

Abstract
Microglia show a rich repertoire of activation patterns regulated by a complex ensemble of surface ion channels, receptors, and transporters. We and others have investigated whether microglia vary their K+ channel expression as a means to achieve functional diversity. However, most of the prior studies were conducted using in vitro models such as BV2 cells, primary microglia, or brain slices in culture, which may not accurately reflect microglia physiology in adult individuals. Here we employed an in vivo mouse model of selective innate immune activation by intracerebroventricular injection of lipopolysaccharides (ICV-LPS) to determine the role of the voltage-gated Kv1.3 channel in LPS-induced M1-like microglial activation. Using microglia acutely isolated from adult brains, we detected Kv1.3 and Kir2.1 currents, and found that ICV-LPS increased the current density and RNA expression of Kv1.3 but did not affect those of Kir2.1. Genetic knockout of Kv1.3 abolished LPS-induced microglial activation exemplified by Iba-1 immunoreactivity and expression of pro-inflammatory mediators such as IL-1β, TNF-α, IL-6 and iNOS. Moreover, Kv1.3 knockout mitigated the LPS-induced impairment of hippocampal long-term potentiation (hLTP), suggesting that Kv1.3 activity regulates pro-inflammatory microglial neurotoxicity. Pharmacological intervention using PAP-1, a small molecule that selectively blocks homotetrameric Kv1.3 channels, achieved anti-inflammatory and hLTP-recovery effects similar to Kv1.3 knockout. We conclude that Kv1.3 is required for microglial M1-like pro-inflammatory activation in vivo. A significant implication of our in vivo data is that Kv1.3 blockers could be therapeutic candidates for neurological diseases where microglia-mediated neurotoxicity is implicated in the pathogenesis.
Keywords: microglia, potassium channel, Kv1.3, Kir2.1, PAP-1, pro-inflammatory
Graphical abstract
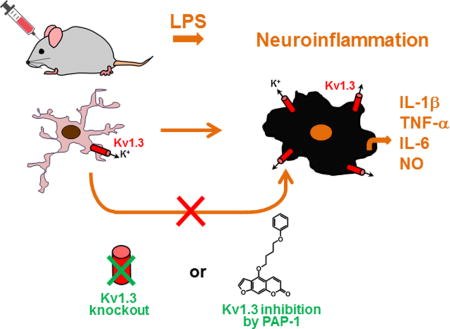
INTRODUCTION
Neuroinflammation driven by microglia activation plays a significant pathological role in several neurological disorders. Microglia, the resident macrophages of the brain, continuously survey their microenvironment and rapidly respond to changes in brain tissue homeostasis or injuries (Wolf et al. 2017). It has long been conceptualized that microglia, similar to macrophages, respond in two major patterns: “classically or M1” activated and “alternatively or M2” activated, as exemplified by the phenotypes induced by activation with interferon-γ and interleukin-4 (IL-4), respectively. The M1 state is usually associated with a pro-inflammatory and neurotoxic role of microglia, while the M2 state has been linked to a reparative and neuroprotective role. However, several recent lines of evidence indicate vast heterogeneity of microglial activation states including those with concurrent M1/M2 features (Kim et al. 2016; Vogel et al. 2013), thus questioning the “simple” microglial M1/M2 polarization hypothesis (Hanisch and Kettenmann 2007; Ransohoff 2016). The functional versatility is aided by a complex ensemble of ion channels, receptors, and transporters on the microglial surface that regulate intracellular signaling and gene expression (Kettenmann et al. 2011). Activities of these surface molecules in principle can be modulated selectively in order to harness microglial activation.
K+ channels are encoded by a super-family of 78 genes (Harmar et al. 2009) and are involved in diverse physiological and pathological processes. K+ channels accordingly already serve as drug targets for cardiac arrhythmia and type-2 diabetes and have been proposed as targets for immunosuppression, cancer and various neurological disorders (Wulff et al. 2009). The voltage-gated Kv1.3 channel is expressed in T lymphocytes, macrophages and microglia and has been suggested as a novel target for immunomodulation in autoimmune diseases such as multiple sclerosis, type-1 diabetes, and psoriasis (Beeton et al. 2006; Arnoux et al. 2013; Kundu-Raychaudhuri et al. 2014; Visentin et al. 2001). Kv1.3 plays an important role in immune cell activation by modulating Ca2+ signaling (Feske et al. 2015; Wulff et al. 2007). Through K+ efflux, Kv1.3 helps maintain a negative membrane potential, which provides the driving force for Ca2+ entry through store-operated inward-rectifier calcium channels like the Ca2+ release activated Ca2+ channel ORAI or transient receptor potential cation channels. We previously demonstrated that cultured primary microglia activated by lipopolysaccharides (LPS), which induces a classic M1-like activation state, exhibited high Kv1.3 current densities but virtually no activities of the other major microglial K+ channel, the inward-rectifier Kir2.1 (Nguyen et al. 2017). A systems pharmacology-based study identified functional roles for Kv1.3 in pro-inflammatory microglial activation (Rangaraju et al. 2017) while electrophysiological studies by our group demonstrated upregulated Kv1.3 expression in microglia acutely isolated from the infarct areas of mice subjected to experimental stroke (Chen et al. 2016). Kv1.3 blockers have further been demonstrated to inhibit microglia mediated neurotoxicity in culture (Fordyce et al. 2005) and to protect mice from microglia mediated radiation-induced brain injury (Peng et al. 2014).
Although Kv1.3 appears to be a suitable target for microglia-targeted modulation, a definite in vivo demonstration of the role of Kv1.3 in neuroinflammation is missing, as nearly all prior studies were conducted using BV2 cells, primary microglia, or brain slices in culture (Charolidi et al. 2015; Fordyce et al. 2005; Khanna et al. 2001; Kotecha and Schlichter 1999; Küst et al. 1999; Nguyen et al. 2017; Rangaraju et al. 2017; Schilling and Eder 2003; Schilling and Eder 2011). It is now realized that BV2 cells significantly differ from cultured or in vivo microglia in many aspects (Butovsky et al. 2014; Henn et al. 2009; Kettenmann et al. 2011). In the widely used culture systems of microglia derived from neonatal rodents, the cells generally assume an amoeboid morphology and are highly proliferative, resembling microglia in injured tissue (Bohlen et al. 2017). The validity of using these cultures to represent adult microglia needs to be carefully verified. Brain slice culture allows preservation of interactions between microglia, neurons, and astrocytes, but in these cultures microglia rapidly lose their characteristic ramified morphology and mature marker expression (Bohlen et al. 2017). The above downsides of various in vitro systems are particularly significant when ion channels such as Kv1.3 are being studied, as much of the physiological recordings in vitro are from moderately to fully activated microglial cells and do not fully represent the microglial physiology in vivo (Kettenmann et al. 2011). For example, our previous study showed that microglia acutely isolated from normal adult mouse brains exhibited very small K+ currents, smaller than what was observed in unstimulated cultured neonatal mouse microglia (Chen et al. 2016). Similarly, microglia, tissue-printed from the hippocampus of 5- to 14-days old rats, were initially not proliferating and showed little Kv1.3 current. However, after several days in culture the microglia became highly proliferative and many cells exhibited a prominent Kv current that was indistinguishable from Kv1.3 (Kotecha and Schlichter 1999). Therefore, the in vivo significance of microglial Kv1.3 remains undetermined based on currently available literature replete with in vitro studies.
In the current study, we employed an in vivo model of selective innate immune activation by intracerebroventricular (ICV) injection of LPS (Maezawa et al. 2006). LPS specifically activates cluster of differentiation (CD) 14/toll-like receptor (TRL) 4 co-receptors, expressed on glia and especially microglia, to induce M1-like pro-inflammatory activation. Here we report evidence supporting that Kv1.3 is required for pro-inflammatory response of microglia in vivo.
METHODS
Mice
All protocols involving mouse models were approved by the Institutional Animal Care and Use Committee of the University of California Davis. C57BL/6 mice were originally purchased from Jackson laboratory. The Kv1.3−/− (Kv1.3KO) line on the C57BL/6 background was a gift from Dr. Leonard Kaczmarek at Yale University (Fadool et al. 2004).
Intracerebroventricular injection of LPS (ICV-LPS)
LPS (E. coli O55:B5, Millipore) dissolved in phosphate-buffered saline (PBS) or vehicle PBS only was ICV administrated to mice stereotactically in a total volume of 2 μL per side of the ventricle. Briefly, mice were anesthetized by 3% isoflurane and then restrained onto a stereotaxic apparatus. A small incision was made to expose the skull and a small burr hole was drilled using a surgical drill. LPS, or vehicle, was injected using a Hamilton syringe with a 27-gauge needle (Hamilton, Reno, NV, USA) into the lateral ventricles via the coordinates: −1 mm posterior to bregma, 1.3 mm lateral to sagittal suture, and 2 mm in depth. The incision was closed using a surgical suture (Ethicon Inc, Somerville NJ), and the mice were placed on an isothermal pad at 36°C and continuously observed following surgery until recovery. Immediately after ICV injection, mice received either an intraperitoneal injection of PAP-1 (40 mg/kg) or the vehicle (Miglyol-812, Neobee M5®, Spectrum Chemicals, Gardena, CA) as control. Twenty-four hours later, mice were euthanized and the brain tissues were processed for LTP, isolation of microglia, or immunostaining. PAP-1 was synthesized in our laboratory as previously described (Schmitz et al. 2005).
Acute isolation of microglia from adult brains
Microglia were acutely isolated from adult brains without culturing as we described (Jin et al. 2015). Briefly, brains were dissociated enzymatically with a Neural Tissue Dissociation Kit (Miltenyi Biotec). Microglia were subsequently purified by the magnetic-activated cell sorting (MACS) using anti-CD11b magnetic beads (Miltenyi Biotec). The whole procedure took about 60 minutes.
Electrophysiology
Patch-clamp of acutely isolated microglia
Acutely isolated microglia were immediately plated on poly-Lysine-coated glass coverslips. All electrophysiological recordings started after cells were incubated at 37° C for 10 min to allow them to attach. Currents were recorded using the whole-cell configuration of the patch-clamp technique at room temperature with an EPC-10 HEKA amplifier. External normal Ringer solution contained 160 mM NaCl2, 4.5 mM KCl, 2 mM CaCl2, 1 mM MgCl2, 10 mM HEPES, pH 7.4, 300 mOsm. Patch pipettes were pulled from soda lime glass (micro-hematocrit tubes, Kimble Chase, Rochester, NY) to resistances of 2-3 MΩ when submerged in the bath solution and filled with an internal solution containing 160 mM KF, 2 mM MgCl2, 10 mM HEPES, and 10 mM EGTA, pH 7.2, 300 mOsm. K+ currents were elicited with 200-ms voltage ramps from 120 to 40 mV at a frequency of 0.1 Hz. Inward rectifier (Kir) currents were measured as peak inward currents at −120 mV and Kv1.3 currents were measured as PAP-1-sensitive and/or use-dependent inactivating outward currents at +40 mV from the same voltage ramp protocol. Access resistance and cell capacitance, a direct measurement of cell surface area, was monitored continuously throughout all recordings. Kir and Kv1.3 current densities were determined by dividing their current amplitudes in pico-amperes (pA) at −120 mV (Kir) or +40 mV (Kv1.3) by the cell capacitance measured in pico-farads (pF). Whole-cell patch-clamp data are presented as mean ± S.D. and statistical significance was determined using paired Student’s t-Test.
Induction of hippocampal long-term potentiation (hLTP) by high frequency stimulation
The preparation of mouse hippocampal slices, and the induction of hLTP by high frequency stimulation of the Schaffer collateral afferents were conducted as we previously described (Maezawa 2017). Coronal slices (300 μm) of mouse hippocampus were prepared from WT and Kv1.3KO mice 24 hrs after ICV injection. The animals were subjected to deep anesthesia with isoflurane and decapitated. The brain was rapidly removed and transferred to a modified artificial cerebrospinal fluid (HI-ACSF) containing (in mM): 220 sucrose, 2 KCl, 0.2 CaCl2, 6 MgSO4, 26 NaHCO3, 1.3 NaH2PO4, and 10 D-glucose (pH 7.4, set by aeration with 95% O2 and 5% CO2). Coronal brain slices were cut in ice-cold modified artificial cerebrospinal fluid (ACSF) with the use of a DTK-1000 D.S.K Microslicer (TedPella, Inc., Redding, CA, USA). The slices were immediately transferred into an ACSF solution containing (in mM) : 126 NaCl, 3 KCl, 2 CaCl2, 1 MgCl2, 26 NaHCO3, 1.25 NaH2PO4, and 10 D-glucose (pH 7.4, set by aeration with 95% O2 and 5% CO2) for at least 40 min at controlled temperature of 35°C. After subsequent incubation for at least 1 hour at room temperature, hemi-slices were transferred to the recording chamber, which was perfused with standard ACSF at a constant flow rate of ~2ml/min. Recordings of field excitatory postsynaptic potentials (fEPSPs) were obtained from the stratum radiatum of the CA1 region of the hippocampus after stimulation of the Schaffer collateral afferents. Extracellular recording electrodes were prepared from borosilicate capillaries with an outer diameter of 1.5 μm (Sutter Instruments) and were filled with 3 M NaCl (resistance, 1-2 MΩ). Baseline stimulation rate was 0.05Hz. fEPSPs were filtered at 2 kHz and digitized at 10 kHz with a Multiclamp 700B amplifier (Molecular Devices, Sunnyvale, CA). Data were collected and analyzed with pClamp 10.3 software (Molecular Devices). Slope values of fEPSPs were considered for quantitation of the responses. For the input-output curves, stimulation current intensity ranged from 0 to 1 mA with steps of 0.1 mA. After 10 min of stable baseline recording of fEPSPs evoked every 20s by application of a constant current pulse of 0.2-0.4 mA with a duration of 60 μs at the current intensity set to evoke 50-60% of the maximal response, LTP was elicited by high-frequency stimulation (HFS), consisting of 2 trains of 100-Hz (1 sec) stimulation with the same intensity and pulse duration used in sampling of baseline fEPSPs (Nicholson and Kullmann 2017). Recording was then continued for 60 min with stimulation of fEPSPs every 20s.
qPCR
Total RNA from acutely isolated microglia was extracted using RNeasy Plus Mini Kit (Qiagen), reverse-transcribed and pre-amplified with Ovation PicoSL WTA System V2 kit (NuGen, San Carlos, CA). The following forward/reverse primer pairs were used: IL-1β (il1β):5’-CCCCAAGCAATACCCAAAGA-3’/5’-TACCAGTTGGGGAACTCTG-3’; TNF-α(tnfα):5’-GACGTGGAACTGGCAGAAGAG-3′/5′-TGCCACAAGCAGGAATGAGA-3′ IL-6 (il6): 5′-GTTCTCTGGGAAATCGTGGA-3′/5-TTCTGCAAGTGCATCATCGT-3′; iNOS (inos2):5′-CGGATAGGCAGAGATTGGAG-3′/5′-GTGGGGTTGTTGCTGAACTT-3′ KV1.1(kcna1):5′-GAGAATGCGGACGAGGCTTC-3′/5′-CCGGAGATGTTGATTACTACG-3′ KV1.2(kcna2):5′-GGTTGAGGCGACCTGTGAAC-3′/5′-TCTCCTAGCTCATAAAACCGGA-3′ KV1.3(kcna3):5′-ATCTTCAAGCTCTCCCGACCA-3′/5′-CGAATCACCATATACTCCGAC-3′ KV3.1(kcnc1):5′-TCGAGGACCCCTACTCATCC-3′/5′-CGATTTCGGTCTTGTTCACG-3′ KCa2.3(kcnn3): 5′-CCCATCCCTGGAGAGTACAA-3′/5′-TTGCTATGGAGCAGCATGAC-3′
For KCa3.1 and Kir2.1, the commercially available primer sets from Bio-Rad were used. For β-actin the commercially available primer set Mouse-ACTB from Applied Biosystems was used. Relative cDNA levels for the target genes were analyzed by the 2-ΔΔCt method using Actb as the internal control for normalization.
Immunofluorescence staining and quantification
Immunofluorescence staining and quantification was performed as previously described (Jin et al. 2015). Briefly, frozen brain sections (20 μm) were fixed in 4% paraformaldehyde and stained with anti-Iba1 (1:200; Wako Chemical) overnight at 4°C, followed by counterstain with DAPI. Photomicrographs of Iba1-immunostain at the cortical and hippocampal CA1 region were taken from three cortical and hippocampal sections per animal. The images were then transformed to 8 bit grayscale and the immunofluorescence intensity analyzed by the ImageJ program. The photography and analysis of immunoreactivity were conducted in a fashion in which the operator did not know the experimental condition of the animal.
Tissue homogenate preparation and Western blot analysis
Brain tissues were homogenized in lysis buffer (150mM NaCl, 10mM NaH2PO4, 1mM EDTA, 1% TritonX100, 0.5% SDS) with protease inhibitor cocktail and phosphatase inhibitor (Sigma). Equivalent amounts of protein were analyzed by 4-15%Tris-HCl gel electrophoresis (Bio-Rad). Proteins were transferred to polyvinylidene difluoride membranes and probed with antibodies. Visualization was enabled using enhanced chemiluminescence (GE Healthcare Pharmacia). The following primary antibodies (dilutions) were used: anti-CD68 (1:1000, BioRad), anti-CD11b (1:1000, Abcam), anti-Iba1 (1:1000, Millipore), anti-Phopspho-p38MAPK (1:1000, Cell Signaling), anti-p38MAPK (1:1000, Cell Signaling) and β-actin (1:2000, Cell Signaling). Secondary antibodies were HRP-conjugated anti-rabbit or anti-mouse antibody (1:1000, GE Healthcare).
ELISA quantification
ELISA quantification of cytokines was conducted as previously described (Maezawa et al. 2011). Briefly, Tissue sample were homogenized in lysis buffer (100 mM TRIS, pH 7.4; 150 mM NaCl; 1 mM EGTA; 1 mM EDTA; 1% Triton X-100; 0.5% Sodium deoxycholate; proteinase inhibitor mix), and centrifuged for 20 min at 15,000 × rpm at 4°C. The supernatants were directly used for the total cytokine (IL-1ß and TNF-α). Concentrations of IL-1ß and TNF-α were measured using the Quantikine sandwich ELISA kit (R&D systems, Minneapolis, MN).
Statistical analysis
Paired Student’s t-test and two-way analysis of variance (ANOVA) with post hoc Bonferroni test, as appropriate, were conducted using the SigmaStat 3.1 (Systat Inc. Point Richmond, CA) or StatView program (version 5.0.1, SAS Institute Inc., Cary, NC). The significance level for the two-sided analysis was set at p<0.05.
RESULTS
LPS increases Kv1.3 current densities on microglia in vivo
We injected 3-month-old WT and Kv1.3 KO (Kv1.3−/−) mice with intracerebroventricular LPS (ICV-LPS) and 24-hrs later acutely isolated microglia for whole-cell voltage-clamp and cytokine measurements. Microglia were isolated using CD11b-based immunopanning as previously described (Bohlen et al. 2017; Jin et al. 2015). The procedure took approximately one hour and the cells, without further culturing, were immediately used for subsequent studies. We elicited Kv currents by voltage ramps from −120 to +40 mV as previously described (Nguyen et al. 2017; Wulff et al. 2003). In order to avoid contributions from calcium-activated K+ channels or chloride currents we used a KF-based pipette solution. While Kv current amplitudes were very low or often barely detectable in microglia isolated from mice receiving intracerebral injection of PBS (ICV-PBS) (Fig. 1A), ICV-LPS induced outwardly rectifying currents with biophysical properties characteristic of homotetrameric Kv1.3 channels typically expressed on immune cells, based on their sensitivity to the specific Kv1.3 blocker PAP-1 (Fig. 1B) and their characteristic use-dependent current decrease upon repeated depolarization at an interval of 1 sec (Fig. 1C). Recordings from Kv1.3 KO microglia showed mostly no detectable Kv1.3 currents with ICV-PBS (Fig. 1D) or ICV-LPS (Fig. 1E), further confirming that Kv1.3 indeed carries the Kv current in LPS-stimulated wild-type (WT) microglia. In addition, in a small subset of WT and Kv1.3 KO microglia we observed some remaining current that was carried by TRP (transient receptor potential) channels or other Kv channels (Fig. 1A, B, and F), which were not further characterized in this study.
Figure 1. LPS upregulates microglial Kv1.3 in vivo.
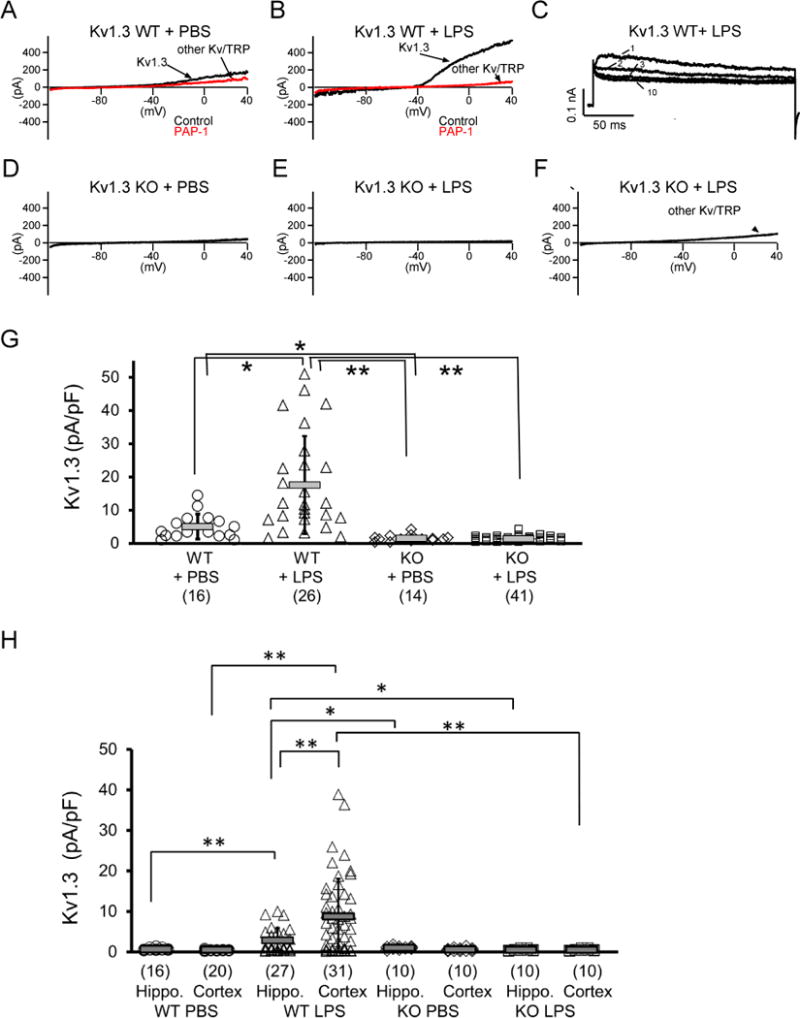
WT (Kv1.3 WT) and Kv1.3−/− (Kv1.3KO) mice at three months of age received ICV injection of LPS or vehicle (PBS). Twenty-four hours after injection, microglia were acutely isolated by immunopanning and immediately studied by whole-cell patch clamp. (A-F) Representative currents elicited by a voltage-ramp from −120 mV to +40 mV. Currents in WT mice are PAP-1 sensitive (A, B) and exhibit use-dependent C-type inactivation elicited by repetitive depolarization from −80 to + 40 mV (1 pulse/s for 10 pulses), as demonstrated by the 1st, 2nd, 3rd, and 10th pulses in (C), thus verifying the functional expression and LPS-induced enhanced activity of surface Kv1.3 channels. KO mouse microglia exhibited no detectable Kv1.3 currents (D, E) but sometimes exhibit other Kv/TRP-like currents (F). (G) Scatterplot showing Kv1.3 current density measured on microglia acutely isolated from Kv1.3 WT and Kv1.3 KO mice: Kv1.3 WT + PBS (5.06 + 3.77 pA/pF, n = 16), Kv1.3 WT + LPS (17.58 + 14.72 pA/pF, n = 26), Kv1.3 KO + PBS (1.51 + 0.96 pA/pF, n = 14) and Kv1.3 KO + LPS (1.40 + 0.76 pA/pF, n = 41). Data are presented as mean ± S.D., *p <0.05 and **p<0.001. Statistical significance was determined using paired Student’s t-test. (H) Scatterplot of data from a set of experiments independent from those shown in (G) showing Kv1.3 current density measured on microglia acutely isolated from either hippocampus or cortex. *p <0.05 and **p<0.005.
We quantified Kv1.3 current densities as PAP-1 sensitive outward current (for example, the difference between the black and red tracings in Fig. 1B) at +40 mV. The scatterplot in Fig. 1G shows that, as expected, Kv1.3 KO diminished the measurable Kv1.3 current density, in contrast to the two WT groups. ICV-LPS significantly increased Kv1.3 current densities on WT microglia, but had no effect on Kv1.3 KO microglia. Interestingly, in additional independent experiments in which cerebral cortex and hippocampus were dissected before microglia isolation, it was found that Kv1.3 was upregulated in hippocampal microglia in response to ICV-LPS, but this response was significantly less robust (p<0.001) than that in cortical microglia (Fig. 1H). This result adds to previous evidence supporting location-dependent heterogeneity of microglia properties (Horiuchi et al. 2017; McCarthy 2017).
Microglial Kir2.1 is not activated by LPS in WT mice but is upregulated in Kv1.3 KO mice
We also detected inward rectifier current in microglia, measured as peak inward currents at −120 mV, which were identified as Kir2.1 based on Ba2+ sensitivity and the presence of Kir2.1 message, as previously described (Nguyen et al. 2017). Consistent with our previous in vitro data (Nguyen et al. 2017), LPS stimulation in vivo did not lead to any increases in Kir2.1 current density in WT microglia (Fig. 2A). Interestingly, in contrast to WT microglia, Kv1.3 KO microglia showed a significant increase in Kir2.1 current density following ICV-LPS (Fig. 2A-C). Quantitative PCR conducted using RNA extracted from acutely isolated microglia showed that Kv1.3 knockout did not affect the expression of other K+ channels previously described to be expressed in microglia (Kettenmann et al. 2011) (Fig. 2D, white bars). However, Kv1.3 knockout did alter the pattern of microglial K+ channel expression following LPS stimulation, as exemplified by altered responses of Kv1.1, Kir2.1, and KCa3.1 (Fig. 2D, black bars). The increased Kir2.1 transcript level in response to ICV-LPS is consistent with the increased Kir2.1 current density (Fig. 2A). Interestingly, the KCa3.1 transcript level was reduced by LPS in WT microglia, consistent with prior in vitro data (Nguyen et al. 2017), but was upregulated by LPS in Kv1.3 KO microglia (Fig. 2D).
Figure 2. Kv1.3 knockout alters microglial response to LPS exemplified by K+ channel expression.
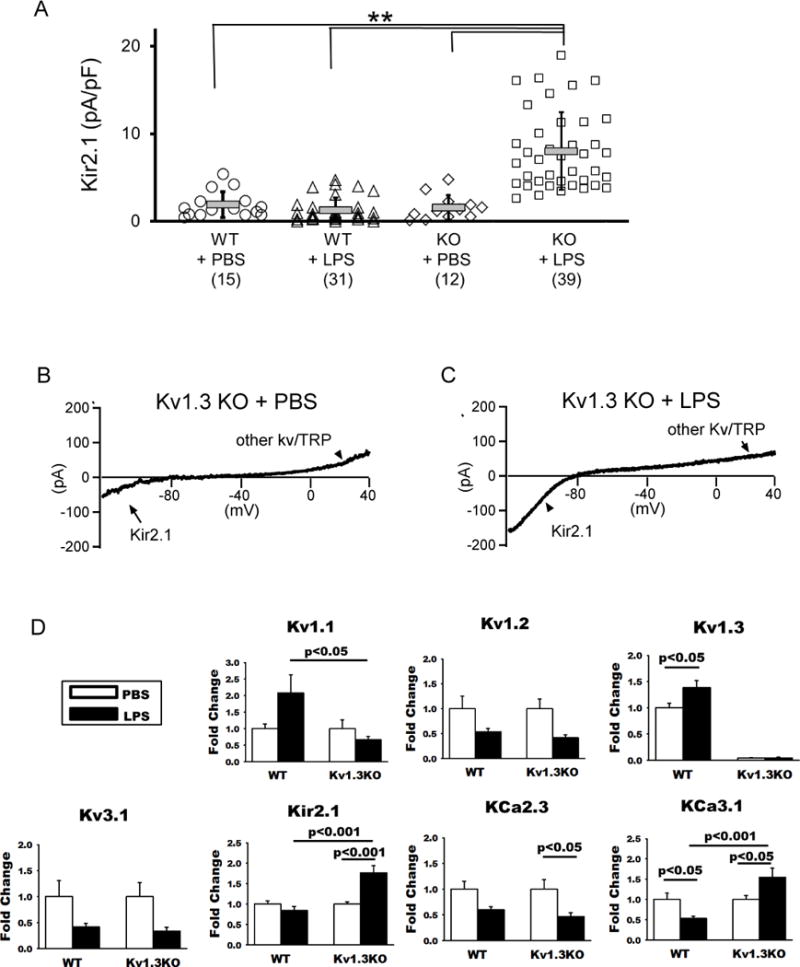
Kv1.3WT and Kv1.3KO mice at three months of age received ICV injection of LPS or vehicle (PBS). Twenty-four hours after injection, microglia were acutely isolated by immunopanning and immediately studied by whole-cell patch clamp and qPCR quantification of RNA. (A) Scatterplot showing Kir2.1 current density measured on microglia acutely isolated from WT and Kv1.3 KO mice: Kv1.3 WT + PBS (1.92 + 1.48 pA/pF, n = 15), Kv1.3 WT + LPS (1.26 + 1.40 pA/pF, n = 31), Kv1.3 KO + PBS (1.55 + 1.41 pA/pF, n = 12) and Kv1.3 KO + LPS (8.02 + 4.43 pA/pF, n = 39). Data are presented as mean ± S.D., **p<0.001. Statistical significance was determined using paired Student’s t-test. (B) A representative current tracing of a microglia acutely isolated from a Kv1.3 KO mouse receiving ICV-PBS showing peak inward currents at −120 mV, characteristic of Kir2.1. (C) The Kir2.1 currents were augmented in a microglia acutely isolated from a Kv1.3 KO mouse receiving ICV-LPS. (D) RNA was isolated from WT mice receiving ICV-PBS (n = 10) and ICV-LPS (n = 10), and Kv1.3 KO mice receiving ICV-PBS (n = 11) and ICV-LPS (n = 12). Shown are qPCR results of indicated microglial K+ channel genes. Data are presented as mean ± SEM. Statistical analysis was performed by two-way ANOVA with Bonferroni correction for multiple comparisons.
Kv1.3 knockout reduces microglial pro-inflammatory response after ICV-LPS
As expected, WT mice receiving ICV-LPS exhibited increased Iba-1 immunoreactivities, illustrated by the photomicrographs of cortical and hippocampal CA1 region (Fig. 3A and B), indicating enhanced microglial activation. Consistent with the immunostaining result, Western blotting showed increased levels of microglial markers (CD11b and Iba-1) and microglial activation markers (CD68 and phosphorylated p38MAPK) in brain extracts (Fig. 3C and D). Such increases were not seen in Kv1.3 KO mice receiving ICV-LPS (Fig. 3A-D), indicating suppressed microglial activation. Likewise, ELISA performed on brain extracts showed that while WT brains responded to ICV-LPS with increased cerebral levels of pro-inflammatory cytokines IL-1β and TNF-α, Kv1.3 knockout substantially subdued these responses (Fig. 4A). In parallel, qPCR performed on RNA purified from acutely isolated microglia showed that Kv1.3 knockout suppressed the LPS-induced over-expression of the pro-inflammatory modulator genes IL-1β, TNF-α, IL-6, and iNOS (Fig. 4B). ANOVA analysis shows that ICV-LPS failed to induce significant increases in pro-inflammatory modulators in Kv1.3 KO mice, and that values for the Kv1.3KO/ICV-LPS group are significantly lower than for the WT/ICV-LPS group (Fig. 4B).
Figure 3. LPS-induced microglial activation requires Kv1.3 expression in vivo.
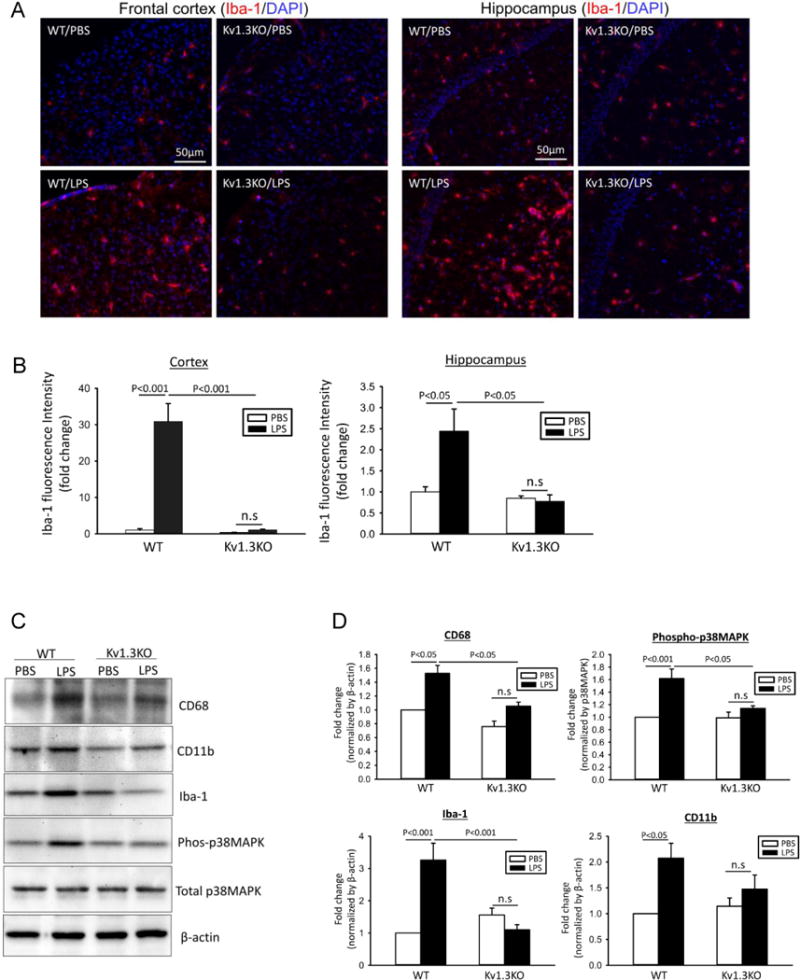
(A-B) Hippocampal sections of Kv1.3WT or Kv1.3KO mice at 24 hours after ICV-PBS or ICV-LPS were co-stained with Iba-1 (red) and DAPI (blue). (A) Shown are representative photomicrographs from frontal cortex and hippocampus. (B) Quantification of Iba-1 immunoreactivity was performed in three sections per animal in WT/PBS (n = 10), WT/LPS (n = 10), KO/PBS (n = 11), and KO/LPS (n = 12) mice. (C-D) The levels of indicated proteins in brain homogenates were evaluated by Western blotting. The increased expression microglial markers (CD11b and Iba-1) and activation markers (CD68 and phosphorylated p38MAPK) were curbed by Kv1.3KO. A representative Western blot is presented in (C). The band intensities were quantified and expressed as fold change of values obtained from WT/PBS in (D). Data are presented as mean ±SEM, with n=5 in all groups. Statistical analysis was performed by two-way ANOVA with Bonferroni correction for multiple comparisons
Figure 4. LPS-induced pro-inflammatory phenotype of microglia requires Kv1.3 expression in vivo.
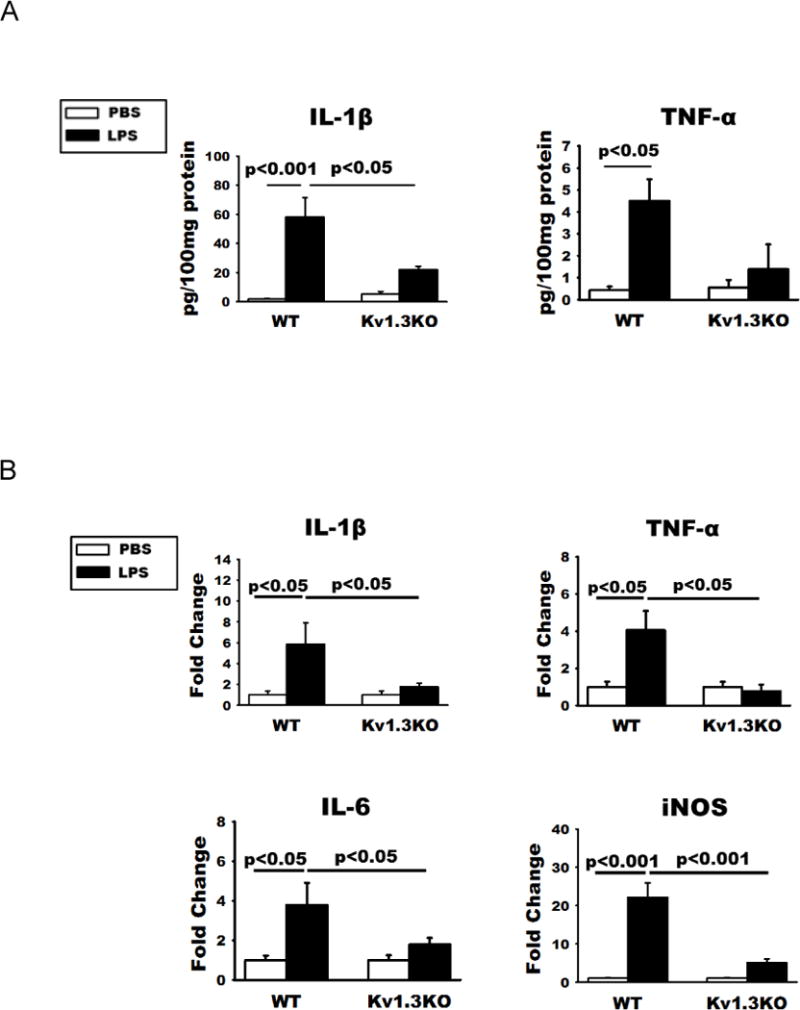
(A) ELISA quantification of IL-1β and TNF-α levels in brain homogenates of these mice showed that increased production of the two pro-inflammatory cytokines were curbed by Kv1.3 knockout. (B) qPCR quantification of the RNA of indicated molecules showed increases of pro-inflammation cytokines and iNOS mRNA in microglia acutely isolated from WT mice at 24h after ICV-LPS. These increases were absent in Kv1.3KO mice. Data are presented as means ± SEM. There are no statistical differences in all measures between Kv1.3KO/PBS and Kv1.3KO/LPS groups. RNA was isolated from WT mice receiving ICV-PBS (n =10) and ICV-LPS (n = 10), and Kv1.3 KO mice receiving ICV-PBS (n = 11) and ICV-LPS (n = 12). Statistical analysis was performed by two-way ANOVA follow by Bonferroni correction for multiple comparisons.
Kv1.3 knockout mitigates microglial activation-related hippocampal long-term potentiation deficits
Previous evidence demonstrates that aberrant immune processes in the brain produce detrimental effects on neuronal plasticity, exemplified by the reduction of hippocampal long-term potentiation (hLTP) induction and maintenance (O’Connor and Coogan 1999; Yirmiya and Goshen 2011). Because pro-inflammatory microglia, especially through their increased production of TNF-α, IL-1β, and nitric oxide (NO), impair hLTP (Costello et al. 2011; Lynch 2015; Wang et al. 2004), we tested if blocking the development of the pro-inflammatory state by Kv1.3 knockout may protect this form of synaptic plasticity. Hippocampal slices acutely obtained from WT mice receiving ICV-LPS showed a deficit of hLTP in Schaffer-commissural synapses where the response, quantified between 50 to 60 min after induction, was substantially reduced compared to that of WT mice receiving ICV-PBS. Kv1.3 knockout completely mitigated this hLTP deficit (Fig. 4A and 4B). ICV-LPS itself did not affect basal transmission as the input/output curves recorded prior to LTP induction did not show any differences between groups (Fig. 5C). This result suggests that Kv1.3 activity regulates pro-inflammatory microglial neurotoxicity.
Figure 5. LPS-induced neurotoxic phenotype of microglia requires Kv1.3 expression in vivo.
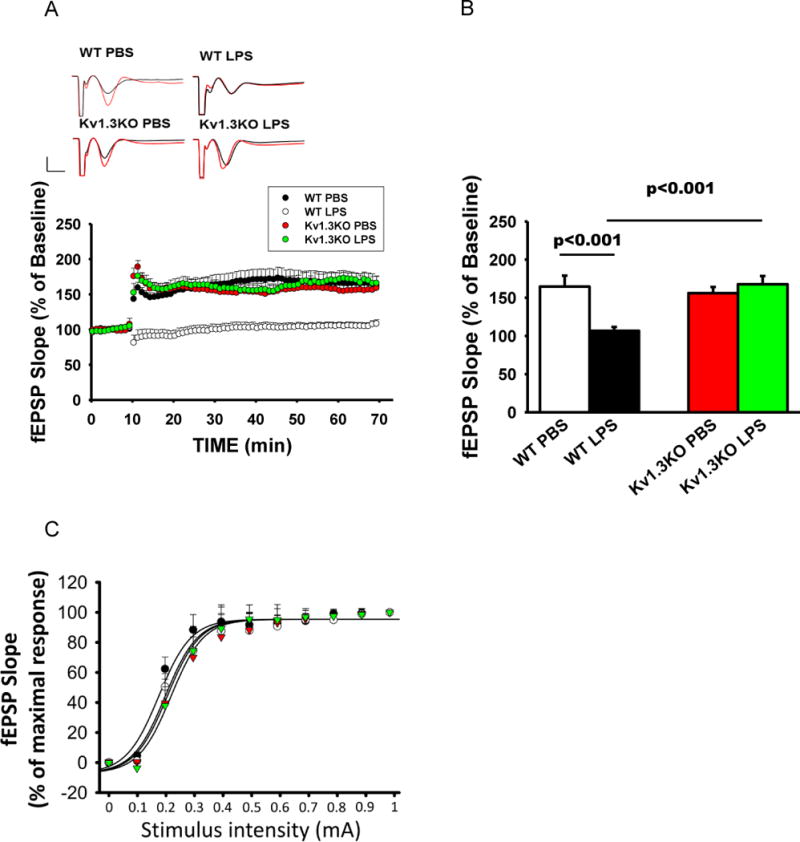
Kv1.3WT and Kv1.3KO mice at three months of age received ICV injection of LPS or PBS. Twenty-four hours after injection, hippocampal slices were used for hLTP measurement. (A) Traces and time course of hLTP induced with high frequency stimulation showing that ICV-LPS injection reduced the amplitude of LTP but this reduction was completely mitigated by Kv1.3 knockout. (B) Summary bar graphs showing the average fEPSP slope between 50 and 60 minutes after high frequency stimulation. Data were compiled from recordings using slices obtained from the groups of WT/control (15 slices from 4 mice), WT/LPS (16 slices from 4 mice), Kv1.3KO/PBS (15 slices from 4 mice), and Kv1.3KO/LPS (14 slices from 4 mice), and are presented as mean percent change in fEPSP slope ± SEM from baseline. Bar scale: 0.5mV/5ms. Two-way ANOVA follow by Bonferroni post hoc test. (C) Input/output relationship shows no difference in basal transmission before LTP induction; the curves were constructed by increasing stimulation current intensity ranged from 0 to 1 mA with steps of 0.1mA.
Pharmacological inhibition of microglial Kv1.3 reduces microglial pro-inflammatory response and mitigates hLTP deficit after ICV-LPS
Although published (Charolidi et al. 2015; Nguyen et al. 2017; Rangaraju et al. 2015; Rangaraju et al. 2017; Rus et al. 2005) and present data clearly showed that in the brain, Kv1.3 is mainly expressed in activated microglia, it has been described in neurons, most prominently in mitral cells of the olfactory bulb (Fadool et al. 2004) and presynaptic terminals of brain stem auditory neurons (Gazula et al. 2010), and also in cortical interneurons and in oligodendrocyte progenitor cells but at low levels (Vautier et al. 2004; Duque et al. 2013), raising the possibility that the above in vivo effects of Kv1.3 knockout may be secondary to Kv1.3 hypomorph in neurons. To address this possibility, we used a chemical biology approach, as in neurons Kv1.3 is typically part of hetereomultimers with Kv1.1, Kv1.2 and Kv1.6 subunits (Helms et al. 1997) and therefore has a different pharmacology from the Kv1.3 homotetramers or Kv1.3/Kv1.5 heterotetramers found in T cells, microglia and macrophages (Chandy et al. 2004; Feske et al. 2015; Wulff et al. 2009). We previously developed a small molecule Kv1.3 blocker called PAP-1 (Schmitz et al. 2005) that selectively targets homotetrameric Kv1.3 channels which are most commonly found on immune cells (Feske et al. 2015). PAP-1 inhibits Kv1.3 with an IC50 of 2 nM (Schmitz et al. 2005), has a half-life of 3 hours in rodents, is orally available, brain penetrant and does not exhibit any long-term toxicity in rodents or primates (Azam et al. 2007; Beeton et al. 2006; Pereira et al. 2007). We administered PAP-1 (40 mg/kg) by intraperitoneal injection at one hour after ICV-LPS and retrieved the brain for study at 24 hrs. Similar to Kv1.3 knockout, PAP-1 treatment reduced the increased cerebral levels of the pro-inflammatory cytokines IL-1β and TNF-α induced by ICV-LPS (Fig. 6A). In parallel, qPCR performed on RNA purified from acutely isolated microglia showed that PAP-1 treatment suppressed the LPS-induced over-expression of the pro-inflammatory modulator genes IL-1β, TNF-α, IL-6, and iNOS (Fig. 6B). Moreover, like Kv1.3 knockout, intraperitoneal PAP-1 treatment of the mice receiving ICV-LPS mitigated the LPS-induced hLTP deficit (Fig. 7A and B), while showing no significant effect on mice receiving ICV-PBS. ICV-LPS and PAP-1 treatment did not affect basal transmission as the input/output curves recorded prior to LTP induction did not show any differences between groups (Fig. 7C). Interestingly, in contrast to the effect of Kv1.3 KO (Fig 5A), which rescued both LPS-induced LTP as well short-term plasticity (STP) deficits, PAP-1 treatment failed to affect the STP deficit (Fig. 7A).
Figure 6. PAP-1 treatment reduces the expression of pro-inflammatory mediators following ICV-LPS.
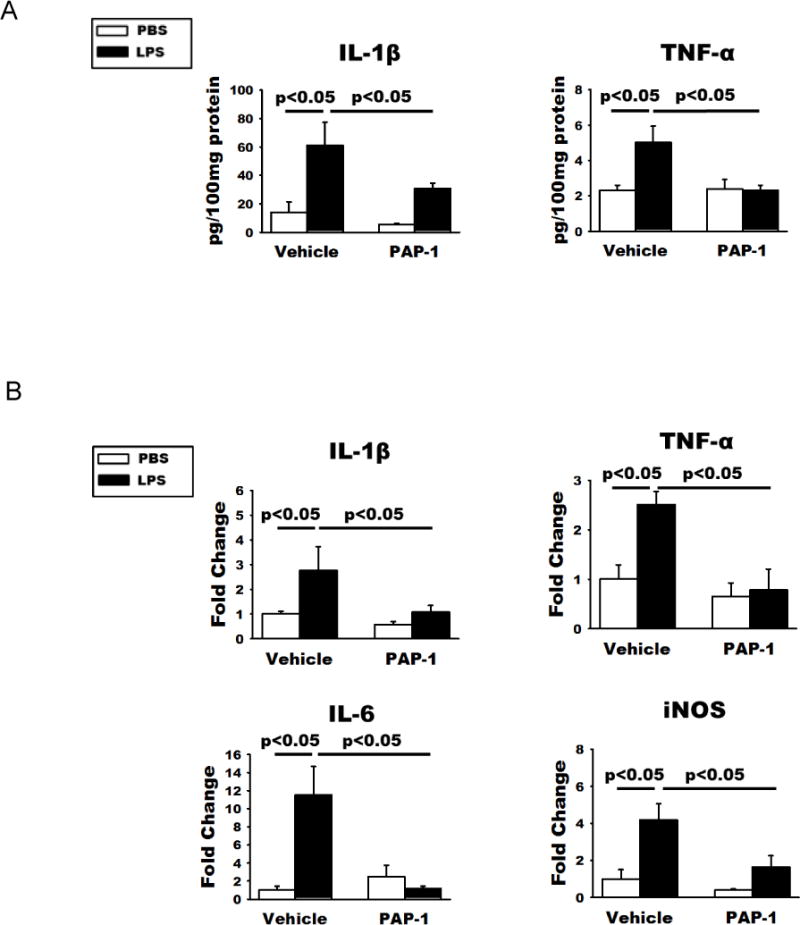
WT mice first received ICV-PBS or ICV-LPS and one hour later received intraperitoneal injection of PAP-1 (40 mg/kg) or the vehicle Miglyol-812. Twenty-four hours later, the brains were bisected and used for either homogenization or acute microglia isolation. (A) ELISA quantification of IL-1β and TNF-α levels in brain homogenates of these mice showed that increased production of the two pro-inflammatory cytokines were curbed by PAP-1 treatment. (B) qPCR quantification of the RNA of indicated molecules showed increases of pro-inflammation cytokines and iNOS mRNA in microglia acutely isolated from WT mice at 24h after ICV-LPS. These increases were curbed by PAP-1 treatment. There are no statistical differences in all measures between PBS/PAP-1 and LPS/PAP-1 groups. Statistical analysis was performed by two-way ANOVA with Bonferroni correction for multiple comparisons. Data are presented as means ± SEM, n = 6/group for qPCR and n = 5/group for ELISA studies.
Figure 7. PAP-1 treatment reverses the LPS-induced hLTP deficit.
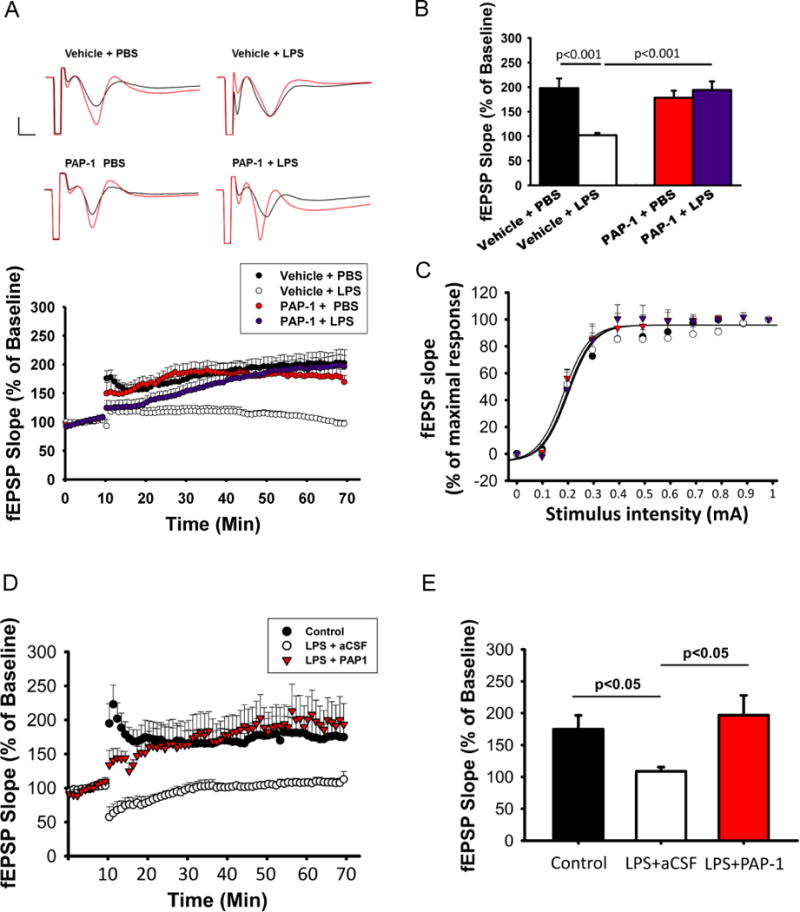
WT mice received ICV-PBS or ICV-LPS and one hour later received intraperitoneal injection of PAP-1 (40 mg/kg) or the vehicle (Miglyol-812). Twenty-four hours later, hippocampal slices were used for hLTP measurement. (A) Time course of hLTP induced with high frequency stimulation showing that ICV-LPS injection reduced the amplitude of LTP but this reduction was mitigated by PAP-1 treatment. (B) Summary bar graphs showing the average fEPSP slope between 50 and 60 minutes after high frequency stimulation. Data were compiled from recordings using slices obtained from the groups of Vehicle/PBS (10 slices from 3 mice), Vehicle/LPS (12 slices from 3 mice), PAP-1/PBS (10 slices from 3 mice), and PAP-1/LPS (12 slices from 3 mice) and are presented as mean percent change in fEPSP slope ± SEM from baseline. Bar scale: 0.5mV/5ms. Two-way ANOVA follow by Bonferroni post hoc test. (C) Input/output relationship shows no difference in basal transmission before LTP induction; the curves were constructed by increasing stimulation current intensity ranged from 0 to 1 mA with steps of 0.1mA. (D-E) WT mice at three months of age received ICV injection of LPS or PBS. Twenty-four hours after injection, hippocampal slices were used for hLTP measurement. Slices from mice receiving ICV-LPS were preincubated with PAP-1 (1 μM) or aCSF for 45 min before LTP induction. PAP-1 was also continuously perfused during the recording. (D) Traces and time course of fEPSP slope. (E) Summary bar graphs showing the average fEPSP slope between 50 and 60 minutes after high frequency stimulation. Data were compiled from recordings using slices obtained from the groups of control (7 slices from 3 mice), LPS (4 slices from 2 mice), and LPS/PAP-1 (4 slices from 2 mice), and are presented as mean percent change in fEPSP slope ± SEM from baseline. Two-way ANOVA follow by Bonferroni post hoc test.
To test if acute administration of PAP-1 on hippocampal slices, which allows a more direct action of the drug on microglial Kv1.3, would also rescue the LPS-induced hLTP deficit, we administered PAP-1 by continuous perfusion, from 45 minutes prior to LTP induction to the end of recording, to hippocampal slices from mice 24 hours after receiving ICV-LPS. This acute treatment of PAP-1 also recovered hLTP to the control level (Fig 7D and E). However, similar to in vivo administration of PAP-1 (Fig. 7A), the LPS-induced STP deficit was not rescued. Taken together, our data suggest that LPS-stimulated microglial pro-inflammatory response and associated neurotoxicity require microglial Kv1.3 activity in vivo.
DISCUSSION
Based on data obtained from both genetic knockout and pharmacological intervention, we conclude that Kv1.3 is required for microglial M1-like pro-inflammatory activation in vivo. A first in vivo suggestion of such a role of Kv1.3 came from a recent report showing that systemic inflammation induced by intravenous administration of LPS to adult mice resulted in increased ShK-F6CA (a fluorescein-conjugated Kv1.3-specific peptide inhibitor) labeling of microglia and infiltrating macrophages in the brain (Rangaraju et al. 2017). In the current study, we stereotactically injected LPS into the ventricles (Maezawa et al. 2006). This approach eliminates the confounding involvement of peripheral macrophages or lymphocytes as well as the disruption of the blood-brain barrier seen with peripheral LPS injection or in stroke models (Chen et al. 2016), thus allowing a better assessment of the direct effect of LPS on microglia in vivo. Moreover, we verified the identity and functional expression of microglial Kv1.3 based on its signature biophysical properties and its selective pharmacological inhibition by PAP-1. We found that LPS stimulation increased microglial surface Kv1.3 current density (Fig. 1), which is likely in part due to increased Kv1.3 transcript levels (Fig. 2D). The Kv1.3 enhancement was much more robust in cortical microglia than in hippocampal microglia, supporting the location-dependent heterogeneity of microglia properties (Horiuchi et al. 2017; McCarthy 2017). Furthermore, both genetic knockout (Fig. 3–5) and pharmacological inhibition (Fig. 6 and 7) of Kv1.3 abolished LPS-induced pro-inflammatory microglial activation and hLTP impairment, supporting an essential role of Kv1.3 in such responses in vivo.
While Kv1.3 knockout did not affect basal-level expressions of other microglial K+ channels tested, LPS stimulation of Kv1.3 KO microglia teased out different responses of some K+ channels (Fig. 2D), reflecting an altered reactivity. In contrast to WT microglia, Kv1.3KO microglia showed a significant increase in Kir2.1 currents as well as RNA expression following ICV-LPS. This, in combination with a study implicating Kir2.1 activity in microglia under “resting” and “anti-inflammatory states” (Lam and Schlichter 2015), our previous observation that Kir2.1 upregulation is characteristic of IL-4 (an anti-inflammatory cytokine)-stimulated microglia (Nguyen et al. 2017), and a report that Kv1.3 deletion biases T cells towards an “immunosuppressive” phenotype with increased IL-10 production (Gocke et al. 2012), suggests that Kv1.3 reduction may skew microglial activation towards a M2-like or anti-inflammatory response. Kv1.3 knockout also abolished the LPS-increased Kv1.1 expression and reversed the LPS-reduced KCa3.1 expression (Fig. 2D); the in vivo significance of these findings awaits further studies. We previously conducted a study on a mouse ischemia/reperfusion model, in which the microglia/macrophages acutely isolated from the infarcted area were found to exhibit higher densities of K+ currents with the biophysical and pharmacological properties of Kv1.3, KCa3.1 and Kir2.1 (Chen et al. 2016), suggesting a mixed activation pattern following an ischemic insult to the brain. A recent article reported that neocortical microglia isolated and cultured from human epilepsy patients responded to LPS with an increase in TNF-α production but no change in KCa3.1 expression. In contrast, IL-4 slightly increased the KCa3.1 current per cell, although no significant change in channel density was observed (Blomster et al. 2016). Thus, although previous studies using cultured neonatal microglia suggest a pro-inflammatory role of KCa3.1 (Kaushal et al. 2007; Maezawa et al. 2011; Schlichter et al. 2010), its actual functional roles in adults appear complicated and need to be verified by in vivo experiments as exemplified by this study.
It is intriguing to hypothesize that selectively blocking microglial Kv1.3 activity while preserving or even enhancing Kir2.1 activity would mitigate the neurotoxicity accompanying the pro-inflammatory activation while preserving the anti-inflammatory and tissue repair functions associated with IL-4-like effects. In this study, we used mouse hLTP as a model to test how LPS-induced M1-like microglia activation causes deterioration of neural plasticity as a form of neurotoxicity. This notion becomes particularly intriguing in view of the recent identification of LPS in adult human brains, and the reported significantly higher levels of LPS in Alzheimer’s disease brains, notably in the hippocampus (Zhan et al. 2016; Zhao et al. 2017). Among several inflammatory mediators produced by microglia that may detrimentally affect hLTP, the best studied are nitric oxide (NO), TNF-α, and IL-1β (Costello et al. 2011; Lynch 2015; Wang et al. 2004). Both Kv1.3 knockout and PAP-1 treatment effectively reduced TNF-α and IL-1β expression and production, and reduced the expression of iNOS coding for the inducible NO synthase, which in combination may underlie the complete recovery of hLTP to the normal WT levels. A significant implication of our in vivo validation, therefore, is that selective Kv1.3 blockers could be therapeutic candidates for diseases in which neurotoxic microglial activation or dysfunction is implicated, such as Alzheimer’s disease, Parkinson’s disease, amyotrophic lateral sclerosis, stroke, epilepsy, and multiple sclerosis (Wolf et al. 2017).
We found that ICV-LPS impaired both short-term plasticity (STP) and long-term potentiation in the hippocampal Schaffer-commissural synapses (Fig. 5 and 7). Interestingly, while both PAP-1 and Kv1.3KO mitigated LPS-induced hLTP deficit, only Kv1.3KO, but not PAP-1 mitigated LPS-induced STP deficit. This result suggests that the acute effect of LPS on STP is not likely mediated by microglial Kv1.3, as both intraperitoneal injected and bath-applied PAP-1 failed to rescue STP (Fig. 7A and D). We speculate that Kv1.3KO (or chronic Kv1.3 inhibition), but not acute inhibition of Kv1.3 by PAP-1, may cause a long-lasting change of synaptic release mechanism, which endows the Schaffer collateral-CA1 synapses with resistance to LPS-mediated acute effect on the STP. Further studies are needed to clarify the mechanism.
One limitation of the current study is that, by global Kv1.3 knockout and systemic application of PAP-1, we cannot completely exclude the possible contributions of peripheral monocyte-derived macrophages and effector memory T cells, which also express Kv1.3 (Feske et al. 2015; Wulff et al. 2003). Peripheral inhibition of Kv1.3 via our interventions may indirectly affect brain microglia, as peripheral immune modulation may impact neuroinflammation or brain function via humoral or cellular factors (Prinz and Priller 2017; Villeda et al. 2014). For example, heterochronic parabiosis studies showed that increasing peripheral CCL11 chemokine levels in vivo in young mice decreased adult neurogenesis and impaired learning and memory (Villeda et al. 2011), and systemic administration of young blood plasma into aged mice improved age-related cognitive impairments (Villeda et al. 2014). Myeloid cells and lymphocytes were also found in the newly identified meningeal lymphatics (Louveau et al. 2015), through which the trafficking of immune cells might influence brain pathology. It is important in future studies to employ Cre recombinase-guided cell targeting to selectively knockout Kv1.3 in brain microglia, peripheral macrophages/monocytes, or T cells, in order to parse out their relative contributions to neuroinflammation.
Main Points.
This in vivo study of the role of Kv1.3 in microglial activation shows that Kv1.3 is required for microglial M1-like pro-inflammatory activation and neurotoxicity.
Selective Kv1.3 blockers could be therapeutic candidates for neurological diseases in which neurotoxic microglial activation is implicated.
Acknowledgments
This work was supported by National Institute of Aging awards AG043788 (to I.M.) and AG038910 (to L-W.J.), a National Institute of Neurological Disease and Stroke award NS100294 (to H.W.), and an Alzheimer’s Association award NIRG-10-174150 (to I.M.).
Footnotes
Conflict of interest statement: The authors have no financial conflict of interest.
References
- Arnoux I, Hoshiko M, Mandavy L, Avignone E, Yamamoto N, Audinat E. Adaptive phenotype of microglial cells during the normal postnatal development of the somatosensory “Barrel” cortex. Glia. 2013;61:1582–94. doi: 10.1002/glia.22503. [DOI] [PubMed] [Google Scholar]
- Azam P, Sankaranarayanan A, Homerick D, Griffey S, Wulff H. Targeting Effector Memory T Cells with the Small Molecule Kv1.3 Blocker PAP-1 Suppresses Allergic Contact Dermatitis. J Invest Dermatol. 2007;127:1419–1429. doi: 10.1038/sj.jid.5700717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beeton C, Wulff H, Standifer NE, Azam P, Mullen KM, Pennington MW, Kolski-Andreaco A, Wei E, Grino A, Counts DR, et al. Kv1.3 channels are a therapeutic target for T cell-mediated autoimmune diseases. Proc Natl Acad Sci U S A. 2006;103:17414–9. doi: 10.1073/pnas.0605136103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blomster LV, Strobaek D, Hougaard C, Klein J, Pinborg LH, Mikkelsen JD, Christophersen P. Quantification of the functional expression of the Ca2+ -activated K+ channel KCa 3.1 on microglia from adult human neocortical tissue. Glia. 2016;64:2065–2078. doi: 10.1002/glia.23040. [DOI] [PubMed] [Google Scholar]
- Bohlen CJ, Bennett FC, Tucker AF, Collins HY, Mulinyawe SB, Barres BA. Diverse Requirements for Microglial Survival, Specification, and Function Revealed by Defined-Medium Cultures. Neuron. 2017;94:759–773 e8. doi: 10.1016/j.neuron.2017.04.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butovsky O, Jedrychowski MP, Moore CS, Cialic R, Lanser AJ, Gabriely G, Koeglsperger T, Dake B, Wu PM, Doykan CE, et al. Identification of a unique TGF-beta-dependent molecular and functional signature in microglia. Nat Neurosci. 2014;17:131–43. doi: 10.1038/nn.3599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandy KG, Wulff H, Beeton C, Pennington M, Gutman GA, Cahalan MD. Potassium channels as targets for specific immunomodulation. Trends Pharmacol Sci. 2004;25:280–289. doi: 10.1016/j.tips.2004.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charolidi N, Schilling T, Eder C. Microglial Kv1.3 Channels and P2Y12 Receptors Differentially Regulate Cytokine and Chemokine Release from Brain Slices of Young Adult and Aged Mice. PLoS One. 2015;10:e0128463. doi: 10.1371/journal.pone.0128463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YJ, Nguyen HM, Maezawa I, Grossinger EM, Garing AL, Kohler R, Jin LW, Wulff H. The potassium channel KCa3.1 constitutes a pharmacological target for neuroinflammation associated with ischemia/reperfusion stroke. J Cereb Blood Flow Metab. 2016;36:2146–2161. doi: 10.1177/0271678X15611434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costello DA, Lyons A, Denieffe S, Browne TC, Cox FF, Lynch MA. Long Term Potentiation Is Impaired in Membrane Glycoprotein CD200-deficient Mice: A role for Toll-like receptor activation. Journal of Biological Chemistry. 2011;286:34722–34732. doi: 10.1074/jbc.M111.280826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duque A, Gazula VR, Kaczmarek LK. Expression of Kv1.3 potassium channels regulates density of cortical interneurons. Dev Neurobiol. 2013;73:841–55. doi: 10.1002/dneu.22105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fadool DA, Tucker K, Perkins R, Fasciani G, Thompson RN, Parsons AD, Overton JM, Koni PA, Flavell RA, Kaczmarek LK. Kv1.3 channel gene-targeted deletion produces “Super-Smeller Mice” with altered glomeruli, interacting scaffolding proteins, and biophysics. Neuron. 2004;41:389–404. doi: 10.1016/s0896-6273(03)00844-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feske S, Wulff H, Skolnik EY. Ion Channels in Innate and Adaptive Immunity. Annual Review of Immunology. 2015;33:291–353. doi: 10.1146/annurev-immunol-032414-112212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fordyce CB, Jagasia R, Zhu X, Schlichter LC. Microglia Kv1.3 channels contribute to their ability to kill neurons. J Neurosci. 2005;25:7139–49. doi: 10.1523/JNEUROSCI.1251-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gazula VR, Strumbos JG, Mei X, Chen H, Rahner C, Kaczmarek LK. Localization of Kv1.3 channels in presynaptic terminals of brainstem auditory neurons. J Comp Neurol. 2010;518:3205–20. doi: 10.1002/cne.22393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gocke AR, Lebson LA, Grishkan IV, Hu L, Nguyen HM, Whartenby KA, Chandy KG, Calabresi PA. Kv1.3 deletion biases T cells toward an immunoregulatory phenotype and renders mice resistant to autoimmune encephalomyelitis. J Immunol. 2012;188:5877–86. doi: 10.4049/jimmunol.1103095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanisch UK, Kettenmann H. Microglia: active sensor and versatile effector cells in the normal and pathologic brain. Nat Neurosci. 2007;10:1387–94. doi: 10.1038/nn1997. [DOI] [PubMed] [Google Scholar]
- Harmar AJ, Hills RA, Rosser EM, Jones M, Buneman OP, Dunbar DR, Greenhill SD, Hale VA, Sharman JL, Bonner TI, et al. IUPHAR-DB: the IUPHAR database of G protein-coupled receptors and ion channels. Nucleic Acids Res. 2009;37:D680–5. doi: 10.1093/nar/gkn728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helms LM, Felix JP, Bugianesi RM, Garcia ML, Stevens S, Leonard RJ, Knaus HG, Koch R, Wanner SG, Kaczorowski GJ, et al. Margatoxin binds to a homomultimer of K(V)1.3 channels in Jurkat cells. Comparison with K(V)1.3 expressed in CHO cells. Biochemistry. 1997;36:3737–44. doi: 10.1021/bi962351p. [DOI] [PubMed] [Google Scholar]
- Henn A, Lund S, Hedtjam M, Porzgen P, Leist M. The suitability of BV2 cells as alternative model system for primary microglia cultures or animal experiments of brain inflammation. European Journal of Cell Biology. 2009;88:72–72. doi: 10.14573/altex.2009.2.83. [DOI] [PubMed] [Google Scholar]
- Horiuchi M, Smith L, Maezawa I, Jin LW. CX3CR1 ablation ameliorates motor and respiratory dysfunctions and improves survival of a Rett syndrome mouse model. Brain Behav Immun. 2017;60:106–116. doi: 10.1016/j.bbi.2016.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin LW, Horiuchi M, Wulff H, Liu XB, Cortopassi GA, Erickson JD, Maezawa I. Dysregulation of Glutamine Transporter SNAT1 in Rett Syndrome Microglia: A Mechanism for Mitochondrial Dysfunction and Neurotoxicity. J Neurosci. 2015;35:2516–2529. doi: 10.1523/JNEUROSCI.2778-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaushal V, Koeberle PD, Wang Y, Schlichter LC. The Ca2+-activated K+ channel KCNN4/KCa3.1 contributes to microglia activation and nitric oxide-dependent neurodegeneration. J Neurosci. 2007;27:234–44. doi: 10.1523/JNEUROSCI.3593-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kettenmann H, Hanisch UK, Noda M, Verkhratsky A. Physiology of microglia. Physiological Reviews. 2011;91:461–553. doi: 10.1152/physrev.00011.2010. [DOI] [PubMed] [Google Scholar]
- Khanna R, Roy L, Zhu X, Schlichter LC. K+ channels and the microglial respiratory burst. Am J Physiol Cell Physiol. 2001;280:C796–806. doi: 10.1152/ajpcell.2001.280.4.C796. [DOI] [PubMed] [Google Scholar]
- Kim CC, Nakamura MC, Hsieh CL. Brain trauma elicits non-canonical macrophage activation states. J Neuroinflammation. 2016;13 doi: 10.1186/s12974-016-0581-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotecha SA, Schlichter LC. A Kv1.5 to Kv1.3 switch in endogenous hippocampal microglia and a role in proliferation. J Neurosci. 1999;19:10680–93. doi: 10.1523/JNEUROSCI.19-24-10680.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kundu-Raychaudhuri S, Chen YJ, Wulff H, Raychaudhuri SP. Kv1.3 in psoriatic disease: PAP-1, a small molecule inhibitor of Kv1.3 is effective in the SCID mouse psoriasis - Xenograft model. Journal of Autoimmunity. 2014;55:63–72. doi: 10.1016/j.jaut.2014.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Küst BM, Biber K, Van Calker D, Gebicke-Haerter PJ. Regulation of K+ channel mRNA expression by stimulation of adenosine A2a-receptors in cultured rat microglia. Glia. 1999;25:120–30. [PubMed] [Google Scholar]
- Lam D, Schlichter LC. Expression and contributions of the Kir2.1 inward-rectifier K(+) channel to proliferation, migration and chemotaxis of microglia in unstimulated and anti-inflammatory states. Front Cell Neurosci. 2015;9:185. doi: 10.3389/fncel.2015.00185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louveau A, Smirnov I, Keyes TJ, Eccles JD, Rouhani SJ, Peske JD, Derecki NC, Castle D, Mandell JW, Lee KS, et al. Structural and functional features of central nervous system lymphatic vessels. Nature. 2015;523:337–41. doi: 10.1038/nature14432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch MA. Neuroinflammatory changes negatively impact on LTP: A focus on IL-1beta. Brain Res. 2015;1621:197–204. doi: 10.1016/j.brainres.2014.08.040. [DOI] [PubMed] [Google Scholar]
- Maezawa I, Zaja-Milatovic S, Milatovic D, Stephen C, Sokal I, Maeda N, Montine TJ, Montine KS. Apolipoprotein E isoform-dependent dendritic recovery of hippocampal neurons following activation of innate immunity. J Neuroinflammation. 2006;3:21. doi: 10.1186/1742-2094-3-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maezawa I, Zimin PI, Wulff H, Jin LW. Amyloid-beta Protein Oligomer at Low Nanomolar Concentrations Activates Microglia and Induces Microglial Neurotoxicity. Journal of Biological Chemistry. 2011;286:3693–3706. doi: 10.1074/jbc.M110.135244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maezawa I, Zou B, Di Lucente J, Cao W, Pascual C, Weerasekara S, Zhanf M, Xie XS, Hua DH, Jin L-W. The Anti-Aβ and Neuroprotective Properties of A Novel Tricyclic Pyrone Molecule. J Alzheimers Dis. 2017;58:559–574. doi: 10.3233/JAD-161175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy MM. Location, Location, Location: Microglia Are Where They Live. Neuron. 2017;95:233–235. doi: 10.1016/j.neuron.2017.07.005. [DOI] [PubMed] [Google Scholar]
- Nguyen HM, Grossinger EM, Horiuchi M, Davis KW, Jin LW, Maezawa I, Wulff H. Differential Kv1.3, KCa3.1, and Kir2.1 expression in “classically” and “alternatively” activated microglia. Glia. 2017;65:106–121. doi: 10.1002/glia.23078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson E, Kullmann DM. T-type calcium channels contribute to NMDA receptor independent synaptic plasticity in hippocampal regular-spiking oriens-alveus interneurons. J Physiol. 2017;595:3449–3458. doi: 10.1113/JP273695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connor JJ, Coogan AN. Actions of the pro-inflammatory cytokine IL-1 beta on central synaptic transmission. Exp Physiol. 1999;84:601–14. [PubMed] [Google Scholar]
- Peng Y, Lu K, Li Z, Zhao Y, Wang Y, Hu B, Xu P, Shi X, Zhou B, Pennington M, et al. Blockade of Kv1.3 channels ameliorates radiation-induced brain injury. Neuro Oncol. 2014;16:528–39. doi: 10.1093/neuonc/not221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira LE, Villinger F, Wulff H, Sankaranarayanan A, Raman G, Ansari AA. Pharmacokinetics, toxicity, and functional studies of the selective Kv1.3 channel blocker 5-(4-phenoxybutoxy)psoralen in rhesus macaques. Exp Biol Med (Maywood) 2007;232:1338–54. doi: 10.3181/0705-RM-148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prinz M, Priller J. The role of peripheral immune cells in the CNS in steady state and disease. Nat Neurosci. 2017;20:136–144. doi: 10.1038/nn.4475. [DOI] [PubMed] [Google Scholar]
- Rangaraju S, Gearing M, Jin LW, Levey A. Potassium Channel Kv1.3 Is Highly Expressed by Microglia in Human Alzheimer’s Disease. J Alzheimers Dis. 2015;44:797–808. doi: 10.3233/JAD-141704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rangaraju S, Raza SA, Pennati A, Deng Q, Dammer EB, Duong D, Pennington MW, Tansey MG, Lah JJ, Betarbet R, et al. A systems pharmacology-based approach to identify novel Kv1.3 channel-dependent mechanisms in microglial activation. J Neuroinflammation. 2017;14:128. doi: 10.1186/s12974-017-0906-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ransohoff RM. A polarizing question: do M1 and M2 microglia exist? Nat Neurosci. 2016;19:987–91. doi: 10.1038/nn.4338. [DOI] [PubMed] [Google Scholar]
- Rus H, Pardo CA, Hu L, Darrah E, Cudrici C, Niculescu T, Niculescu F, Mullen KM, Allie R, Guo L, et al. The voltage-gated potassium channel Kv1.3 is highly expressed on inflammatory infiltrates in multiple sclerosis brain. Proc Natl Acad Sci U S A. 2005;102:11094–9. doi: 10.1073/pnas.0501770102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schilling T, Eder C. Effects of kinase inhibitors on TGF-beta induced upregulation of Kv1.3 K+ channels in brain macrophages. Pflugers Arch. 2003;447:312–5. doi: 10.1007/s00424-003-1155-3. [DOI] [PubMed] [Google Scholar]
- Schilling T, Eder C. Amyloid-beta-induced reactive oxygen species production and priming are differentially regulated by ion channels in microglia. Journal of Cellular Physiology. 2011;226:3295–302. doi: 10.1002/jcp.22675. [DOI] [PubMed] [Google Scholar]
- Schlichter LC, Kaushal V, Moxon-Emre I, Sivagnanam V, Vincent C. The Ca2+ activated SK3 channel is expressed in microglia in the rat striatum and contributes to microglia-mediated neurotoxicity in vitro. J Neuroinflammation. 2010;7:4. doi: 10.1186/1742-2094-7-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz A, Sankaranarayanan A, Azam P, Schmidt-Lassen K, Homerick D, Hansel W, Wulff H. Design of PAP-1, a selective small molecule Kv1.3 blocker, for the suppression of effector memory T cells in autoimmune diseases. Mol Pharmacol. 2005;68:1254–70. doi: 10.1124/mol.105.015669. [DOI] [PubMed] [Google Scholar]
- Vautier F, Belachew S, Chittajallu R, Vittorio G. Shaker-type potassium channel subunits differentially control oligodendrocyte progenitor proliferation. Glia. 2004;48:337–45. doi: 10.1002/glia.20088. [DOI] [PubMed] [Google Scholar]
- Villeda SA, Luo J, Mosher KI, Zou BD, Britschgi M, Bieri G, Stan TM, Fainberg N, Ding ZQ, Eggel A, et al. The ageing systemic milieu negatively regulates neurogenesis and cognitive function. Nature. 2011;477:90–U157. doi: 10.1038/nature10357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villeda SA, Plambeck KE, Middeldorp J, Castellano JM, Mosher KI, Luo J, Smith LK, Bieri G, Lin K, Berdnik D, et al. Young blood reverses age-related impairments in cognitive function and synaptic plasticity in mice. Nat Med. 2014;20:659–663. doi: 10.1038/nm.3569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visentin S, Renzi M, Levi G. Altered outward-rectifying K+ current reveals microglial activation induced by HIV-1 Tat protein. Glia. 2001;33:181–90. doi: 10.1002/1098-1136(200103)33:3<181::aid-glia1017>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- Vogel D, Vereyken E, Glim J, Heijnen P, Moeton M, van der Valk P, Amor S, Teunissen C, van Horssen J, Dijkstra C. Macrophages in Inflammatory Multiple Sclerosis Lesions Have an Intermediate Activation Status. Glia. 2013;61:S182–S183. doi: 10.1186/1742-2094-10-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Rowan MJ, Anwyl R. Beta-amyloid-mediated inhibition of NMDA receptor-dependent long-term potentiation induction involves activation of microglia and stimulation of inducible nitric oxide synthase and superoxide. J Neurosci. 2004;24:6049–56. doi: 10.1523/JNEUROSCI.0233-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf SA, Boddeke HW, Kettenmann H. Microglia in Physiology and Disease. Annu Rev Physiol. 2017;79:619–643. doi: 10.1146/annurev-physiol-022516-034406. [DOI] [PubMed] [Google Scholar]
- Wulff H, Calabresi PA, Allie R, Yun S, Pennington M, Beeton C, Chandy KG. The voltage-gated Kv1.3 K(+) channel in effector memory T cells as new target for MS. J Clin Invest. 2003;111:1703–13. doi: 10.1172/JCI16921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wulff H, Castle NA, Pardo LA. Voltage-gated potassium channels as therapeutic targets. Nat Rev Drug Discov. 2009;8:982–1001. doi: 10.1038/nrd2983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wulff H, Kolski-Andreaco A, Sankaranarayanan A, Sabatier JM, Shakkottai V. Modulators of small- and intermediate-conductance calcium-activated potassium channels and their therapeutic indications. Curr Med Chem. 2007;14:1437–57. doi: 10.2174/092986707780831186. [DOI] [PubMed] [Google Scholar]
- Yirmiya R, Goshen I. Immune modulation of learning, memory, neural plasticity and neurogenesis. Brain, Behavior, and Immunity. 2011;25:181–213. doi: 10.1016/j.bbi.2010.10.015. [DOI] [PubMed] [Google Scholar]
- Zhan X, Stamova B, Jin LW, DeCarli C, Phinney B, Sharp FR. Gram-negative bacterial molecules associate with Alzheimer disease pathology. Neurology. 2016;87:2324–2332. doi: 10.1212/WNL.0000000000003391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao YH, Jaber V, Lukiw WJ. Secretory Products of the Human GI Tract Microbiome and Their Potential Impact on Alzheimer’s Disease (AD): Detection of Lipopolysaccharide (LPS) in AD Hippocampus. Frontiers in Cellular and Infection Microbiology. 2017;7 doi: 10.3389/fcimb.2017.00318. [DOI] [PMC free article] [PubMed] [Google Scholar]