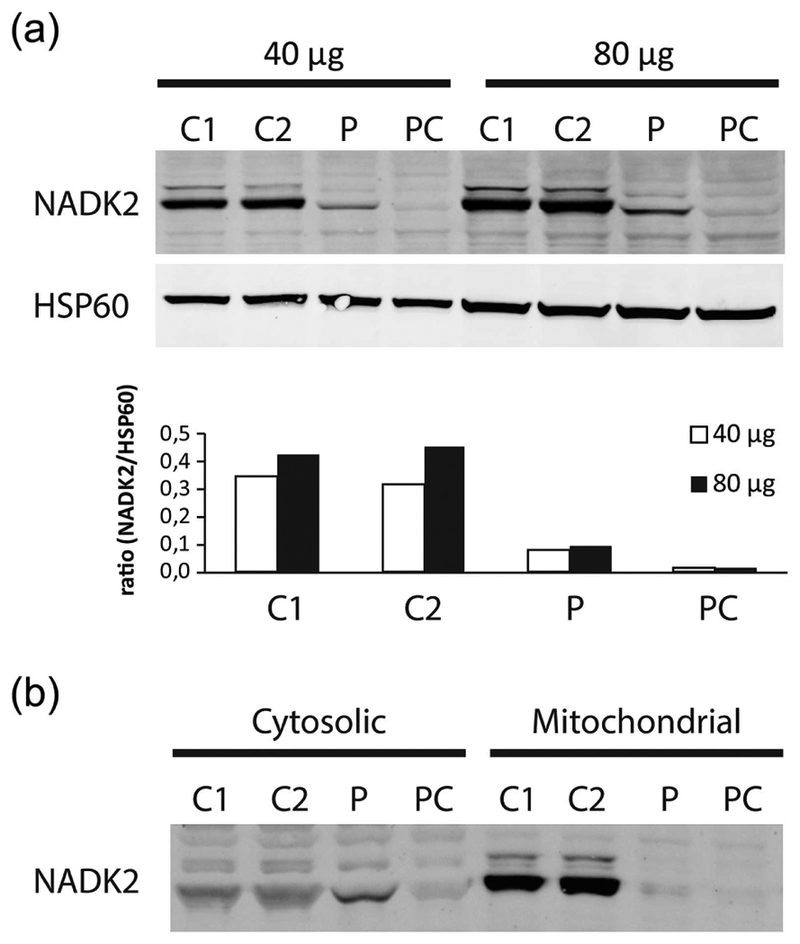
(a) Western blot analysis of fibroblast homogenates of two controls (C1–C2), our patient (P), NADK2D patient control (Houten et al., 2014) (PC) for NADK2 at 40 mg (Left) and 80 mg (Right). Western blot analysis for HSP60 was performed as loading control. (b) NADK2 band for the patient (P) and patient control (PC) appears in the cytosolic fraction rather than, as expected, in the mitochondrial fraction, in contrast to controls (C1–C2)
3 |. DISCUSSION
Our patient has a novel start loss mutation in the NADK2 gene. Our case is phenotypically and molecularly distinct from what has previously been described as DECRD, a severe and fatal inborn error of polyunsaturated fatty acid degradation (Houten et al., 2014; Roe et al., 1990; Tort et al., 2016). NADK2 encodes mitochondrial NAD kinase, a crucial enzyme required for NADP biosynthesis (Zhang, 2013). Previous studies have shown that the deficiency of mitochondrial NADPH that is associated with NADK2 deficiency can lead to reduced function of both DECR1 and AASS (Table 1, Figure 3). The phenotypes of all four patients with NADK2 deficiency (Table 1) are consistent with a complex neuro-metabolic disorder involving a combination of impaired fatty acid and lysine degradation, as well as, other NADPH dependent processes. Our patient’s phenotype is much milder relative to the three prior patients in terms of biochemical derangements, neurological morbidity, and mortality (Table 1).
FIGURE 3.
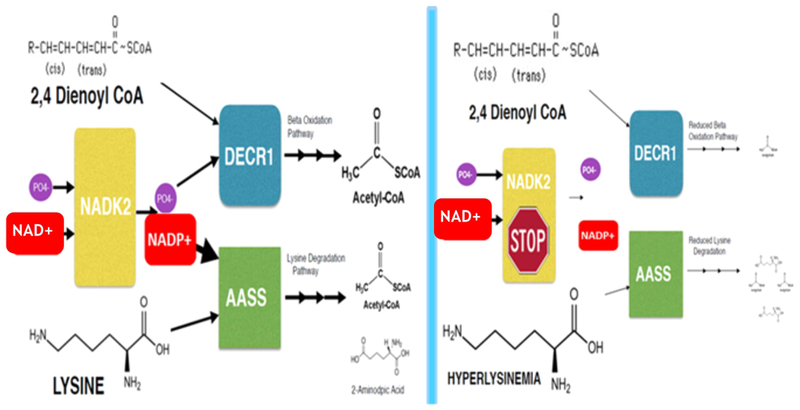
Model of metabolic dysfunction in NADK2 deficiency (right) compared to normal (left). Color figure can be viewed at wileyonlinelibrary.com
We hypothesize that the less severe phenotype of our patient results from a start loss mutation in her NADK2 gene that confers some residual functionality on the protein product (Figures 1 and 2). She is homozygous for a g.36241900 A>G (p.Met1Val) start loss mutation in her NADK2 gene (Figure 1). Interestingly, 31/37 (84%) of documented cases of reported Start-loss mutations in human disorders yielded a non-functional protein. Only 6/37 (16%) cases produce a partially functional protein (Calva-Cerqueira et al., 2009; Felgentreff et al., 2015, 201; Pittis et al., 2004; Rasmussen et al., 2014; Saeki et al., 2011). In these six partially functional cases, use of a downstream Met codon has been demonstrated or hypothesized. However in our case, use of the next in frame Met codon which is embedded in a very weak Kozak sequence would result in a 279 amino acid isoform that is predicted to be nonfunctional (Figure 1). Since immunoblot analysis showed a reduced level but still clearly present NADK2 protein band (see Figure 2), we hypothesize that our patient retains residual enzymatic function through a novel but undetermined mechanism, including the possible alternative use of an upstream AUG codon rescuing translation of the NADK2 RNA as well as the possible translation of non-AUG codons (Barbosa, Peixeiro, & Romão, 2013; Wethmar, 2014; Wethmar, Smink, & Leutz, 2010).
We infer that the secondary deficiencies of DECR1 and AASS may not be the only causes of the complex phenotype seen in NADK2 hypomorphs. Indeed, the Decr1 knockout mouse displayed increased levels of C10:2-carnitine and fasting-induced hypoglycemia, a symptom common in other mitochondrial fatty acid beta-oxidation defects, but appeared to be otherwise healthy (Miinalainen et al., 2009). Also AASS mutation dependent hyperlysinemia is generally considered to be clinically benign (Houten et al., 2013). Thus, other NAD+/NADPH dependent enzymes, including those causing hyper-prolinemia, may also contribute to the complex phenotype of NADK2 deficiency (Table 1). Indeed proline oxidase & 1-pyrroline-5-carboxylate dehydrogenase, the enzymes associated with Hyper-prolinemia Type I (OMIM: 239500), and Hyperprolinemia Type II (239510), are NAD+Dependent Enzymes (Hancock, Liu, Alvord, & Phang, 2016; Srivastava et al., 2012). Though hyperprolinemia has not been demonstrated in other cases of NADK2 Deficiency, we infer that due to our patients prolonged survival with likely decreased NADK2 activity and increased cytosolic NAD+, this benign condition was able to present itself.
Other additional NADPH dependent enzymes may include the Aldose Reductase dependent reduction of glucose into sorbitol, the malic enzyme dependent reductive carboxylation of pyruvate, the isocitrate dehydrogenase dependent reductive carboxylation in the citric acid cycle, and the glutathione reductase dependent mitochondrial metabolism of reactive oxygen species (Love et al., 2015; Tedeschi et al., 2016). Symptoms of other mitochondrial disorders can range from clinically mild, like lactic acidosis, peripheral neuropathy and optic neuropathy to more severe symptoms such as renal tubular acidosis, hypotonia, developmental delay, progressive encephalopathy, and failure to thrive (Zhang, 2015). For example, the NADPH dependent enzyme, Aldo-Ketoreductase, B1 (AKRB1, OMIM# 103880), is located along the optic nerves, peripheral myelin sheaths and kidneys, suggesting a possible role of AKRB1 in the clinical heterogeneity seen in NADK2 deficiency. We postulate that our patient has milder phenotypic findings, presumably due to residual NADK2 activity and less severe NADPH deficiency in the mitochondrion due, perhaps to rescue of translation of the NADK2 mRNA by means of the use of an alternative AUG start codon.
DECRD can be detected in newborn screening (NBS) using C10: 2-carnitine as the screening analite (Houten, Violante, Ventura, & Wanders, 2016; McHugh et al., 2011). Since our patient does not have elevated C10:2-carnitine levels she would not have been detected. Interestingly, no cases of DECRD have been detected by NBS to date, although DECR has been included in screening programs since at least 2006 (Houten et al., 2016). While our patient may have had elevated lysine and proline levels as a neonate these analyses are not included in the current ACMG NBS panel. Thus, our case suggests that NADK2 deficiency should be considered when unexplained elevations of lysine and proline are found.
Previous therapies for NADK2 have included carnitine supplementation and low lysine diets to manage the biochemical disruptions of DECRD and hyperlysinemia (Table 1). Our patient presumably does not have DECRD because she has normal C10:2 Levels, but she does have hyperlysinemia. The efficacy of a low lysine diet is not clear in treating the more severe patients of NADK2 deficiency (Houten et al., 2014; Roe et al., 1990; Tort et al., 2016). We think that these treatment modalities may be focusing on downstream effects of the disease rather than its primary cause, NADPH deficiency. Thus, we propose that potential therapeutic options, which can more explicitly target the production of NADPH, should be considered: NADH supplementation.
NAD+ is an essential cofactor required for a diversity of intracellular processes including electron transfer for ATP synthesis. It is also phosphorylated by NADK2 to produce NADPH (Love et al., 2015). The instability of NADPH, as well as its limited permeability across cellular membranes, suggests that it is unlikely to be beneficial as a nutrient supplement (Wu, Wu, & Knight, 1986). However, we hypothesize that supplementation with NADH, or other NADPH precursors such as niacin or nicotinamide riboside that may cross cellular membranes better, may be effective in maximizing the reduced efficiency of our patient’s enzyme by ensuring that higher levels of substrate are available for conversion to NADPH (Tedeschi et al., 2016). We further hypothesize that our patient has some residual NADK2 because of her lower lysine levels and absence of C10:2. This residual NADK2 activity could lead to increased production of NADPH. Furthermore, since no side effects have been reported from the use of NADH supplementation, we propose that it should be considered for evaluation in the treatment of milder cases of NADK2 deficiency. Our preliminary clinical data from initiating NADH supplementation are encouraging.
In conclusion, we describe a fourth patient with NADK2 Deficiency and the first NADK2 deficient patient with a milder hypomorphic phenotype presumably without DECRD (Table 1). We also have characterized the sixth known case of a Start-Loss mutation with apparent residual enzyme activity (Calva-Cerqueira et al., 2009; Felgentreff et al., 2015, 2011; Pittis et al., 2004; Rasmussen et al., 2014; Saeki et al., 2011). The clinical presentation of our case further expands the phenotype of NADK2 mutations, suggesting that molecular pathogenesis in this patient may not be due to hyper-lysinemia and DECRD, but rather to other NADPH dependent processes. Further, molecular and clinical investigations will be required to elucidate the diversity of the NADPH dependent enzymes affected in NADK2 deficiency and the potential for NADPH synthesis as a treatment modality in this complex disease.
ACKNOWLEDGMENT
This work was supported in part by NIH/NHGRI grant UO1HG007674 (JAP, JH, and RH).
Funding information
John A Phillips III, John H. Newman & Rizwan Hamid, Grant number: NIH/NHGRI grant UO1HG007674
REFERENCES
- Barbosa C, Peixeiro I, & Romão L (2013). Gene expression regulation by upstream open reading frames and human disease. PLoS Genetics, 9(8), e1003529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calva-Cerqueira D, Chinnathambi S, Pechman B, Bair J, Larsen-Haidle J, & Howe JR (2009). The Rate of germline mutations and large deletions of SMAD4 and BMPR1A in juvenile polyposis. Clinical Genetics, 75(1), 79–85. [DOI] [PubMed] [Google Scholar]
- Felgentreff K, Lee YN, Frugoni F, Du L, van der Burg M, Giliani S, … Sleckman BP (2015). Functional analysis of naturally occurring DCLRE1C mutations and correlation with the clinical phenotype of ARTEMIS deficiency. Journal of Allergy and Clinical Immunology, 136(1), 140–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felgentreff K, Perez-Becker R, Speckmann C, Schwarz K, Kalwak K, Markelj G, … Ehl S (2011). Clinical and immunological manifestations of patients with atypical severe combined immunodeficiency. Clinical Immunology, 141(1), 73–82. [DOI] [PubMed] [Google Scholar]
- Hancock CN, Liu W, Alvord WG, & Phang JM (2016). Co-regulation of mitochondrial respiration by proline dehydrogenase/oxidase and succinate. Amino Acids, 48(3), 859–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houten SM, Denis S, Te Brinke H, Jongejan A, Van Kampen AH, Bradley EJ, … Frazier DM (2014). Mitochondrial NADP (H) deficiency due to a mutation in NADK2 Causes dienoyl-CoA reductase deficiency with hyperlysinemia. Human Molecular Genetics, 23(18), 5009–5016. [DOI] [PubMed] [Google Scholar]
- Houten SM, Te Brinke H, Denis S, Ruiter JP, Knegt AC, de Klerk JB, … Zschocke J (2013). Genetic basis of hyperlysinemia. Orphanet Journal of Rare Disease, 8(1), 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houten SM, Violante S, Ventura FV, & Wanders RJ (2016). The biochemistry and physiology of mitochondrial fatty acid β-oxidation and its genetic disorders. Annual Review of Physiology, 78, 23–44. [DOI] [PubMed] [Google Scholar]
- Love NR, Pollak N, Dölle C, Niere M, Chen Y, Oliveri P, & Ziegler M (2015). NAD kinase controls animal NADP biosynthesis and is modulated via evolutionarily divergent calmodulin-dependent mechanisms. Proceedings of the National Academy of Sciences USA, 112(5), 1386–1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McHugh DM, Cameron CA, Abdenur JE, Abdulrahman M, Adair O, Al Nuaimi SA, … Au S (2011) Clinical validation of cutoff target ranges in newborn screening of metabolic disorders by tandem mass spectrometry: A worldwide collaborative project. Genetics in Medicine, 13(3):230–254. [DOI] [PubMed] [Google Scholar]
- Miinalainen IJ, Schmitz W, Huotari A, Autio KJ, Soininen R, Ver Loren van TE, … Hiltunen JK (2009). Mitochondrial 2,4-dienoyl-CoA reductase deficiency in mice results in severe hypoglycemia with stress intolerance and unimpaired ketogenesis. PLoS Genetics, 5(7): e1000543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohashi K, Kawai S, Murata K (2012). Identification and characterization of a human mitochondrial NAD kinase. Nature Communications 3: 1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pittis MG, Ricci V, Guerci VI, Marcais C, Ciana G, Dardis A, … Bembi B (2004). Acid sphingomyelinase: Identification of nine novel mutations among italian niemann pick type b patients and characterization of in vivo functional in-frame start codon. Human Mutation, 24(2):186–187. [DOI] [PubMed] [Google Scholar]
- Rasmussen TB, Nissen PH, Palmfeldt J, Gehmlich K, Dalager S, Jensen UB, … Baandrup UT (2014). Truncating plakophilin-2 mutations in arrhythmogenic cardiomyopathy are associated with protein haploinsufficiency in both myocardium and epidermis. Circulation: Cardiovascular Genetics, 7(3):230–240. [DOI] [PubMed] [Google Scholar]
- Roe CR, Millington DS, Norwood DL, Kodo N, Sprecher H, Mohammed BS, & McVie R (1990). 2, 4-dienoyl-coenzyme a reductase deficiency: A possible new disorder of fatty acid oxidation. Journal of Clinical Investigation, 85(5): 1703–1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saeki N, Saito A, Choi IJ, Matsuo K, Ohnami S, Totsuka H, … Kook MC (2011). A functional single nucleotide polymorphism in mucin 1, at chromosome 1q22, determines susceptibility to diffuse-type gastric cancer. Gastroenterology, 140(3):892–902. [DOI] [PubMed] [Google Scholar]
- Srivastava D, Singh RK, Moxley MA, Henzl MT, Becker DF & Tanner JT (2012) The three-dimensional structural basis of type ii hyperprolinemia. Journal of Molecular Biology, 420(3):176–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tedeschi PM, Bansal N, Kerrigan JE, Abali EE, Scotto KW, & Bertino JR (2016). Nad ± kinase as a therapeutic target in cancer. Clinical Cancer Research, 22(21):5189–5195. [DOI] [PubMed] [Google Scholar]
- Tort F, Ugarteburu O, Torres MA, García-Villoria J, Girós M, Ruiz A, & Ribes A (2016). Lysine restriction and pyridoxal phosphate administration in a NADK2 patient. Pediatrics, 138(5): e20154534. [DOI] [PubMed] [Google Scholar]
- Wethmar K (2014). The regulatory potential of upstream open reading frames in eukaryotic gene expression. Wiley Interdisciplinary Reviews: RNA, 5(6):765–768. [DOI] [PubMed] [Google Scholar]
- Wethmar K, Smink JJ & Leutz A (2010). Upstream open reading frames: Molecular switches in (patho) physiology. Bioessays, 32(10):885–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu JT, Wu LH, & Knight JA (1986). Stability of NADPH: Effect of various factors on the kinetics of degradation. Clinical Chemistry, 32(2), 314–319. [PubMed] [Google Scholar]
- Zhang R (2013). MNADK, a novel liver-enriched mitochondrion-localized NAD kinase. Biology Open, 2(4):432–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang R (2015). MNADK, a long-awaited human mitochondrion-localized NAD kinase. Journal of Cellular Physiology, 230(8):1697–1701. [DOI] [PubMed] [Google Scholar]