

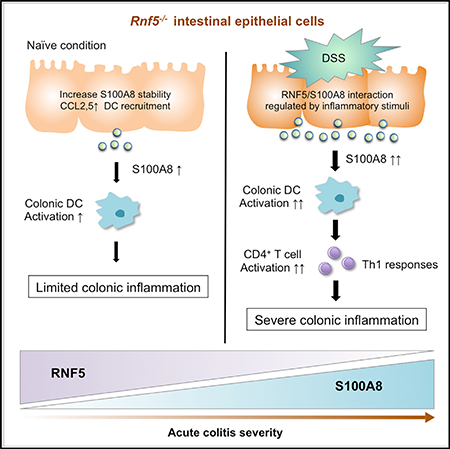
SUMMARY
Inflammatory bowel disease (IBD) is prevalent, but the mechanisms underlying disease development remain elusive. We identify a role for the E3 ubiquitin ligase RNF5 in IBD. Intestinal epithelial cells (IECs) express a high level of RNF5, while the colon of Rnf5−/− mice exhibits activated dendritic cells and intrinsic inflammation. Rnf5−/− mice exhibit severe acute colitis following dextran sodium sulfate (DSS) treatment. S100A8 is identified as an RNF5 substrate, resulting in S100A8 ubiquitination and proteasomal-dependent degradation that is attenuated upon inflammatory stimuli. Loss of RNF5 from IECs leads to enhanced S100A8 secretion, which induces mucosal CD4+ T cells, resulting in Th1 pro-inflammatory responses. Administration of S100A8-neutralizing antibodies to DSS-treated Rnf5−/− mice attenuates acute colitis development and increases survival. An inverse correlation between RNF5 and S100A8 protein expression in IECs of IBD patients coincides with disease severity. Collectively, RNF5-mediated regulation of S100A8 stability in IECs is required for the maintenance of intestinal homeostasis.
In Brief
Fujita et al. show that RNF5 regulation of S100A8 stability in intestinal epithelial cells defines the degree of pro-inflammatory response, culminating in severe intestinal inflammation following DSS treatment to Rnf5−/− mice. Neutralizing S100A8 antibodies attenuates acute colitis phenotypes, and inverse RNF5/S100A8 expression coincides with clinical severity in IBD patients.
Graphical Abstract
INTRODUCTION
Ulcerative colitis and Crohn’s disease, the two main types of inflammatory bowel disease (IBD), are multifactorial immune-mediated disorders characterized by chronic relapsing inflammation of the gastrointestinal tract (Cohen and Sachar, 2017; de Souza and Fiocchi, 2016). Common to both is that they are incurable, often diagnosed at a young age, and associated with significant morbidity (de Souza and Fiocchi, 2016). The precise etiology of IBD remains unclear, but genetic susceptibility, environmental factors, microbial flora, and dysregulation of the immune response are thought to be involved (Halfvarson et al., 2017; Ni et al., 2017; Peters et al., 2017). In particular, the innate and adaptive immune systems are implicated as major contributors to the development and progression of IBD, predominantly through an imbalance between the pro-inflammatory responses of T helper (Th) 1, Th2, and Th17 versus the anti-inflammatory response of regulatory T cells (Canavan et al., 2016; de Souza and Fiocchi, 2016). Indeed, the use of immunosuppressive agents and biologics helped reduce the use of corticosteroids and to augment response in IBD (Cohen and Sachar, 2017; Neurath, 2017). Currently, various therapeutic approaches, including integrin and cytokine inhibitors, are exploited in IBD treatment (Yadav et al., 2016). Nonetheless, limited effectiveness in clinical trials highlights the need for better understanding of the complex biological processes that underlie the pathogenesis of IBD.
One of the current focuses in IBD research attends to a group of immunogenic endogenous damage-associated molecular patterns (DAMPs). These include S100 proteins and high-mobility group box-1 (HMGB1), which are released during tissue damage and bind to pattern recognition receptors (PRRs) on innate immune cells, eliciting inflammatory responses. Notably, DAMPs are increasingly recognized to play a role in the etiology of IBD (Boyapati et al., 2016). Several DAMPs, including the calprotectin complex of S100A8/S100A9 proteins, are present at high levels in the serum and corresponding intestinal tissues during inflammation, and some have been established as valuable biomarkers for a number of colonic inflammatory conditions (Sands, 2015). Correspondingly, the level of calprotectin in fecal samples is largely considered a sensitive and specific biomarker, though inter- and intra-personal variation has been reported (Sands, 2015). S100A8 and S100A9 are expressed primarily in neutrophils and monocytes and induced under inflammatory conditions in keratinocytes and epithelial cells (Henke et al., 2006; Mørk et al., 2003). Although the role of DAMPs as active mediators of inflammation points to their possible consideration as therapeutic target in human inflammatory diseases, the mechanisms underlying their regulation in the course of intestinal inflammation are poorly understood. Here we identify the ubiquitin ligase RNF5 as a regulator of the S100A8 protein.
Originally found in Arabidopsis thaliana as suppressor of the yeast secretory mutant SEC15 gene (Matsuda and Nakano, 1998), the RING finger E3 ubiquitin ligase RNF5 is anchored to the endoplasmic reticulum (ER) membrane and is part of the ER-associated degradation (ERAD) machinery. RNF5 was shown to play an important role in the control of misfolded proteins implicated in cystic fibrosis (Younger et al., 2006) and inclusion body myositis (Delaunay et al., 2008). In tumor cells, RNF5 controls the stability of the glutamine carrier proteins SLC1A5 and SLC38A2, which limits glutamine uptake and renders the tumor cells more sensitive to ER stress-inducing chemotherapy (Jeon et al., 2015). Under normal growth conditions, RNF5 was shown to control the level of the ATG4B protein, which is important for LC3 maturation and autophagosome formation and thus controls degree of group A streptococcus infection (Kuang et al., 2012). RNF5 was also implicated in the regulation of viral and bacterial infection, through control of immune sensing mechanism (Zhong et al., 2009), pointing to a possible role RNF5 may play in inflammatory diseases. Here, using the Rnf5−/− mouse model, we identify and characterize its role in intestinal inflammatory disorders.
RESULTS
Rnf5−/− Mice Exhibit Intestinal Inflammation and Infiltration of Activated Leukocytes
In wild-type (WT; C57BL/6) mice, RNF5 is widely expressed in the small intestine and colon, as demonstrated by immunoblot and immunohistochemical (IHC) analysis, and its expression is particularly high in intestinal epithelial cells (IECs) (Figures 1A and1B). Given the importance of IECs in maintaining intestinal homeostasis (Peterson and Artis, 2014), we asked whether loss of RNF5 affects the integrity and/or function of these cells. Histological analysis of colonic sections from Rnf5−/− mice revealed mild inflammation, with some prominent focal areas of immune cell infiltration compared with WT mice (Figure 1C). Immunofluorescence staining of CD45+ cells in colon sections revealed that leukocytes were significantly more abundant in the colons of Rnf5−/− mice compared with WT mice (Figure 1D). Flow cytometric analysis of lamina propria CD4+ and CD8+ T cells, NK1.1+ natural killer cells, colonic macrophages (CD45+ CD11c+ F4/80+ CD103-), neutrophils (CD45+ Ly6G+), and inflammatory monocytes (CD45+ Ly6C+) did not identify marked differences (Figure 1E), except for a modest, albeit significant, increase in the frequency of total colonic dendritic cells (DCs) (CD45+ CD11c+ CD103+ F4/80-) in colonic lamina propria cells from Rnf5–/– mice compared with WT mice (Figure 1E). In addition, the expression of MHC classes I and II was higher on colonic DCs from Rnf5–/– mice than from WT mice (Figure 1F), consistent with increased DC activation in the colon. Finally, qRT-PCR analysis of isolated colonic extracts demonstrated that expression of pro-inflammatory cytokines (II1β, II6, II12p35, and Tslp) was elevated in colons obtained from Rnf5–/– compared with WT genotype (Figure 1G). These data suggest that Rnf5–/– mice exhibited basal intestinal inflammation and infiltration of activated DCs compared with WT mice. To further examine the involvement of RNF5 in intestinal inflammation, we assessed changes in gene expression in the colon tissues of Rnf5–/– and WT mice. RNA sequencing (RNA-seq) analysis identified 156 significantly dysregulated (87 up- and 69 downregulated) genes in colon tissues of Rnf5–/– compared with WT mice (Figure 1H). Ingenuity Pathway Analysis (IPA) pointed to the enrichment of genes involved in active antigen presentation in Rnf5–/– colon tissues (Table S1). Increased expression of H2-Bl, H2-T10, Gm8909, and Ly6c2 mRNA in Rnf5–/– colon tissues implies that the infiltration of activated DCs may underlie colonic inflammation seen in Rnf5–/– mice (Figure S1A).
Figure 1. RNF5 Is Crucial for the Maintenance of Intestinal Homeostasis.

(A) Representative immunoblot of RNF5 in IECs from different intestinal regions of WT and Rnf5−/− mice: duodenum (D), jejunum (J), ileum (I), and colon (C).
(B) Representative IHC staining of RNF5 in small intestine and colon sections of WT and Rnf5−/− mice. Scale bars, 50 μm.
(C) Representative H&E staining of WT and Rnf5−/− colon sections (red triangles indicate infiltrating lymphocytes). Scale bars, 50 μm.
(D) Representative images of immunofluorescence staining of CD45 in WT and Rnf5−/− colon sections and quantification of CD45+ cells (n = 10, two fields each). Scale bars, 50 μm.
(E) Flow cytometric analysis of the frequencies of CD4+, CD4+CD44+, CD4+CD25+, CD8+, and CD8+CD44+ T cells, colonic DCs (CD45+ CD11c+ CD103+ F4/80-), colonic macrophages (CD45+ CD11c+ CD103- F4/80+), inflammatory monocytes (CD45+ CD11c- Ly6C+), neutrophils (CD45+ CD11c- Ly6G+), and NK1.1+ NK cells among gated CD45+ cells isolated from the lamina propria of colons from WT and Rnf5−/− mice (n = 6).
(F) (F) Mean fluorescence intensity (MFI) of MHC classes I and II on colonic DCs cells isolated from the lamina propria of colons from WT and Rnf5−/− mice (n = 6). (G) qRT-PCR analysis of elative mRNA expression of the indicated cytokines in WT and Rnf5−/− colon tissues (n = 6).
(G) (H) Heatmap analysis of RNA-seq data performed from three naive WT or Rnf5−/− colon tissues per group (left). One hundred fifty-six significantly dysregulated genes were identified in Rnf5−/− colon tissues compared with naive Rnf5−/− colon tissues (false discovery rate [FDR] adjusted p value < 0.05 by Benjamini-Hochberg method). Right table shows the top 10 upregulated genes identified in Rnf5−/− colon tissues.
(H) All data are representative of three independent experiments. Graphs show mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001 by two-tailed t test (D–G).
To determine whether RNF5 plays a role in the activation of general DC population or specific intestinal DC subsets, we sorted splenic DCs (CD11c+) and generated bone marrow-derived dendritic cells (BMDCs) from WT and Rnf5–/– mice. No difference in the expression of MHC classes I and II or in the production of IL-12p70 and IL-1β after LPS stimulation was identified (Figures S1B–S1E), suggesting that loss of RNF5 results in the selective activation of DCs in the intestinal microenvironment.
DSS-Induced Acute Colitis Is Exacerbated in Rnf5−/− Mice
To investigate whether RNF5 deficiency influences the development of IBD, we used the dextran sodium sulfate (DSS) model of acute colitis. DSS induces inflammation throughout the gut, with particularly prominent damage in the colon, and is a commonly used model of human colitis. Rnf5–/– mice and WT littermates were administered to 2.5% DSS in drinking water for 7 days and were monitored for an additional 48 hr. Rnf5–/– mice lost significantly more weight than control mice, with an estimated difference of 13%, which coincided with more severe shortening of the colon on day 9 (Figures 2A and2B). Correspondingly, Rnf5–/– mice exhibited more severe colitis symptoms, including abundant bloody diarrhea and rectal bleeding, which resulted in a higher clinical activity index and earlier death, compared with WT mice (Figures 2C and2D). Histological analysis revealed larger areas of ulceration and crypt loss, more extensive inflammatory cell infiltration reaching the submucosa, and thickening of the mucosa with abundant edema in the colon of the Rnf5−/− compared with that of WT mice, analyzed on day 9 after DSS administration (Figure 2E). Changes associated with enhanced inflammation in the colonic tissue include altered proliferation and cell death programs. Indeed, a lower level of the proliferation marker Ki67 and a higher level of the apoptotic marker cleaved caspase-3 were seen in colonic sections from Rnf5−/− compared with WT mice (Figures 2F and S2A), suggesting that loss of RNF5 impaired the regenerative response. In addition, CD4+ T cells, F4/80+ macrophages, and Ly6G/C+ neutrophils, but not CD8+ T cells or CD11c+ DCs, were more abundant in colonic sections obtained 9 days following DSS treatment of Rnf5−/− compared with WT mice (Figures 2F and S2A). Lastly, colonic sections from DSS-treated Rnf5−/− animals had markedly fewer goblet cells compared with WT mice, as indicated by periodic acid-Schiff/Alcian blue staining (PAS/AB) (Figures 2F and S2A). Taken together, these data demonstrate that the absence of RNF5 not only exacerbates colonic damage during DSS-induced acute colitis leading to increased mouse mortality but also impairs the subsequent regenerative process.
Figure 2. DSS-Induced Colitis Is Exacerbated in Rnf5−/− Mice.

(A) Initial weight (left) and percentage body weight after DSS treatment (right) of WT and Rnf5−/− mice. DSS (2.5%) was administered in drinking water for 7 days followed by a 48 hr recovery period (n = 10).
(B) Representative photographs of colon and cecum (left) and quantification of colon length (right) from WT and Rnf5−/− mice on day 9 after initiation of DSS treatment (n = 10).
(C and D) Disease activity index (C) and survival by Kaplan-Meier method (D) of DSS-treated WT and Rnf5−/− mice (n = 10).
(E) Representative H&E staining (left) and histological score (right) for colon sections from WT and Rnf5−/−mice on day 9 after DSS administration (n = 8, two fields each).
(F) Representative images of Ki67, cleaved caspase-3, F4/80, Ly6G/C, CD11c, CD4, CD8, and periodic acid-Schiff/Alcian blue (PAS/AB) staining in colonsections of WT and Rnf5−/− mice on day 9 after DSS administration. Scale bars, 50 μm.
All data are representative of three independent experiments. Graphs show mean ± SEM. *p < 0.05 and **p < 0.01 by two-tailed t test (A–C and E) or log rank test (D).
Gut Microbiota Does Not Affect Acute Colitis in Rnf5–/– Mice
Deregulation of intestinal homeostasis and susceptibility to inflammation are often associated with alterations in the commensal bacterial population (Zhang et al., 2017). Therefore, we set to test whether co-housing of Rnf5–/– and WT mice, a condition in which the mice exchange their gut microbiota, prior to DSS treatment, would affect the susceptibility to develop acute colitis in either the WT and Rnf5–/– mice. Notably, although co-housing did not increase the degree of inflammation seen following DSS administration in the WT mice, it also did not diminish the degree of acute colitis (body weight loss, clinical activity index, colon length, and histological score), seen in the Rnf5–/– mice (Figures S2B–S2E). These data suggest that the acute colitis seen in DSS-treated Rnf5–/– mice does not depend on altered gut microbiota composition, further pointing to altered inflammation as a key driver of this pathology.
Identification of S100A8 as an RNF5 Substrate
To understand the mechanism by which RNF5 contributes to the maintenance of intestinal homeostasis, we used liquid chromatography-tandem mass spectrometry to identify proteins that were co-immunoprecipitated with RNF5. Focusing on proteins that may play a role in inflammation, we selected to examine S100A8, one of RNF5 interacting proteins identified in this analysis. S100A8 is a component of the calcium-binding calprotectin complex, which has been shown to play a role as a gut mucosal DAMP involved in tissue inflammation (Bertheloot and Latz, 2017). To confirm interaction of S100A8 with RNF5, ectopic expression of Flag-tagged RNF5 and V5-tagged S100A8 in HEK293T cells, followed by anti-Flag immunoprecipitation, confirmed that the two proteins interact (Figure 3A). Moreover, co-expression of hemagglutinin (HA)-tagged ubiquitin together with RNF5 and S100A8 proteins identified that S100A8 is ubiquitinated by RNF5 (Figure 3B). To verify these findings, we expressed control or RNF5-targeted short hairpin RNA (shRNA) in MODE-K cells, a mouse IEC line. Analysis of the endogenous proteins in these cells revealed that the ubiquitination of S100A8 was lower upon RNF5 knockdown (KD) (Figure 3C). Treatment of cells with the proteasomeinhibitorMG132 increased the steady-state levels of S100A8, partially attenuating the effect of RNF5 co-expression (Figure 3D). These observations suggest that RNF5-mediated ubiquitination of S100A8 targets it for proteasomal degradation. Indeed, treatment of shRNF5-expressing MODE-K cells with the translational inhibitor cycloheximide extended the half-life of S100A8 from 6.3 to 11.9 hr (Figure 3E). Conversely, ectopic expression of Flag-RNF5 decreased the half-life of S100A8 from 3.6 to 1.3 hr, compared with cells expressing the corresponding the control vectors (Figures S3A and S3B). Notably, interaction of endogenous RNF5 and S100A8 proteins was confirmed in MODE-K cells (Figure 3F). Collectively, these findings suggest that by its ubiquitination and proteasomal mediated degradation, RNF5 association with S100A8 determines its stability.
Figure 3. S100A8 Is a Substrate of RNF5.

(A) Immunoprecipitation and immunoblot analysis of the RNF5-S100A8 interaction in HEK293T cells ectopically expressing Flag-tagged RNF5 or empty vector (EV) and V5-tagged S100A8.
(B) Immunoprecipitation and immunoblot analysis of HEK293T cells co-expressing V5-S100A8, HA-ubiquitin (HA-Ub), plus Flag-RNF5. Cells were treated withMG132 (10 μM) for 4 hr prior to lysis.
(C) Anti-S100A8 immunoprecipitation and anti-Ub immunoblot of MODE-K cells transfected with EV or RNF5-targeting shRNA and treated with MG132 (10 μM) for 4 hr before lysis.
(D) Immunoblot analysis of HEK293T cells expressing Flag-EV or Flag-RNF5 and treated with medium or 10 μM MG132 for 4 hr before lysis.
(E) Immunoblot analysis of MODE-K cells expressing EV or shRNF5. Cells were treated with cycloheximide (CHX) as indicated (n = 3).
(F) Immunoprecipitation and immunoblot analysis of the interaction between endogenous RNF5 with S100A8 analyzed in MODE-K cells treated with MG132 (10 μM) for 4 hr.
(G) Immunoprecipitation and immunoblot analysis of cell lysates prepared from MODE-K cells treated with 10 ng/mL TNF-α for the indicated times followed by MG132 (10 μM) for 4 hr before lysis.
Data are representative of three independent experiments. Graphs show mean ± SEM.
To determine whether RNF5 regulation of S100A8 in IECs was affected by inflammation, we examined RNF5-S100A8 interaction in MODE-K cells exposed to the pro-inflammatory cytokine TNF. Interestingly, the interaction between S100A8 and RNF5 decreased within 6–12 hr of TNF treatment and then increased over the next 24–48 hr (Figure 3G). Consistent with these observations, RNF5 expression in WT colon tissues did not change following DSS treatment (Figure S3C), supporting changes in the activity of this ubiquitin ligase following inflammatory stimuli. These results suggest that inflammatory stimuli determine RNF5 interaction and hence control of S100A8 stability. We thus set to test the possibility that increased level of S100A8 derived from RNF5-deficient IECs may activate innate immune receptors and drive intestinal inflammatory responses.
Increased Level of S100A8 Secreted by RNF5-Deficient IECs Affects Host Immune Responses in a Paracrine and Autocrine Manner
S100A8 and S100A9 are highly expressed by neutrophils, monocytes, and epithelial cells and are found at high levels in the extracellular milieu at inflammatory sites. Consistent with this, S100A8 and S100A9 were both expressed in the MODE-K IEC line; however, expression of S100A8, but not S100A9, was increased upon RNF5 KD (Figures 4A and S4A). Similarly, S100A8 was markedly upregulated in colonic epithelial cells from Rnf5−/− mice compared with WT mice, whereas S100A9 expression was largely unaffected (Figure 4B). Secretion of S100A8 by MODE-K cells was also elevated following RNF5 KD and reduced upon KD of both RNF5 and S100A8 (Figures 4C and4D). To gain insight into the physiological significance of S100A8 secreted by IECs, we next asked whether RNF5-regulated S100A8 could activate DCs. To this end, we incubated BMDCs with conditioned medium (CM) collected from cultures of MODE-K, MODE-K-shRNF5, and MODE-KshRNF5/shS100A8cells.The effects of the MODE-K CM, medium alone, or recombinant S100A8 on BMDCs were then assessed by measuring the secretion of cytokines and chemokines and cell-surface expression of MHC classes I and II. Indeed, production of IL-12p70, production of IL-1β, and MHC expression by BMDCs were strongly induced by CM from MODE-K cells and by recombinant S100A8. Notably, these effects were further enhanced in the presence of CM from cells expressing shRNF5 compared with empty vector (EV) or shRNF5/shS100A8 (Figures 4E and4F), supporting the direct stimulatory component in CM as RNF5-regulated S100A8. To further assess the contribution of RNF5-regulated S100A8 to DC activation seen in these studies, we cultured BMDCs in the presence of CM that were supplemented with a specific S100A8-blocking antibody or an immunoglobulin G (IgG) control antibody. The presence of S100A8-neutralizing antibodies, but not IgG control antibodies, diminished the effects of CM from the MODE-K shRNF5 cells on BMDCs (Figures S4B and S4C). These findings establish that RNF5-regulated S100A8 is directly responsible for stimulation of BMDCs.
Figure 4. Increased Level of S100A8 Secreted by RNF5-Deficient IECs Affects Host Immune Responses.

(A) Immunoblot analysis of endogenous proteins in MODE-K cells expressing empty vector (EV) or two RNF5-targeting shRNAs.
(B) Representative images of IHC staining for S100A8 and S100A9 (left) and quantification of IHC scores (right) in colon sections from WT and Rnf5−/− mice (n = 6, two fields each). Scale bars, 50 μm. ****p < 0.0001 by two-tailed t test.
(C) Immunoblot analysis of MODE-K cells expressing EV, shRNF5, or shRNF5 plus shS100A8.
(D) ELISA quantification of S100A8 levels in the culture supernatants of MODE-K cells expressing EV, shRNF5, or shRNF5 plus shS100A8 (n = 3).
(E) ELISA quantification of IL-12p70 and IL-1β levels in the culture supernatants of bone marrow-derived dendritic cells (BMDCs) fromWT mice incubated for 18 hr with medium alone (no stimulation), recombinant S100A8 (1 ng/mL), or conditioned medium (CM) from MODE-K cells expressing EV, shRNF5, or shRNF5 plus shS100A8 (n = 3).
(F) MFI of MHC classes I and II on CD11c+ BMDCs incubated as in (E) (n = 3).
(G) Relative luciferase activity of MODE-K-EV, MODE-K-shS100A8, MODE-K-shRNF5, or MODE-K-shRNF5/shS100A8 cells transiently transfected with the NF-κB-dependent firefly luciferase reporter plasmid NF-κB-luc and Renilla luciferase plasmid pRL-TK (n = 3).
All data are representative of three independent experiments. Graphs show mean ± SEM. n.s., not significant (p > 0.05). *p < 0.01, **p < 0.001, and ***p < 0.0001 by one-way ANOVA followed by Tukey’s multiple comparison test (D–G).
In addition to S100A8, the CM of MODE-K cells contained a number of pro-inflammatory cytokines and chemokines. In particular, MODE-K-shRNF5 cells and their CM contained higher levels of CCL2 and CCL5 mRNA and protein, respectively, compared with control MODE-K cells and their CM (Figures S4D–S4F). As CCL2 and CCL5 have been implicated in the progression of IBD (Turner et al., 2014), our finding points to a possible mechanism by which DCs may be recruited to the inflamed intestinal microenvironment in Rnf5−/− mice. Of note, CM from MODE-K-shRNF5 cells showed decreased levels of CCL11, which has been implicated in eosinophil recruitment, while the levels of major pro-inflammatory cytokines and chemokines were not significantly altered (Figures S4D–S4F). Collectively, these data indicate that increased level of S100A8, secreted by MODE-K IECs subjected to RNF5 KD, effectively activate DCs to produce pro-inflammatory cytokines and chemokines.
S100A8 can activate receptor for advanced glycation end products (RAGE) and Toll-like receptor (TLR)-4, which also result in the activation of NF-κB and the upregulation of CCL2 and CCL5, involved in cellular inflammatory responses (Zackular et al., 2015). S100A8 proteins are secreted or released, affecting immune responses via activation of the corresponding receptors (Bresnick et al., 2015). Consistent with these, MODE-K-shRNF5 cells exhibited higher activation of the NF-κB pathway (Figure 4G). Furthermore, KD of both RNF5 and S100A8 abrogated NF-κB induction, seen upon RNF5 KD alone, suggesting that NF-κB activation in RNF5 KD cells is S100A8-dependent. In all, our findings suggest that extracellular S100A8, which is secreted from RNF5-deficient IECs, can stimulate host innate immune responses in the intestinal microenvironment in a paracrine and autocrine manner.
Elevated Th1 Cytokine Production and CD4+ T Cell Activation in DSS-Treated Rnf5−/− Mice
Having established that RNF5 KD elevates S100A8 production by IECs in vitro and secreted S100A8 can stimulate BMDCs, we next asked whether RNF5-regulated S100A8 contributes to the exacerbation of DSS-induced colitis in Rnf5−/− mice compared with WT mice. For this, we isolated colon sections from mice treated with DSS for 3, 6, and 9 days and then analyzed the tissues for S100A8 expression and the supernatants for secreted S100A8 and cytokines. In WT colons, S100A8 expression peaked at day 3 after initiation of DSS treatment and declined thereafter, while RNF5 expression did not change following DSS treatment (Figure 5A), consistent with the temporal regulation of S100A8 by RNF5. In contrast, S100A8 levels were higher in colon tissue from Rnf5−/− mice on day 0, as noted (Figure 4B), but were further increased by DSS and remained high until the end of the analysis at day 9 (Figure 5A). Analysis of S100A8, cytokine, and chemokine levels in the culture supernatants showed that Rnf5−/− colonic tissues produced significantly higher levels of S100A8 and TNF than did tissue from WT mice (Figure 5B), which was also seen for other Th1 cytokines and chemokines (IFN-γ, IL-12p70, CCL2, CCL3, CCL4, and CCL5) (Figures S5A and S5B). In agreement, qRTPCR analysis of colonic extracts isolated on day 9 following DSS administration confirmed elevated expression of pro-inflammatory cytokines and chemokines (II12p35, Tnf, Ifnγ, Ccl2, Ccl3, Ccl4, and Ccl5) in colons obtained from Rnf5–/– compared with WT mice (Figure S5C). Likewise, analysis of serum from DSS-treated mice identified higher levels of S100A8, Th1 cytokines, and chemokine (IFN-γ, TNF-α, IL-12p70, CCL2, CCL3, CCL4, and CCL5) in Rnf5−/− compared with WT mice (Figures 5C, S5D, and S6A). These observations suggest that the production of pro-inflammatory cytokines correlated strongly with the severity of disease and leukocyte infiltration seen in the colons of DSS-treated Rnf5−/− mice. Of note, the levels of IL-6, IL-23, and IL-17A, cytokines involved in the differentiation and function of Th17 lymphocytes, were similar in both the colon tissues and serum of DSS-treated WT and Rnf5−/− mice (Figures S5A and S5D). Taken together, these results suggest that DSS induces a Th1-type inflammatory response in the colon that is exacerbated upon elevated levels of S100A8, because of the absence of its E3 ubiquitin ligase RNF5.
Figure 5. RNF5 Regulates the Development of DSS-Induced Colitis through S100A8-Mediated CD4+ T Cell Activation.

(A) Representative immunoblots of S100A8 and RNF5 in protein extracts of the distal colon from WT and Rnf5−/− mice on the indicated days after DSS administration.
(B) ELISA quantification of S100A8 and TNF-α in the culture supernatants after 24 hr incubation of distal colonic explants isolated from WT and Rnf5−/− mice on the indicated days after DSS administration (n = 3 mice/group).
(C) ELISA quantification of S100A8 levels in the serum from WT and Rnf5−/− mice isolated on day 0 and day 9 after DSS administration (n = 4/group).
(D) Flow cytometric analysis of the frequencies of CD4+, CD4+CD25+, CD4+CD44+, CD8+, and CD8+CD44+ T cells, colonic DCs, colonic macrophages, neutrophils, monocytes, and NK cells in gated CD45+ cells isolated from the lamina propria of colons from WT and Rnf5−/− mice on the indicated days after DSS administration (n = 3–6 mice/group).
(E) Intracellular IFN-γ and TNF-α staining of the CD4+ T cells from LCMV-specific TCR transgenic SMARTA mice. BMDCs were generated from WT mice and incubated for 18 hr with conditioned medium (CM) derived from MODE-K cells expressing EV, shS100A8, shRNF5, or shRNF5 plus shS100A8 treated with 0.5% DSS for 24 hr. BMDCs were then incubated for 72 hr with CFSE-labeled SMARTA CD4+ T cells (shown in Figure S5H) in the presence of 2 μg/mL GP61–80 peptide. Right plot shows quantification of intracellular IFN-γ+ and TNF-α+ CD4+ T cells (n = 3 wells/condition).
(F) Body weight of WT and Rnf5−/− mice administered 2.5% DSS and treated with control rat IgG2b or anti-CD4 neutralizing antibody (200 μg, intraperitoneal [i.p.]) on days –1, 3, and 7, relative to the start of DSS treatment on day 0 (n = 8 mice/group), as shown by red arrows (on day 9: ****p < 0.0001; Rnf5−/− +isotype control versus Rnf5−/− +anti-CD4, ****p < 0.0001; WT + isotype control versus Rnf5−/− + isotype control, n.s.; WT + anti-CD4 versus Rnf5−/− + anti-CD4).
(G) Representative images (top) of colon and cecum and quantification (bottom) of colon length from anti-CD4- or isotype control-treated WT or Rnf5−/− mice as described for (F) on day 9 after 2.5% DSS treatment (n = 8).
(H) Disease activity index (see STAR Methods) of anti-CD4- or isotype control-treated WT or Rnf5/ mice after 2.5% DSS treatment as described for (F) (n = 8) (on day 9: ****p < 0.0001; Rnf5−/− isotype control versus Rnf5−/− + anti-CD4, ****p < 0.0001; WT + isotype control versus Rnf5−/− +isotype control, n.s.; WT + anti-CD4 versus Rnf5−/− + anti-CD4).
All data are representative of two or three independent experiments. Graphs show mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001 by two-tailed t test (B and D). *p < 0.01, **p < 0.001, and ***p < 0.0001 by one-way ANOVA followed by Tukey’s multiple comparison test (C and E). ****p < 0.0001 by two-way ANOVA followed by Tukey’s multiple comparison test (F–H).
To further substantiate these findings, we extracted immune cells from the colonic lamina propria of DSS-treated mice after DSS treatment. We identified increased number of total CD4+, activated CD4+ CD44+ T cells, colonic DCs, colonic macrophages, monocytes, and neutrophils on days 3, 6, and 9 in Rnf5−/− compared with WT mice (Figure 5D). This observation suggests that lack of RNF5 expression in IECs increases the recruitment of antigen-presenting cells and CD4+ T cells to the inflamed intestines of DSS-treated mice, leading to increased production of Th1 type inflammatory cytokines.
RNF5 Controls the Severity of DSS-Induced Colitis through S100A8-Mediated CD4+ T Cell Activation
We next investigated the involvement of S100A8 in the exacerbation of DSS-induced colitis in the absence of RNF5. We first examined the effects of DSS on S100A8 secretion by MODE-K cells expressing EV or shRNF5 and then determined the effects of MODE-K cell CM on BMDC activation, as described earlier. Notably, DSS alone elevated S100A8 secretion by MODE-K cells, and KD of RNF5 further increased the amount of S100A8 that was secreted (Figure S6B). Moreover, the CM from DSS-treated MODE-K and MODE-K-shRNF5 further increased the expression of MHC classes I and II on BMDCs compared with CM from untreated MODE-K cells (Figure S6C), indicating that DSS treatment enhances IEC-mediated activation of DCs.
This observation raised the possibility that DSS-treated MODE-K-shRNF5 cells may increase antigen presentation by DCs to T cells in the intestinal microenvironment. IECs can exert their influence through the priming of both cellular and humoral adaptive immune responses through a continuous dialog with antigen-presenting cells. To test this, we treated BMDCs with CM from DSS-treated MODE-K and MODE-K-shRNF5 cells and monitored the activation of CD4+ T cells purified from the GP61–80-specific CD4+ TCR transgenic (SMARTA) mice. Proliferation (measured by carboxyfluorescein succinimidyl ester [CFSE] dilution) and IFN-γ and TNF-α production (intracellular staining) by CD4+ T cells were significantly higher after co-incubation with BMDCs stimulated by CM from DSS-treated MODEK-shRNF5 cells compared with MODE-K or MODE-K-shRNF5/ shS100A8 cells (Figures 5E and S6D). These data further support the notion that loss of RNF5 from IECs leads to enhanced secretion of S100A8, which consequently activates DCs and enhances antigen-specific CD4+ T cell proliferation and effector responses. Importantly, the reversal of these effects by simultaneous KD of both RNF5 and S100A8 in MODE-K cells confirms that these effects result from RNF5-mediated control of S100A8 ubiquitination and degradation in IECs.
To substantiate the possible role of CD4+ T in the DSS-induced colitis, we monitored level and possible contribution of CD4 cells to the severe colitis phenotype identified in the Rnf5−/− mice. A notable increase in the number of CD4+/ Ki67+ T cells was identified in the colon of Rnf5−/− mice after DSS treatment (Figure S6E), consistent with its expected role in promoting the intestinal inflammatory phenotype. Significantly, administration of neutralizing antibodies against CD4+ T cells abolished severe acute colitis phenotypes in Rnf5−/− mice including body weight loss, colon length, and disease activity index (Figures 5F–5H). These data establish the causative role of CD4+ T cell in the acute colitis phenotype driven by the RNF5-S100A8 axis.
Neutralizing Antibodies to S100A8 Attenuate Acute Colitis in DSS-Treated Rnf5−/− Mice
To directly assess the significance of elevated S100A8 expression to the acute colitis phenotype, identified in the Rnf5−/− mice, we evaluated the therapeutic potential of S100A8 blockade in the DSS-induced colitis model. Intraperitoneal injection of S100A8-neutralizing antibodies (Raquil et al., 2008; Vandal et al., 2003) or a control rabbit IgG on days −1, +1, and +3 relative to the initiation of DSS treatment (day 0) ameliorated body weight loss, colon shortening, and clinical disease activity index (based on the severity of diarrhea, stool blood, and weight loss) and elevated survival in DSS-treated Rnf5−/− mice, whereas it only modestly improved the disease phenotypes in WT mice (Figures 6A–6D). Histological analysis confirmed that administration of S100A8-neutralizing antibody significantly reduced the DSS-induced colon damage, histological scores, and CD4+ T cell, F4/80+ cell, and Ly6G/C+ cell infiltration in Rnf5−/− mice (Figures 6E, 6F, S7A, and S7B). These data suggest that high levels of S100A8 seen with absence of RNF5 in the colon underlie the severe acute colitis phenotype seen upon DSS stimuli. Furthermore, using neutralizing S100A8 antibodies was also effective in WT mice that developed severe colitis following treatment with higher concentration of DSS (4.5%) (Figures S7C–S7E), pointing to the therapeutic potential of neutralizing S100A8 for IBD patients with severed colitis phenotypes associated with high S100A8 levels.
Figure 6. Neutralizing Antibodies to S100A8 Attenuate Acute Colitis in DSS-Treated Rnf5−/− Mice.

(A) Body weight of WT and Rnf5−/− mice administered 2.5% DSS and treated with control rabbit IgG or rabbit anti-S100A8-neutralizing antibody (200 μg, i.p.) on days –1, 1, and 3, relative to the start of DSS treatment on day 0 (n = 6 mice/group), as shown by red arrows (on day 9: ****p < 0.0001; Rnf5−/− + control IgG versus Rnf5−/− + anti-S100A8, ****p < 0.0001; WT + control IgG versus Rnf5−/− +control IgG, n.s.; WT + anti-S100A8 versus Rnf5−/− +anti-S100A8).
(B) Disease activity index (see STAR Methods) of anti-S100A8- or control rabbit IgG-treated WT or Rnf5−/− mice after 2.5% DSS treatment as described for (A) (n = 5) (on day 9: *p < 0.05; Rnf5−/− + control IgG versus Rnf5−/− + anti-S100A8, *p < 0.05; WT + control IgG versus Rnf5−/− +control IgG, n.s.; WT + anti-S100A8 versus Rnf5−/− + anti-S100A8).
(C) Survival of anti-S100A8- or control IgG-treated WT and Rnf5−/− mice after 2.5% DSS treatment (n = 6 mice/group) by Kaplan-Meier method.
(D) Representative images (left) of colon and cecum and quantification (right) of colon length from anti-S100A8- or control rabbit IgG-treated WT or Rnf5−/− mice as described for (A) on day 9 after 2.5% DSS treatment (n = 5).
(E) Representative images of H&E-stained and anti-CD4-stained colon sections from WT and Rnf5−/− mice treated as described for (A). Sections are from mice sacrificed on day 9. Scale bars, 50 μm.
(F) Quantification of histological score and CD4+ cells (per 203 field) from WT and Rnf5−/− mice treated with anti-S100A8 antibody or control rabbit IgG after 2.5% DSS treatment for (E) (n = 3 mice/group, three fields each).
(G) Body weight of WT and Rnf5−/− mice administered 2.5% DSS for 5 days and treated with control rabbit IgG or rabbit anti-S100A8-neutralizing antibody (200 μg, i.p.) on days 5, 7, and 9 (n = 6 mice/group), as shown by red arrows (on day 9: **p < 0.01; Rnf5−/− + control IgG versus Rnf5−/− + anti-S100A8, **p < 0.01; WT + control IgG versus Rnf5−/− +control IgG, n.s.; WT + anti-S100A8 versus Rnf5−/− +anti-S100A8).
All data are representative of two or three independent experiments. Graphs show mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001 by two-way ANOVA followed by Tukey’s multiple comparison test (A, B, and G) or one-way ANOVA (D, F). Survival by log rank test (C).
Given the ability to attenuate the colitis phenotype upon administration of the neutralizing antibodies prior and during DSS treatment, we asked whether administration of the S100A8-neutralizing antibodies after DSS administration will be also effective. Administration of the neutralizing antibody on day 5, 7, and 9 after 5 days of 2.5% DSS treatment effectively attenuated the progression of acute colitis in the Rnf5−/− mice (Figure 6G). These results highlight the role of S100A8 in the etiology of acute colitis and signify the importance of its regulation by RNF5, while pointing to the therapeutic potential of targeting the RNF5-S100A8 axis for IBD treatment.
Expression of S100A8 and RNF5 in Samples from IBD Patients
To further understand the potential biological significance of RNF5/S100A8 protein expression in IBD pathogenesis, IHC stains were performed on 17 healthy colon sections and 19 colon sections from un-inflamed and from inflamed areas of ulcerative colitis patients (Table S2). RNF5 was highly expressed in IECs and stroma of healthy controls and in colon sections from the un-inflamed area of colitis patients (Figure 7A). This suggests that RNF5 could be involved in maintaining human intestinal homeostasis. Notably, reduced level of RNF5 expression in IECs and stroma was observed in the damaged area of colitis patients (Figures 7A, 7B, and 7D). Conversely, S100A8 was highly detected in IECs and stromal infiltrating lymphocytes of colon sections from inflamed area of colitis patients (Figures 7A, 7B, 7D). Quantification of RNF5 and S100A8 expression in IECs and stroma confirmed a significant degree of inverse correlation (Figures 7C and7E; p = 0.0395, p = 0.0419). Noteworthy, pathological severity significantly correlated with low RNF5 and high S100A8 expression in the inflamed IECs, not stroma (Figures 7F and7G), suggesting that the control of S100A8 by RNF5 is occurring primarily in IECs and the importance of RNF5-S100A8 regulatory axis in IECs involved in IBD development and disease progression.
Figure 7. Clinical Relevance of S100A8 by RNF5 in Samples from IBD Patients.

(A) Representative images of IHC staining for RNF5 and S100A8 in colon sections from healthy control and IBD patients (un-inflamed and inflamed area). Scale bars, 50 μm.
(B) Quantification of RNF5 and S100A8 IHC scores of IECs in colon sections from healthy control (n = 17) and IBD patients (un-inflamed and inflamed area) (n = 19).
(C) Scatterplots between the IHC score (IECs) of RNF5 and S100A8 in colitis from inflamed area of IBD patients (n = 19).
(D) Number of RNF5- or S100A8-positive cells (403) in stroma of colon sections from healthy control (n = 17) and IBD patients (un-inflamed and inflamed area) (n = 19).
(E) Scatterplots between number of RNF5- and S100A8-positive cells in stroma of colitis from inflamed area of IBD patients (n = 19).
(F) RNF5 IHC score (IECs) and S100A8 IHC score (IECs) in the pathological severity (PS)-low (n = 4) or PS-high (n = 12) colitis from inflamed area of IBD patients.
(G) Number of RNF5- or S100A8-positive cells (stroma) in the PS-low (n = 4) or PS-high (n = 12) colitis from inflamed area of IBD patients.
Graphs show mean ± SEM. *p < 0.01, **p < 0.001, and ***p < 0.0001 by one-way ANOVA followed by Tukey’s multiple comparison test (B and D). Pearson’s correlation (C and E). n.s., not significant (p > 0.05). *p < 0.05 and ***p < 0.001 by two-tailed t test (F and G).
Low RNF5 Expression in IBD Patients Who Are Non-responders to Anti-TNF Therapy
Cell-centered meta-analysis from mucosal gene expression data has recently revealed that the triggering receptor expressed on myeloid cells 1 (TREM1)-CCL7-CCR2 axis is upregulated in IBD patients who are non-responders to anti-TNF treatment, thus pointing to blood TREM-1 expression as a predictor of non-response to anti-TNF treatment (Gaujoux et al., 2018). Notably, comparing the expression of TREM1-CCL7-CCR2 axis with that of RNF5 revealed a negative correlation. A low level of RNF5 transcription coincided with a high level of TREM1 axis-related transcripts in non-responders (Figure S8A) both prior and 2 weeks following anti-TNF treatment (Figure S8B). Although at the transcript level, this observation points to the importance of RNF5 in an orthogonal inflammatory axis for IBD patients receiving anti-TNF therapy.
DISCUSSION
The ubiquitin ligase RNF5 regulates the degradation of proteins important for a number of cellular processes with pathophysiological implications. Here we identify that RNF5 plays a key role in the regulation of DSS-induced acute colitis development through its regulation of S100A8 stability. We demonstrate that colonic tissue of Rnf5−/− mice has higher basal levels of inflammation compared with WT mice, although this is not sufficient to result in a notable inflammatory phenotype, unless challenged with DSS. The acute and severe colitis induced by DSS in the Rnf5−/− mice was associated with excessive inflammation, reduced colon size, bleeding, and diarrhea. Notably, phenotypes seen in the Rnf5−/− mice resemble those seen in mice mutant for IkB kinase/NF-κB signaling components and TLR ligands (Fukata and Arditi, 2013), key immune regulatory molecules, pointing to common or shared regulatory pathways. Mechanistically, our data demonstrate that the stability of S100A8 protein is regulated by RNF5-mediated ubiquitination-dependent proteasomal degradation. Correspondingly, elevated levels of S100A8 were identified in the colon of Rnf5−/− mice and in MODE-K IECs that were subjected to KD of RNF5. Post-translational modifications are expected to determine the binding between S100A8 and RNF5, prior to and 24 hr after TNF stimuli. The window of 24 hr, allowing temporal yet limited increase in S100A8 levels, seen following cytokine stimuli, is expected to define the severity of colitis.
In IBD, the inflamed gut mucosa represents an enriched source of local and systemic DAMPs, including S100A8/ S100A9. We found that S100A8 had both cell intrinsic effects (NF-κB) and extrinsic effects (DCs and Th1 cells) that influenced colitis development. Strikingly, S100A8-neutralizing antibodies were able to significantly attenuate the colitis phenotype, suggesting that the secreted form of S100A8 underlies the severe colitis phenotypes. Neutralization of S100A8 was shown to offer an effective therapeutic strategy in some animal models of inflammatory disorders associated with neutrophil migration in response to lipopolysaccharide stimuli (Vandal et al., 2003), streptococcal pneumonia (Raquil et al., 2008), and lung metastasis (Hiratsuka et al., 2006). Our data suggest that specific blockade of S100A8 may offer a therapeutic strategy for human intestinal inflammatory diseases, including IBD. Of note, blocking S100A9, which is not subject to regulation by RNF5, was also reported to inhibit inflammation in models of systemic lupus erythematosus, streptococcal pneumonia, and acute gouty arthritis. Because the S100A8/S100A9 complex is a highly abundant DAMP that is implicated in mediating severe inflammation, our findings suggest that it may be sufficient to inhibit only S100A8. Our findings are corroborated by analyses of IECs and stroma sections of colon biopsies from IBD patients who exhibited a negative correlation between RNF5 and S100A8 protein expression. Additionally, pathological severity correlated with low RNF5 and high S100A8 expression in the inflamed IECs (but not in stroma of colon sections), further support a role for the RNF5-S100A8 axis in IECs in the development and progression of IBD. Notably, RNF5 expression was negatively correlated with S100A8 and with the TREM1-CCL7-CCR2 axis, which is upregulated in IBD patients who are non-responders to anti-TNFα treatment. That RNF5-S100A8 axis can be linked with the therapeutic responses to anti-TNFα treatment further substantiate the importance of this regulatory pathway for therapeutic considerations of intestinal inflammatory disorders.
In all, our study provides important conceptual advances to current understanding of S100A8 role in intestinal inflammatory disorders. Our findings also raise the possibility that RNF5 may contribute to the etiology of other inflammatory and/or autoimmune disorders. While neutralizing antibodies to S100A8 may provide a therapeutic modality for RNF5 in low expressing intestinal inflammatory diseases, our findings also points to therapeutic opportunities along pathways involved in the regulation of this ubiquitin ligase.
STAR★METHODS
Detailed methods are provided in the online version of this paper and include the following:
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER | ||
|---|---|---|---|---|
| Antibodies | ||||
| anti-RNF5 | (Delaunay et al., 2008) | N/A | ||
| anti-S100A8 | (Vandal et al., 2003) | N/A | ||
| anti-b-actin | Santa Cruz Biotechnology | Cat#sc-47778; RRID: AB_626632 | ||
| anti-HSP90 | Santa Cruz Biotechnology | Cat#sc-13119; RRID: AB_675659 | ||
| anti-CD45 | BD Biosciences | Cat#550539; RRID: AB_2174426 | ||
| anti-CD11c | Abcam | Cat#ab11029; RRID: AB_297683 | ||
| anti-V5 | Invitrogen | Cat# 46–0705; RRID: AB_2556564 | ||
| anti-FLAG | Sigma-Aldrich | Cat# F1804; RRID: AB_262044 | ||
| anti-HA | Santa Cruz Biotechnology | Cat#sc-805; RRID: AB_631618 | ||
| anti-ubiquitin | Cell Signaling Technology | Cat#3936; RRID: AB_331292 | ||
| anti-S100A8 | Santa Cruz Biotechnology | Cat#sc-48352; RRID: AB_628230 | ||
| anti-S100A8 | Abcam | Cat#ab92331; RRID: AB_2050283 | ||
| anti-S100A9 | Santa Cruz Biotechnology | Cat#sc-53187; RRID: AB_784389 | ||
| anti-Ki67 | Abcam | Cat#ab15580; RRID: AB_443209 | ||
| anti-cleaved caspase-3 | Cell Signaling Technology | Cat#9661; RRID: AB_2341188 | ||
| anti-F4/80 | Abcam | Cat#ab6640; RRID: AB_1140040 | ||
| anti-Ly6G/C | R&D systems | Cat#MAB1037; RRID: AB_2232806 | ||
| anti-CD4 | Abcam | Cat#ab183685; RRID: AB_2686917 | ||
| anti-CD4 | eBioscience | Cat#14–9766-80; RRID: AB_2573007 | ||
| anti-CD8 | Invitrogen | Cat#14–0808; RRID: AB_2572860 | ||
| anti-CD16/CD32 | BioLegend | Clone 93; RRID: AB_312800 | ||
| anti-CD45.2 | BioLegend | Clone 104; RRID: AB_893349 | ||
| anti-CD8α | BioLegend | Clone 53–6.7; RRID: AB_493425 | ||
| anti-CD4 | BioLegend | Clone GK1.5; RRID: AB_2564591 | ||
| anti-CD25 | BioLegend | Clone 3C7; RRID: AB_961210 | ||
| anti-CD44 | BioLegend | Clone IM7; RRID: AB_830785 | ||
| anti-TNF-α | BioLegend | Clone MP6-XT22; RRID: AB_2204356 | ||
| anti-IFN-γ | BioLegend | Clone XMG1.2; RRID: AB_315403 | ||
| anti-CD11c | BioLegend | Clone N418; RRID: AB_313778 | ||
| anti-Ly6G | BioLegend | Clone 1A8; RRID: AB_1186104 | ||
| anti-Ly6C | BioLegend | Clone HK1.4; RRID: AB_1732093 | ||
| anti-CD103 | BioLegend | Clone 2E7; RRID: AB_1227503 | ||
| anti-F4/80 | BioLegend | Clone BM8; RRID: AB_2277848 | ||
| anti-NK1.1 | BioLegend | Clone PK136; RRID: AB_2686977 | ||
| anti-MHC class I | BioLegend | Clone AF6–88.5; RRID: AB_313733 | ||
| anti-MHC class II | BioLegend | Clone M5/114.15.2; RRID: AB_493527 | ||
| anti-CD19 | BioLegend | Clone 1D3/CD19; RRID: AB_2629814 | ||
| anti-Thy1.2 | BioLegend | Clone 53–2.1; RRID: AB_10640729 | ||
| anti-CD4 (GoInVivo™ grade) | BioLegend | Clone GK1.5; RRID: AB_2616832 | ||
| rat LgG2b isotype control (GoInVivo™ grade) | BioLegend | Clone RTK4530; RRID: AB_326560 | ||
| Biological Samples | ||||
| IBD biopsy samples | Rambam Health Care Campus,Gastroenterology Institute | N/A | ||
| Chemicals, Peptides, and Recombinant Proteins | ||||
| Dextran sulfate sodium salt colitis grade | MP Biomedicals | Cat#160110 | ||
| GM-CSF | eBioscience | Cat#14–8331-80 | ||
| Lipopolysaccharide | Enzo Life Sciences | Cat#ALX-581–008-L001 | ||
| Recombinant mouse TNF-α protein | BioLegend | Cat#580102 | ||
| Recombinant mouse S100A8 protein | BioLegend | Cat#765302 | ||
| CFSE (carboxyfluorescein succinimidyl easter) | Thermo Fisher Scientific | Cat# C34554 | ||
| GP61–80 peptide | AnaSpec | Cat#AS-64851 | ||
| Jetprime | PolyPlus | Cat#114–07 | ||
| Critical Commercial Assays | ||||
| RNeasy Plus Mini Kit | QIAGEN | Cat#74134 | ||
| Mouse IL-12p70 DuoSet ELISA | R&D Systems | Cat#DY419 | ||
| Mouse IL-1βDuoSet ELISA | R&D Systems | Cat#DY401 | ||
| Mouse S100A8 ELISA | Abcam | Cat#ab213886 | ||
| LEGENDplex mouse inflammation panel | BioLegend | Cat#740446 | ||
| LEGENDplex mouse proinflammatory chemokine panel | BioLegend | Cat#740451 | ||
| Dual-Luciferase Reporter Assay System | Promega | Cat#E1910 | ||
| Deposited Data | ||||
| Ra and analyzed data (deposited in the Short Read Archive (SRA: http://.ncbi.nlm.nih.gov/sra/) of NCBI under BioProject) | This paper | PRJNA422424 | ||
| Experimental Models: Cell Lines | ||||
| Mouse cell line: MODE-K cells | provided by Dr. Dominique Kaisserlain | N/A | ||
| Human cell line:HEK293T cells | ATCC | CRL3216 | ||
| Experimental Models: Organisms/Strains | ||||
| Mouse: Rnf5−/− | (Delaunay et al., 2008) | N/A | ||
| Mouse: SMARTA mice | provided by Dr. Charles D. Surh | N/A | ||
| Recombinant DNA | ||||
| Plasmid: FLAG-RNF5 | {Jeon et al., 2015 #1787} | N/A | ||
| Plasmid: S100A8-V5 | Arizona State University BiodesignInstitute | HsCD00438374 | ||
| Plasmid: pLKO.1 control vector | Sigma-Aldrich | SHC001 | ||
| Plasmid: shRNF5#1 | Sigma-Aldrich | TRC0000041189 | ||
| Plasmid: shRNF5#2 | Sigma-Aldrich | TRC0000041190 | ||
| Plasmid: NF-κB-luc | N/A | |||
| Softare and Algorithms | ||||
| GraphPad Prism 7 | Graphpad Softare | N/A | ||
| FloJo | Treestar | N/A | ||
| Pannoramic vieer | 3DHISTECH Ltd | N/A | ||
| Other | ||||
| qPCR primer sequences, See Table S3 | This paper | N/A | ||
CONTACT FOR REAGENTS AND RESOURCE SHARING
Further information and requests for reagents should be directed to and will be fulfilled by the Lead contact, Ze’ev Ronai, at zeev@ronailab.net.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human samples
The protocol was approved by the Institutional Review Board at the Rambam Health Care Campus (RHCC), Gastroenterology Institute (IRB no. RMB-0405–17). Colonic mucosal biopsies were obtained with written informed consent and collected from healthy controls (n = 17) and patients with known IBD (ulcerative colitis (UC); n = 19) undergoing endoscopy. Formalin embedded human biopsies from colonoscopies of UC patients and healthy controls were retreated from the pathological archives at RHCC. Biopsies were obtained from UC patients who underwent colonoscopy at RHCC between 2015–2017 and had documented biopsies from both inflamed and un-inflamed tissue were included. Clinical data (age, gender, concomitant UC related therapy) and endoscopic severity (severity defined according to the endoscopic Mayo score, range of 0–3, higher scores indicate higher mucosal inflammation) were recorded. Pathological severity of inflamed tissue was documented according to pathological report by normal, mild, moderate and severe inflammation (score 0–3, in increasing order, when 0 stands for normal tissue and 3 for severe inflammation). The clinical severity scores are categorized as 0–1 (low) and 2–3 (high). Biopsies from healthy persons undergoing screening colonoscopy at RHCC with random biopsies taken during this time period was retrieved to serve as healthy controls. Staining intensity of RNF5 and S100A8 (IECs) was scored using a four tier scale from 0 (no staining) to 3 (strongest staining). Intensity scores were multiplied by the percentage of IECs with positive staining to generate an IHC score (maximum score, 300) by Panoramic viewer (3DHISTECH Ltd). Table S2 shows a summary of the patients and tissues used in this study.
Animal studies
All animal experiments were approved by Sanford Burnham Prebys Medical Discovery Institute’s Institutional Animal Care and Use Committee (AUF 17–001). Animal care was followed according to institutional guidelines. All mouse strains were generated in a C57BL/6 background. Rnf5−/− mice were generated as previously described (Delaunay et al., 2008). SMARTA mice (Oxenius et al., 1998) were from Dr. Charles D. Surh (La Jolla Institute for Allergy and Immunology). C57BL/6 were used as WT and control littermates, respectively. For all experiments, 6–10-week-old mice were used. Mice were maintained under controlled temperature (22.5°C) and illumination (12 h dark/light cycle) conditions. Experimental colitis was induced by the addition of 2.5% DSS (weight/ volume) (molecular weight 36–50 kDa; MP Biomedicals) to the drinking water for 5 or 7 days, followed by regular drinking water through the end of the experiment. DSS-containing water was exchanged every other day. Body weight, water consumption, and the severity of diarrhea and rectal bleeding were monitored. Diarrhea was scored as: 0, normal; 2, loose stools; and 4, watery diarrhea. Blood in stools was scored as: 0, normal; 2, slight bleeding; and 4, gross bleeding. Weight loss was scored as: 0, none; 1, 1%–5%; 2, 5%–10%; 3, 10%–15%; 4, > 15%. Disease activity index was the average of ([diarrhea + stool blood + weight loss scores] / 3) for each animal (Yoshihara et al., 2006). Cecum and colon tissues were removed and cleaned, and sections were taken for tissue culture, flow cytometry, IHC, and histology.
Cell lines
MODE-K (established and kindly provided by Dr. Dominique Kaisserlain) and HEK293T (from ATCC) cells were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) (GE Healthcare Life Sciences) containing 10% fetal bovine serum (FBS) and penicillinstreptomycin. Bone marrow-derived dendritic cells (BMDCs) were differentiated [6 days in GM-CSF (20 ng/ml; eBioscience, #14–8331-80)]. BMDCs and splenic DCs were maintained in RPMI 1640 medium (Corning) containing 10% FBS and penicillin-streptomycin. Cells were incubated at 37°C.
METHOD DETAILS
Antibodies and plasmids
The anti-RNF5 antibody was described previously (Delaunay et al., 2008). Polyclonal rabbit S100A8 blocking antibody was produced as previously described (Vandal et al., 2003). Other antibodies were obtained from the following sources: b-actin (Santa Cruz Biotechnology, sc-47778), HSP90 (Santa Cruz Biotechnology, sc-13119), CD45 (BD Biosciences, 550539), CD11c (Abcam, ab11029), V5 (Invitrogen, 46–0705), FLAG (Sigma-Aldrich, F1804), HA (Santa Cruz Biotechnology, sc-805), ubiquitin (Cell Signaling Technology, 3936), S100A8 (Santa Cruz Biotechnology, sc-48352 and Abcam, ab92331), S100A9 (Santa Cruz Biotechnology, sc-53187), Ki-67 (Abcam, ab15580), cleaved caspase-3 (Cell Signaling Technology, 9661), F4/80 (Abcam, ab6640), Ly6G/C (R&D Systems, MAB1037), CD4 (Abcam, ab183685 and eBioscience, 14–9766-80), and CD8 (Invitrogen, 14–0808). Plasmids expressing FLAG-RNF5 were described previously (Jeon et al., 2015). Plasmids expressing S100A8-V5 were purchased from Arizona State University Biodesign Institute. shRNAs were purchased from Sigma-Aldrich (MISSION shRNA Plasmid DNA). The RNAi Consortium lentiviral pLKO.1 control vector (EV) served as the control for shRNA experiments. Cells were transfected with plasmids or shRNAs using Jetprime (PolyPlus).
RNA extraction and analysis
Total RNA was extracted from cultured cells or colon tissues using QIAzol and RNeasy Mini Kits (QIAGEN) according to the manufacturer’s protocol. The purity and concentration of extracted RNA were checked and quantified by reading A260 nm and A280 nm in a NanoDrop 1000 spectrophotometer (Thermo Scientific). cDNA was synthesized using oligo(dT) and random primers (AB Bioscience), and qPCR analysis was performed with SYBR Green (Roche). Primer sequences are listed in Table S3. The results were normalized to Gapdh mRNA.
RNA-Seq
RNA was extracted from naive WT or Rnf5−/− colon tissues (3 per group). RNA was ribo depleted using the NEBNext rRNA Depletion Kit and barcoded libraries were made using the NEBNext Ultra II Directional RNA Library Prep Kit for Illumina (NEB). Libraries were pooled and single end sequenced (1X75) on the Illumina NextSeq 500 using the high output V2 kit (Illumina Inc). Read data was processed in BaseSpace (basespace.illumina.com). Reads were aligned to Mus musculus genome (mm10) using STAR aligner (code.google.com/p/rna-star/) with default settings. Differential transcript expression was determined using the Cufflinks Cuffdiff package (https://github.com/cole-trapnell-lab/cufflinks). The accession number for the RNA-seq data reported in this paper is Short Read Archive (SRA) of NCBI Bioproject: PRJNA422424. For the analysis of naive WT or Rnf5−/− colon tissues, we select differentially expressed genes based on the criteria of FDR-adjusted P value < 0.05 using Ingenuity Pathway Analysis (IPA, http://www.ingenuity.com).
Immunoprecipitation and immunoblotting
For immunoprecipitations, cell lysates were prepared using lysis buffer (1% Triton X-100 in 50 mM Tris-HCl, pH 7.4, 150 mM NaCl) supplemented with protease and phosphatase inhibitors (Thermo Scientific). Lysates were incubated with the appropriate antibodies and protein A/G agarose beads (Santa Cruz Biotechnology) according to the manufacturer’s protocol. Beads were washed with lysis buffer, boiled in Laemmli buffer, and proteins were resolved by SDS-PAGE and transferred to membranes. To detect endogenous S100A8–RNF5 interactions, MODE-K cells were pretreated with 10 μM MG132 (Selleckchem) for 4 h before lysis. For immunoblotting without immunoprecipitation, cell or tissue lysates were prepared using M-PER buffer (Thermo Scientific) containing protease and phosphatase inhibitors. Equal amount of protein samples were fractionated using SDS-PAGE gels and transferred to PVDF membranes (Millipore, Sigma). After blocking with 5% BSA, the membranes were incubated with primary antibodies overnight at 4°C, followed by 1 h incubation with HRP-conjugated secondary antibodies. Protein signals were visualized using the ECL detection system (Mortsel) or ChemiDoc imaging system (Bio-Rad) according to the manufacturer’s instructions.
Histology, immunohistochemistry, and immunofluorescence
Immediately after mouse sacrifice, the intestines were removed, cut open lengthwise, rinsed, and rolled up from the proximal to distal end to form a Swiss roll. Sections (5 mm) were cut in a Leica Microsystems cryostat and transferred onto Superfrost-Plus slides (Fisher Scientific), and stained with hematoxylin and eosin (H&E). Tissue damage in the colons was scored as follows: epithelium, 0 = normal morphology, 1 = loss of goblet cells, 2 = loss of goblet cells in large areas, 3 = loss of crypts, 4 = loss of crypts in large areas; and infiltration, 0 = no infiltrate, 1 = infiltrate around the crypt basis, 2 = infiltrate reaching the lamina muscularis mucosae, 3 = extensive infiltration reaching the lamina muscularis mucosae and thickening of the mucosa with abundant edema, 4 = infiltration of the submucosa. Total histological score was the sum of the epithelium and infiltration scores.
For immunohistochemistry, the sections were deparaffinized and rehydrated, and antigen was retrieved using Dako target retrieval solution. Endogenous peroxidase activity was quenched by incubation with 3% hydrogen peroxide for 30 min. Specimens were incubated with primary antibodies diluted in Dako antibody diluent overnight at 4°C. Slides were then washed three times with PBS/ 0.03% Tween-20 and incubated with Dako Labeled Polymer-HRP for 1 h at room temperature. After three washes with PBS/ 0.03% Tween-20, the sections were incubated with DAB chromogen and counterstained with hematoxylin. Staining intensity of RNF5, S100A8, and S100A9 was scored using a four tier scale from 0 (no staining) to 3 (strongest staining). Intensity scores were multiplied by the percentage of intestinal cells with positive staining to generate an IHC score (maximum score, 300) by Aperio slide scanner.
For immunofluorescence, sections were incubated with Alexa-conjugated secondary antibodies (Life Technologies), and the slides were covered with a drop of Vectashield with DAPI (H-1200, Vector Laboratories). Slides were visualized using a fluorescence microscope with a Slidebook or Aperio slide scanner. Periodic acid–Schiff (PAS) and alcian blue (AB) staining (Thermo Scientific) were performed according to standard protocols.
Flow cytometry and cell isolation
The intestines were removed, carefully cleaned of the mesentery, opened longitudinally, and washed free of fecal content. The intestines were then cut into 0.5-cm pieces and transferred to 50 mL conical tubes. Lamina propria leukocytes were isolated using the Miltenyi Lamina Propria Dissociation Kit (Miltenyi Biotec, 130–097-410) according to the manufacturer’s instructions. For cell surface staining, 2 × 3 106 cells per sample were resuspended in staining buffer (PBS, 2% FBS, and 0.01% NaN3), pre-incubated in mouse Fc block (anti-CD16/CD32; BioLegend) and then stained for 20 min at 4°C with the following antibodies (BioLegend): CD45.2 (104), CD8α (53–6.7), CD4 (GK1.5), CD25 (3C7), CD44 (IM7), TNF-α (MP6-XT22), IFN-γ (XMG1.2), CD11c (N418), Ly6G (1A8), Ly6C (HK1.4), CD103 (2E7), F4/80 (BM8), NK1.1 (PK136), MHC class I (AF6–88.5), and MHC class II (M5/114.15.2). Cells were washed twice with FACS staining buffer, fixed for 15 min with 1% formaldehyde in PBS, washed twice again, and resuspended in FACS staining buffer for analysis. All data were collected on an LSRFortessa (BD Biosciences) and analyzed using FlowJo software (Treestar). For splenic DC isolation, spleens from WT or Rnf5−/− mice were minced and digested with 1 mg/ml collagenase D (Roche) and 100 μg/ml DNase 1 (Sigma) in 5% CO2 incubator. Cells were passed through a 70-μm cell strainer and erythrocytes were depleted using red cell lysis buffer (Sigma). Cells were then stained using anti-mouse CD19 (1D3/CD19), Thy1.2 (53–2.1), NK1.1, CD11c, and CD45.2 antibodies (Biolegend), and CD11c+ cells (DCs) were sorted using BD FACSAria II. The isolated cells were resuspended in RPMI 1640 medium, then cultured at 37°C in 5% CO2 incubator for ELISA and FACS experiments.
BMDC activation and in vitro CD4+ T cell stimulation assay
To prepare conditioned media, MODE-K-EV, MODE-K-shRNF5, or MODE-K-shRNF5/shS100A8 cells were incubated in DMEM supplemented with 10% FBS for 24 h at 37°C to allow cell attachment. The cells were then treated with medium or 0.5% DSS for 24 h, washed with PBS, and cultured for 48 h in Advanced DMEM (Thermo Fisher) containing 2 mM L-glutamine without serum. The medium was then collected and centrifuged at 2000 × g for 10 min at 4°C to remove cellular debris. The resulting conditioned medium was used for treatment of BMDCs.
BMDCs from WT mice were generated by incubating bone marrow cell suspensions for 6 days with GM-CSF. Cells were then washed and incubated for 18 h with MODE-K conditioned medium (20% v/v) prepared as described above. For S100A8 blockade, cells were incubated conditioned medium in the presence of IgG isotype control (SouthernBiotech) or anti-S100A8 blocking antibody (10 μg/ml). After 18 h, BMDC culture supernatants were removed and IL-12p70 and IL-1β levels were quantified by ELISA.
CD4+ T cells were isolated from the spleens of LCMV-specific CD4 TCR transgenic SMARTA mice by negative bead enrichment (StemCell Technologies). Cells were incubated with 5 μM CFSE for 10 min at 37°C and washed. Cells were then mixed 1:1 with the BMDCs and incubated for 72 h pulsed with 2 μg/ml of GP61–80 peptide (AnaSpec). T cell proliferation was monitored by CFSE dilution using flow cytometry and the division index (a measure of the average number of divisions which includes the undivided cells) was determined using FlowJo software (Treestar). For intracellular cytokine staining, the cells were fixed, permeabilized with Cytofix/ Cytoperm (BD Biosciences) and stained with anti-TNF-α and anti-IFN-γ.
ELISA
S100A8 (Abcam, ab213886), IL-12p70 (R&D Systems, DY419), and IL-1β (R&D Systems, DY401) levels in culture supernatants or in mouse serum were determined by ELISA according to the manufacturers’ protocols. Concentrations were determined by comparison with standard curves. Multiplexed analysis of cytokines and chemokines was performed using LEGENDplex mouse cytokine and mouse proinflammatory chemokine kits (BioLegend). Data were collected on an LSRFortessa and analyzed using LEGENDplex software (BioLegend).
Luciferase reporter assay
MODE-K-EV, MODE-K-shS100A8, MODE-K-shRNF5, or MODE-K-shRNF5/shS100A8 cells were transiently transfected with the NF-κB-dependent firefly luciferase reporter plasmid NF-κB-luc and a Renilla luciferase plasmid pRL-TK using Jetprime according to the manufacturer’s protocol. After 48 h, luciferase was assayed using a dual luciferase kit (Promega) according to the manufacturer’s protocol. Firefly luciferase activity was normalized to Renilla luciferase activity as a control for transfection efficiency.
In vivo antibody treatment
For neutralizing S100A8, mice were injected intraperitoneally (i.p.) with 200 μg of rabbit anti-S100A8 or rabbit IgG isotype control 1 day before and 1 and 3 days after the start of 2.5% DSS treatment. For survival analysis, the mice were injected with antibodies 1 day before and 1, 3, 5, and 7 days after the start of DSS treatment. For the treatment after the colitis induction, mice were injected i.p. with the same amount of each antibody on day 5, 7, and 9 after 5 days of 2.5% DSS treatment. The efficiency of S100A8-depletion was tested by flow cytometry of blood samples. CD4+ T cells were depleted by i.p. injection of 200 mg anti-CD4 (clone GK1.5, GoInVivo™ grade, BioLegend) and rat IgG2b isotype control (GoInVivo™ grade, BioLegend) 1 day before and 3 and 7 days after the start of 2.5% DSS treatment. The efficacy of depletion was confirmed by FACS analysis of blood samples.
Public mucosal IBD gene expression data and targeted correlation analysis
For targeted co-expression analysis we used three IBD datasets of public colonic gene expression data at baseline, available in Gene Expression Omnibus (GEO) database: two cohorts of UC patients including GSE14580 and GSE12251 and additional cohort of CD patients from GSE16879, for which we also analyzed expression 2 weeks post first treatment. Probe set annotation was performed using affycoretools and hgu133plus2cdf.db R packages. Response classification was used as reported, according to endoscopic and histologic findings at 4–8 weeks post first treatment. Pairwise Spearman’s rank correlation coefficients were calculating between the genes of interest, separately for responders and non-responders groups using Hmisc R package and visualized by ComplexHeatmap R package.
QUANTIFICATION AND STATISTICAL ANALYSIS
Differences between two groups were assessed by two-tailed t test, and differences between more than two groups were assessed by ANOVA followed by Tukey’s post hoc multiple comparisons test. Pearson’s correlation was estimated between RNF5 and S100A8 expression. p < 0.05 was considered statistically significant. GraphPad Prism 7 software was used for all analyses.
Supplementary Material
Highlights.
Severe intestinal inflammation resembling acute colitis in DSS-treated Rnf5−/− mice
RNF5 regulates S100A8 stability and pro-inflammatory responses
Neutralizing S100A8 antibodies attenuate acute colitis in DSS-treated Rnf5−/− mice
Inverse expression of RNF5 and S100A8 in IBD patients coincides ith disease severity
ACKNOWLEDGMENTS
We thank Guillermina Garcia and Monica Sevilla at the histology core, Hilda Clarke and Judy Wade at the animal core facility, Yoav Altman at the fluorescence-activated cell sorting (FACS) core facility, and Kang Liu at the genomic core facility for their help in respective experiments. Support through grant P30 CA030199 to core facilities at the Sanford-Burnham-Prebys National Cancer Institute (NCI) Cancer Center is gratefully acknowledged. We also thank Serge Y. Fuchs and members of the Ronai lab for discussions. Funding through NCI Outstanding Investigator Award (OIA) grant R35CA197465 (Z.A.R.) is gratefully acknowledged. Y.F. was support by a Japan Society for the Promotion of Science (JSPS) Postdoctoral Fellowship for Research Abroad.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information includes eight figures and three tables and can be found with this article online at https://doi.org/10.1016/j.celrep.2018.08.057.
DATA AND SOFTWARE AVAILABILITY
All software is commercially available. The RNA-Seq data were deposited in the Short Read Archive (SRA: http://www.ncbi.nlm.nih. gov/sra/) of NCBI under BioProject accession number PRJNA422424. The raw data were deposited to Mendeley and are available at https://doi.org/10.17632/xg9pb3vfb3.1.
REFERENCES
- Bertheloot D, and Latz E (2017). HMGB1, IL-1α, IL-33 and S100 proteins: dual-function alarmins. Cell. Mol. Immunol 14, 43–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyapati RK, Rossi AG, Satsangi J, and Ho GT (2016). Gut mucosal DAMPs in IBD: from mechanisms to therapeutic implications. Mucosal Immunol. 9, 567–582. [DOI] [PubMed] [Google Scholar]
- Bresnick AR, Weber DJ, and Zimmer DB (2015). S100 proteins in cancer. Nat. Rev. Cancer 15, 96–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canavan JB, Scottà C, Vossenkämper A, Goldberg R, Elder MJ, Shoval I, Marks E, Stolarczyk E, Lo JW, Powell N, et al. (2016). Developing in vitro expanded CD45RA+ regulatory T cells as an adoptive cell therapy for Crohn’s disease. Gut 65, 584–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen BL, and Sachar DB (2017). Update on anti-tumor necrosis factor agents and other new drugs for inflammatory bowel disease. BMJ 357, j2505. [DOI] [PubMed] [Google Scholar]
- de Souza HS, and Fiocchi C (2016). Immunopathogenesis of IBD: current state of the art. Nat. Rev. Gastroenterol. Hepatol 13, 13–27. [DOI] [PubMed] [Google Scholar]
- Delaunay A, Bromberg KD, Hayashi Y, Mirabella M, Burch D, Kirkwood B, Serra C, Malicdan MC, Mizisin AP, Morosetti R, et al. (2008). The ER-bound RING finger protein 5 (RNF5/RMA1) causes degenerative myopathy in transgenic mice and is deregulated in inclusion body myositis. PLoS ONE 3, e1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukata M, and Arditi M (2013). The role of pattern recognition receptors in intestinal inflammation. Mucosal Immunol. 6, 451–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaujoux R, Starosvetsky E, Maimon N, Vallania F, Bar-Yoseph H, Pressman S, Weisshof R, Goren I, Rabinowitz K, Waterman M, et al. ; Israeli IBD research Network (IIRN) (2018). Cell-centred meta-analysis reveals baseline predictors of anti-TNFα non-response in biopsy and blood of patients with IBD. Gut, Published online April 4, 2018. 10.1136/gutjnl-2017-315494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halfvarson J, Brislawn CJ, Lamendella R, Vázquez-Baeza Y, Walters WA, Bramer LM, D’Amato M, Bonfiglio F, McDonald D, Gonzalez A, et al. (2017). Dynamics of the human gut microbiome in inflammatory bowel disease. Nat. Microbiol 2, 17004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henke MO, Renner A, Rubin BK, Gyves JI, Lorenz E, and Koo JS (2006). Up-regulation of S100A8 and S100A9 protein in bronchial epithelial cells by lipopolysaccharide. Exp. Lung Res 32, 331–347. [DOI] [PubMed] [Google Scholar]
- Hiratsuka S, Watanabe A, Aburatani H, and Maru Y (2006). Tumour-mediated upregulation of chemoattractants and recruitment of myeloid cells predetermines lung metastasis. Nat. Cell Biol 8, 1369–1375. [DOI] [PubMed] [Google Scholar]
- Jeon YJ, Khelifa S, Ratnikov B, Scott DA, Feng Y, Parisi F, Ruller C, Lau E, Kim H, Brill LM, et al. (2015). Regulation of glutamine carrier proteins by RNF5 determines breast cancer response to ER stress-inducing chemotherapies. Cancer Cell 27, 354–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuang E, Okumura CY, Sheffy-Levin S, Varsano T, Shu VC, Qi J, Niesman IR, Yang HJ, López-Otín C, Yang WY, et al. (2012). Regulation of ATG4B stability by RNF5 limits basal levels of autophagy and influences susceptibility to bacterial infection. PLoS Genet. 8, e1003007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda N, and Nakano A (1998). RMA1, an Arabidopsis thaliana gene whose cDNA suppresses the yeast sec15 mutation, encodes a novel protein with a RING finger motif and a membrane anchor. Plant Cell Physiol. 39, 545–554. [DOI] [PubMed] [Google Scholar]
- Mørk G, Schjerven H, Mangschau L, Søyland E, and Brandtzaeg P (2003). Proinflammatory cytokines upregulate expression of calprotectin (L1 protein, MRP-8/MRP-14) in cultured human keratinocytes. Br. J. Dermatol 149, 484–491. [DOI] [PubMed] [Google Scholar]
- Neurath MF (2017). Current and emerging therapeutic targets for IBD. Nat. Rev. Gastroenterol. Hepatol 14, 269–278. [DOI] [PubMed] [Google Scholar]
- Ni J, Wu GD, Albenberg L, and Tomov VT (2017). Gut microbiota and IBD: causation or correlation? Nat. Rev. Gastroenterol. Hepatol 14, 573–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oxenius A, Bachmann MF, Zinkernagel RM, and Hengartner H (1998). Virus-specific MHC-class II-restricted TCR-transgenic mice: effects on humoral and cellular immune responses after viral infection. Eur. J. Immunol 28, 390–400. [DOI] [PubMed] [Google Scholar]
- Peters LA, Perrigoue J, Mortha A, Iuga A, Song WM, Neiman EM, Llewellyn SR, Di Narzo A, Kidd BA, Telesco SE, et al. (2017). A functional genomics predictive network model identifies regulators of inflammatory bowel disease. Nat. Genet 49, 1437–1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson LW, and Artis D (2014). Intestinal epithelial cells: regulators of barrier function and immune homeostasis. Nat. Rev. Immunol 14, 141–153. [DOI] [PubMed] [Google Scholar]
- Raquil MA, Anceriz N, Rouleau P, and Tessier PA (2008). Blockade of antimicrobial proteins S100A8 and S100A9 inhibits phagocyte migration to the alveoli in streptococcal pneumonia. J. Immunol 180, 3366–3374. [DOI] [PubMed] [Google Scholar]
- Sands BE (2015). Biomarkers of inflammation in inflammatory bowel disease. Gastroenterology 149, 1275–1285.e2. [DOI] [PubMed] [Google Scholar]
- Turner MD, Nedjai B, Hurst T, and Pennington DJ (2014). Cytokines and chemokines: At the crossroads of cell signalling and inflammatory disease. Biochim. Biophys. Acta 1843, 2563–2582. [DOI] [PubMed] [Google Scholar]
- Vandal K, Rouleau P, Boivin A, Ryckman C, Talbot M, and Tessier PA (2003). Blockade of S100A8 and S100A9 suppresses neutrophil migration in response to lipopolysaccharide. J. Immunol 171, 2602–2609. [DOI] [PubMed] [Google Scholar]
- Yadav V, Varum F, Bravo R, Furrer E, Bojic D, and Basit AW (2016). Inflammatory bowel disease: exploring gut pathophysiology for novel therapeutic targets. Transl. Res 176, 38–68. [DOI] [PubMed] [Google Scholar]
- Yoshihara K, Yajima T, Kubo C, and Yoshikai Y (2006). Role of interleukin 15 in colitis induced by dextran sulphate sodium in mice. Gut 55, 334–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Younger JM, Chen L, Ren HY, Rosser MF, Turnbull EL, Fan CY, Patterson C, and Cyr DM (2006). Sequential quality-control checkpoints triage misfolded cystic fibrosis transmembrane conductance regulator. Cell 126, 571–582. [DOI] [PubMed] [Google Scholar]
- Zackular JP, Chazin WJ, and Skaar EP (2015). Nutritional immunity: S100 proteins at the host-pathogen interface. J. Biol. Chem 290, 18991–18998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang SL, Wang SN, and Miao CY (2017). Influence of microbiota on intestinal immune system in ulcerative colitis and its intervention. Front. Immunol 8, 1674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong B, Zhang L, Lei C, Li Y, Mao AP, Yang Y, Wang YY, Zhang XL, and Shu HB (2009). The ubiquitin ligase RNF5 regulates antiviral responses by mediating degradation of the adaptor protein MITA. Immunity 30, 397–407. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.