

Abstract
Glycogen Synthase Kinase-3 (GSK-3) is a highly conserved negative regulator of receptor tyrosine kinase, cytokine, and Wnt signaling pathways. Stimulation of these pathways inhibits GSK-3 to modulate diverse downstream effectors that include transcription factors, nutrient sensors, glycogen synthesis, mitochondrial function, circadian rhythm, and cell fate. GSK-3 also regulates alternative splicing in response to T-cell receptor activation, and recent phosphoproteomic studies have revealed that multiple splicing factors and regulators of RNA biosynthesis are phosphorylated in a GSK-3 dependent manner. Furthermore, inhibition of GSK-3 alters the splicing of hundreds of mRNAs, indicating a broad role for GSK-3 in the regulation of RNA processing. GSK-3 regulated phosphoproteins include SF3B1, SRSF2, PSF, RBM8A, nucleophosmin 1 (NPM1), and PHF6, many of which are mutated in leukemia and myelodysplasia. As GSK-3 is inhibited by pathways that are pathologically activated in leukemia and loss of Gsk3 in hematopoietic cells causes a severe myelodysplastic neoplasm in mice, these findings strongly implicate GSK-3 as a critical regulator of mRNA processing in normal and malignant hematopoiesis.
Graphical/Visual Abstract and Caption
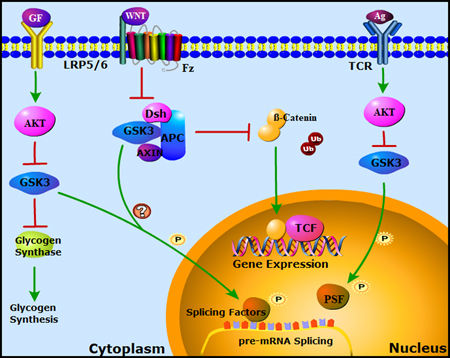
Caption: Signaling Pathways that Regulate GSK-3 and Splicing Factor Phosphorylation
Introduction
Advances in DNA sequencing have revealed that the complexity of gene expression is enhanced by multiple regulatory modalities, including context-dependent changes in mRNA splicing. The identification of splicing variants has improved recently because of advances in sequencing and, importantly, in algorithms to analyze local splice variants. Recent analyses estimate that up to 95% of expressed genes undergo alternative splicing (Heyd & Lynch, 2011; Pan, Shai, Lee, Frey, & Blencowe, 2008; E. T. Wang et al., 2008), an observation that underscores the need to define the functional significance and mechanisms of alternative splicing. Diverse intercellular signaling pathways regulate pre-mRNA splicing through post-translational modifications of splicing factors that include phosphorylation, acetylation, methylation, sumoylation, and hydroxylation (Edmond et al., 2011; Fu & Ares, 2014; Katzenberger, Marengo, & Wassarman, 2009; Lin & Fu, 2007; Lynch, 2007; Lynch & Weiss, 2000; Stamm, 2008). Recent work from several groups has identified glycogen synthase kinase-3 (GSK-3), a core modulator of multiple signaling pathways, as a key regulator of signal-dependent mRNA splicing (Hernandez et al., 2004; Heyd & Lynch, 2010; Martinez et al., 2015; Shinde et al., 2017). This review will focus on the roles of GSK-3 in mRNA splicing, addressing splicing factors that are phosphorylated in a GSK-3-dependent manner, the impact of GSK-3 inhibition on both global and specific mRNA splicing, and possible roles for GSK-3 in aberrant splicing associated with malignancy.
WHAT IS GSK-3? BACKGROUND:
The observations that inhibition of GSK-3 alters the splicing of mRNAs that encode Tau protein in neurons (Hernandez et al., 2004) and CD45 in human T-lymphocytes (Heyd & Lynch, 2010) raised the possibility that GSK-3 could be a broad regulator of alternative splicing in diverse contexts and in a manner that responds to extracellular signaling. This was supported by subsequent observations that pharmacologic and genetic inhibition of GSK-3 alters the splicing of hundreds of mRNAs in a cell-type specific manner (Martinez et al., 2015; Shinde et al., 2017). As GSK-3 functions in diverse signaling pathways and is itself subject to diverse modes of regulation, we will briefly summarize the functions and regulation of GSK-3.
GSK-3 is a constitutively active, ubiquitously expressed protein kinase that regulates multiple signaling pathways, frequently as a pathway antagonist that must be inhibited by upstream signaling (Figure 1) (B. W. Doble & Woodgett, 2003; Kaidanovich-Beilin & Woodgett, 2011; Sutherland, 2011; Valvezan & Klein, 2011). GSK-3 is highly conserved in species from Dictyostelium discoideum to humans. In vertebrates, GSK-3 is typically encoded by two similar genes, Gsk3a and Gsk3b, which encode the proteins, GSK-3α and GSK-3β. The two isoforms share 98% amino acid sequence identity in their catalytic domains and function redundantly in many but not all pathways (for example see (X. Chen et al., 2017; B.W. Doble, Patel, Wood, Kockeritz, & Woodgett, 2007; B. W. Doble & Woodgett, 2003; Hoeflich et al., 2000)). Here we will refer to GSK-3α and GSK-3β collectively as GSK-3 unless otherwise specified.
Figure 1. GSK-3 regulated signaling pathways and splicing factor phosphorylation.

Antigen stimulation of the T cell receptor (TCR) activates the cytosolic protein kinase AKT, which inhibits GSK-3 by phosphorylation at a conserved N-terminal serine. Inhibition of GSK-3 allows the polypyrimidine tract-binding protein (PTB)-associated splicing factor (PSF) to modulate the splicing of CD45 pre-mRNA, as described in detail in Figure 2 (Heyd & Lynch, 2010). Growth factor signaling through receptor tyrosine kinases (RTKs) also activates AKT and inhibits GSK-3, allowing activation of diverse downstream effectors, including glycogen synthase. (Both Akt and GSK-3 have multiple targets not shown here for the sake of clarity). This pathway could also modulate GSK-3-dependent phosphorylation of splicing factors in the cytosol or within the nucleus (Hartmann et al., 2009). The Wnt pathway is also suppressed by GSK-3. In the absence of Wnts, GSK-3 phosphorylates β-catenin, promoting its proteosomal degradation. Wnt signaling inhibits GSK-3 through a distinct, AKT independent mechanism, stabilizing β-catenin, which then activates Wnt target gene expression. Wnts could also regulate splicing factor phosphorylation through inhibition of GSK-3 as well as through other downstream effectors of Wnt signaling (Goncalves et al., 2008; Hartmann et al., 2009).
GSK-3 was first identified as a serine/threonine kinase that phosphorylates and inhibits glycogen synthase (GS), the rate-limiting enzyme in glycogen synthesis (B. W. Doble & Woodgett, 2003; Picton, Woodgett, Hemmings, & Cohen, 1982). Phosphorylation of GS by GSK-3 requires pre-phosphorylation by a priming kinase at a position 4 amino acids C-terminal to the GSK-3 phosphorylation site, representing a frequent but not universal consensus (S/TXXXS/T) for GSK-3 phosphorylation (Fiol, Mahrenholz, Wang, Roeske, & Roach, 1987; Frame, Cohen, & Biondi, 2001; Kaidanovich-Beilin & Woodgett, 2011; Sutherland, 2011; C. Xu, Kim, & Gumbiner, 2009; W. Zhang, DePaoli-Roach, & Roach, 1993). Receptor tyrosine kinase (RTK) signaling (as well as cytokine, T cell receptor, and other signaling pathways) inhibits GSK-3 activity through activation of AKT, which phosphorylates a conserved N-terminal serine in GSK-3α and GSK-3β, creating a pseudosubstrate/autoinhibitory domain that mimics these primed phosphorylation sites (Frame et al., 2001). GSK-3 phosphorylates multiple substrates in addition to GS to regulate diverse processes that include cell growth and survival, cytoskeletal organization, protein stability, circadian rhythm, immune responses, and development (B. W. Doble & Woodgett, 2003; Kaidanovich-Beilin & Woodgett, 2011; Sutherland, 2011; Valvezan & Klein, 2011). N-terminal phosphorylation is therefore one of several mechanisms to block GSK-3 activity and activate downstream signaling through diverse effectors. However, GSK-3 also phosphorylates substrates that lack the +4 priming site, including cyclin D1, Tau, and protein phosphatase-1 inhibitor-2 (Shinde et al., 2017; Sutherland, 2011).
As part of a multiprotein “destruction complex”, GSK-3 also antagonizes the canonical Wnt pathway by phosphorylating ß-catenin and targeting it for proteosomal degradation. Wnt signaling modulates this cytosolic complex to inhibit GSK-3 (Piao et al., 2008; Tran & Polakis, 2012; Valvezan, Zhang, Diehl, & Klein, 2012) and stabilize ß-catenin, which then activates Wnt target genes such as c-myc, cyclin D1, and axin-2 (Polakis, 2000). Wnt-mediated inhibition of GSK-3 also activates other downstream pathways, including mTOR and Hippo signaling (Inoki et al., 2006; Myers, Yousefi, Lengner, & Klein, 2018). Importantly, Wnt inhibition of GSK-3 is mechanistically distinct from RTK/AKT signaling, as Wnt signaling is unaffected by mutating the GSK-3 N-terminal serines to alanine (Ding, Chen, & McCormick, 2000; B.W. Doble et al., 2007; McManus et al., 2005). Nevertheless, direct GSK-3 inhibitors activate signaling downstream of GSK-3 in both pathways, mimicking Wnt and RTK signaling.
Gsk3 loss of function disrupts cell fate in diverse organisms from protozoans like Dictyostelium to invertebrate and vertebrate animals including Drosophila, amphibians, fish, and mice (B. W. Doble & Woodgett, 2003). Gsk3b loss of function mutations in mice are embryonic or neonatal lethal (Hoeflich et al., 2000; Liu, Arron, Stankunas, Crabtree, & Longaker, 2007), and the Gsk3a;Gsk3b double knockout (Gsk3 DKO) is lethal in early embryogenesis (Barrell, Szabo-Rogers, & Liu, 2012; B.W. Doble et al., 2007). Furthermore, DKO mouse embryonic stem cells (mESCs) are unable to differentiate into most embryonic lineages either in vitro or in vivo, and instead maintain an undifferentiated state characterized by the persistent expression of pluripotency markers (B.W. Doble et al., 2007).
Although GSK-3 was initially characterized as a cytosolic protein kinase, nuclear functions for GSK-3β have been well documented, notably for directing the export of NFAT (Beals, Sheridan, Turck, Gardner, & Crabtree, 1997) and cyclin D1 (Alt, Cleveland, Hannink, & Diehl, 2000) as well as for the regulation of transcription factors including Rev-erbα (Yin, Wang, Klein, & Lazar, 2006) and CREB (Sutherland, 2011). These observations identify GSK-3β activity within the nucleus (Beurel, Grieco, & Jope, 2015; Bijur & Jope, 2001), suggesting that it is therefore capable of phosphorylating splicing factors in the nucleus. In contrast to GSK-3β, the nuclear access of GSK-3α appears to be more restricted and dependent on calcium signaling (Azoulay-Alfaguter et al., 2011). However, splicing factors that shuttle between the nucleus and cytoplasm may still be accessible to both GSK-3 isoforms.
SPLICING REGULATION
Signaling pathways that regulate splicing.
Splicing requires multiple protein–protein and protein–RNA interactions and is regulated by trans-acting proteins that are also subject to regulation by post-translational modifications for normal function. Splicing is primarily a co-transcriptional process regulated by the spliceosome, a mega-dalton complex comprising five short nuclear ribonucleoprotein particles (snRNPs), which in turn are composed of specific uridine-rich small nuclear RNAs (U1, U2, U4, U5, and U6 snRNAs), Sm proteins, and hundreds of auxiliary proteins known collectively as splicing factors (reviewed in (Dvinge, Kim, Abdel-Wahab, & Bradley, 2016; Fu & Ares, 2014; Wahl, Will, & Luhrmann, 2009)). Basic mechanisms for constitutive RNA splicing require cis-regulatory elements and trans-acting factors that bind to these elements to promote engagement with the spliceosome and modulate splicing. Exonic and intronic splicing enhancers (ESEs and ISEs) and splicing silencers (ESSs and ISSs) are sequence elements in the pre-mRNA that bind regulatory proteins which in turn modulate the binding and activity of splicing factors at distal splice sites. Splice site recognition is pivotal to determining the final combination of exons included in each complete mRNA transcript. Serine-arginine rich (SR) splicing factors typically bind ESEs to promote exon inclusion (although they may also bind intronic sequences), whereas heterogeneous nuclear ribonucleoproteins (hnRNPs) typically bind ESSs and/or ISSs to promote exon exclusion. However, these patterns are not absolute, and the function of hnRNPs, as well as SR proteins, may vary with cell context (Fu & Ares, 2014). More specific, tissue restricted factors also function as enhancers or suppressors of splicing in a context specific manner (Heyd & Lynch, 2011).
Signal-induced alternative splicing is a primary mechanism for regulating protein isoform expression in response to changing cellular environmental cues (Lynch, 2007; Shin & Manley, 2004). For example, antigen induced signaling through the T-cell receptor (TCR) regulates the splicing of hundreds of genes involved in multiple T-cell effector functions, including the secretion of cytokines and cytotoxins (Martinez & Lynch, 2013; Martinez et al., 2012). Additionally, RTK signaling through AKT induces phosphorylation of the splicing factor SRSF1 to regulate alternative splicing of fibronectin in fibroblasts (Blaustein et al., 2005; White et al., 2010) and caspase-9 in lung cancer cells (Shultz et al., 2010). As discussed below, insulin and Wnt signaling also modulate the splicing of multiple genes in Drosophila S2 cells (Hartmann et al., 2009). However, in most cases, the mechanisms by which signaling pathways regulate pre-mRNA processing remain to be elucidated.
GSK-3 dependent phosphorylation of splicing factors
Pharmacologic inhibition of GSK-3 mimics activation of diverse signaling pathways and alters the splicing of specific mRNAs in lymphocytes, neurons, cardiomyocytes, and embryonic stem cells. GSK-3 also phosphorylates specific splicing factors in these settings, suggesting a direct role for GSK-3 in alternative splicing.
PSF in alternative splicing of CD45
Activation of the T-cell receptor (TCR) by antigen presentation induces alternative splicing of the receptor tyrosine phosphatase CD45, resulting in expression of isoforms that exclude various combinations of exons 4, 5, and/or 6 (Figure 2) (Hermiston, Xu, Majeti, & Weiss, 2002; Heyd & Lynch, 2010). Induction of CD45 lacking exons 4–6 provides negative feedback to reduce sensitivity to additional antigen stimulation (Hermiston et al., 2002). CD45 alternative splicing is regulated by two factors, the polypyrimidine tract-binding protein (PTB)-associated splicing factor (PSF, also known as splicing factor-proline/glutamine rich (SFPQ)) and hnRNP L-like (hnRNP LL). Both factors bind to ESS elements in CD45 pre-mRNA to promote variable exon exclusion. However, TCR activation regulates hnRNPLL by increasing its transcription and regulates PSF by modulating its phosphorylation state. In the absence of TCR signaling, PSF is phosphorylated by GSK-3 at threonine-687, which promotes interaction with thyroid-hormone receptor associated protein 150 (TRAP150) and prevents PSF from binding to ESS elements in CD45 pre-mRNA. TCR activation inhibits GSK-3, alleviating the phosphorylation and sequestration of PSF, allowing it to bind CD45 ESS elements in variable exons 4–6 and repress exon inclusion (Heyd & Lynch, 2010). Small molecule inhibitors of GSK-3 mimic TCR activation by similarly inhibiting phosphorylation of PSF at Thr-687. As PSF has additional functions in the nucleus, including roles in transcription and nuclear retention of RNA (Shav-Tal & Zipori, 2002), the regulation of PSF by GSK3 may influence a wider spectrum of PSF-mediated gene expression events.
Figure 2. Role of GSK-3 in TCR regulation of CD45 splicing.

CD45 encodes a receptor tyrosine phosphatase that serves as a feedback inhibitor of TCR signaling. In the absence of TCR activation, GSK-3 phosphorylates PSF, which is sequestered by the thyroid hormone receptor associated protein (TRAP)-150. Under these conditions, the variable exons 4, 5, and 6 are retained (R456), extending the sequence and reducing the activity of CD45 (only exons 3–7 are shown here). Upon antigen engagement of the TCR, GSK-3 is phosphorylated and inhibited; newly translated PSF binds to exonic splicing silencer (ESS) elements in exons 4, 5, and 6 of the CD45 pre-mRNA and promotes exon exclusion, leading to expression of a more active PTPase (Heyd & Lynch, 2010, 2011). Multiple splice variants are expressed; for clarity only the R0 form, lacking all 3 variable exons, is shown.
SRSF2 and alternative splicing of Tau/MAPT exon 10
Tau is a microtubule-associated protein expressed predominantly in the brain that promotes microtubule stability. Tauopathies are a group of neurodegenerative disorders in which tau protein accumulates in the brain to form neurofibrillary tangles composed of tau protein aggregates organized in paired helical filaments. These neurofibrillary tangles develop through hyperphosphorylation of tau, missense mutations in the gene that encodes tau (MAPT), or aberrant splicing of MAPT pre-mRNA. GSK-3 is one of several protein kinases that phosphorylate tau at sites associated with paired helical filaments, but an additional role for GSK-3 in modulating tau/MAPT splicing has also been described (Hernandez et al., 2004). The MAPT gene contains 16 exons and is subject to extensive alternative splicing that generates 6 tau isoforms in the adult brain. The inherited tauopathy frontotemporal dementia with parkinsonism associated with chromosome-17 (FTDP-17) is caused by both exonic and intronic mutations in MAPT (Poorkaj et al., 1998; Spillantini et al., 1998). Most of the intronic mutations disrupt MAPT pre-mRNA processing in a manner that increases inclusion of exon 10, which encodes one of four repeat sequences involved in microtubule binding and is believed to promote accumulation of paired helical filaments and ultimately neurodegeneration (Hernandez et al., 2004). Inhibition of GSK-3 in cultured neurons increases inclusion of exon 10 in tau mRNA (Hernandez et al., 2004). GSK-3 can also phosphorylate the splicing factor SRSF2 (SC35) (Hernandez et al., 2004; S. Wang et al., 2018) and GSK-3 inhibition was reported to promote colocalization of GSK3 and SRSF2 in the nucleus (Hernandez et al., 2004). Although a causal relationship between GSK-3 activity, SRSF2 phosphorylation, and exon 10 inclusion has not yet been shown, these findings support a role for GSK-3 in regulating tau via alternative splicing.
Phosphoproteomic identification of GSK-3 substrates
A comprehensive analysis of GSK3 substrates recently performed in mouse embryonic stem cells (mESCs) using stable isotope labeling of amino acids in cell culture (SILAC) and mass spectroscopy identified multiple phosphoproteins involved in RNA biosynthesis and processing (Shinde et al., 2017). Previous work using genetic approaches, in vitro kinase assays, and small molecule GSK-3 inhibitors had identified over 100 proposed GSK-3 substrates in diverse cell types, but the spectrum of endogenous GSK-3 substrates in a given cell type had not been determined. mESCs were selected for this analysis because of the availability of a complete knockout (DKO) of both Gsk3a and Gsk3b in a uniform population of cells (B.W. Doble et al., 2007). The comparison of wild-type and Gsk3 DKO ESCs identified multiple splicing factors (Table 1), including RBM8A (RNA binding protein 8A, also known as Y14), SRSF9 (serine-arginine rich splicing factor-9), and the nucleolar proteins NPM1 (nucleophosmin 1) and PHF6 (plant homeodomain finger protein 6) (Shinde et al., 2017).
Table 1. GSK-3-dependent phosphorylation of RNA binding/processing proteins.
RNA processing factors, primarily splicing factors, phosphorylated in a GSK-3 dependent manner. Specific examples include PSF/SFPQ, SRSF2, p40AUF1, and FTO. RNA binding and processing factors identified in mESCs by SILAC are also shown. GSK-3 phosphorylation in vitro was reported for a subset of target proteins, as indicated by “Y” in that column. The asterix indicates proteins that demonstrate multiple sites of GSK-3-dependent phosphorylation in mESCs. Specifically: SRRM2 is phosphorylated at ser-1535 and 1864; RBM8A is phosphorylated at ser-166 and 168; SRSF9 is phosphorylated at ser-190, 205, and 212; Tra2b is phosphorylated at ser-29, 39, 83, 85, 87, 95, 99, 264, 270, 366, and thr-201.
| Protein | Gene name | Function | Phosphorylation by GSK3 in vitro | Reference |
|---|---|---|---|---|
| PSF/SFPQ (T cells) | Sfpq | Splicing | Y | Heyd and Lynch, 2010 |
| SRSF2/SC35 (neurons) | Srsf2 | Splicing | Y | Hernandez et al., 2004 |
| AU-rich element binding protein AUF1/HNRNPD | Hnrnpd | RNA stability | Y | Wilson et al., 2003 |
| Fat mass and obesity-associated protein | FTO | RNA demethylase | Y | Faulds et al., 2018 |
| Gsk3 KO (mESCs)/SILAC: | Shinde et al., 2017 | |||
| Eukaryotic translation initiation factor 3 subunit B | Eif3b | Translation | ||
| PHD finger protein 6 | Phf6 | Nucleolar protein | Y | |
| 40S ribosomal protein S28 | Rps28 | Ribosomal protein | ||
| Protein phosphatase 4 regulatory subunit 2 | Ppp4r2 | Splicing | ||
| 60S ribosomal protein L18 | Rpl18 | Ribosomal protein | ||
| Pre-mRNA 3’-end-processing factor FIP1 | Fip1l1 | RNA processing | ||
| 5’−3’ exoribonuclease 2 | Xrn2 | Ribonuclease | ||
| CUGBP Elav-like family member 1 | Celf1 | Splicing | ||
| 7SK snRNA methylphosphate capping enzyme | Mepce | RNA capping | ||
| Serine/arginine repetitive matrix protein 2 (2 sites)* | Srrm2 | Splicing | ||
| RNA-binding protein 8A (2 sites)* | Rbm8a | Splicing, NMD | Y | |
| Serine/arginine-rich splicing factor 9 (3 sites)* | Srsf9 | Splicing | Y | |
| Transformer-2 protein homolog beta (11 sites)* | Tra2b | Splicing | ||
| Ribosomal L1 domain-containing protein 1 | Rsl1d1 | Ribosomal protein | ||
| Ribosomal RNA processing protein 1 homolog B | Rrp1b | Splicing | ||
| UAP56-interacting factor | Fyttd1 | RNA export | ||
| Nucleophosmin | Npm1 | Nucleolar | Y | |
| Serine/arginine repetitive matrix protein 1 | Srrm1 | Splicing | ||
| ATP-dependent RNA helicase DDX24 | Ddx24 | RNA helicase | ||
| Nuclear cap-binding protein 3 | NCBP3 | RNA export | ||
| WW domain-binding protein 11 | Wbp11 | Splicing | ||
| RNA-binding protein 39 | Rbm39 | Splicing | ||
Although the SILAC approach does not distinguish direct phosphorylation from indirect dependence on GSK-3 activity, direct phosphorylation of RBM8A, SRSF9, PSF, NPM1, and PHF6 was confirmed by in vitro kinase reactions with recombinant GSK-3β. Significant GSK-3-dependent phosphorylation of additional SR proteins, including Tra2b/SFRS10, CELF1 (CUGBP Elav-like family member 1), SRRM1, SRRM2, SRSF7, RBM39, and snRNP70 was also observed, with phosphorylation predominantly in SR domains (Figure 3).
Figure 3. GSK-3 phosphorylation of RS motif.

GSK-3 dependent phosphorylation of serine-arginine (SR) rich splicing factors identified by SILAC/MS comparing wild-type and Gsk3 KO mouse embryonic stem cells (Shinde et al., 2017). Phosphorylated residues are in red. RS motif is in bold.
Additional proteins associated with pre-mRNA processing that demonstrated GSK-3-dependent phosphorylation included PPP4R2, a regulatory subunit of protein phosphatase 4 (PP4) that interacts with the SMN complex and regulates the maturation of spliceosomal snRNPs; WBP11/SIPP1 (splicing factor that Interacts with PQBP-1 and PP1); NCBP3 and UIF, both involved in RNA export; the pre-mRNA 3’-end processing factor FIP1; and the 7SK snRNA methylphosphate capping enzyme (Mepce). A high level of GSK-3 dependent phosphorylation was also observed for the RNA binding proteins eIF3b, Rps28, Rpl18, and Rsl1d1. Although a S/TXXXS/T consensus site has been identified for several well-characterized GSK-3 substrates (B. W. Doble & Woodgett, 2003; Kaidanovich-Beilin & Woodgett, 2011; Sutherland, 2011), this motif was present in a minority of substrates identified in the SILAC analysis (Shinde et al., 2017). As discussed below, Gsk3 DKO also disrupted the splicing of ~190 pre-mRNAs.
A recent study identified site-specific S-nitrosylation as a novel post-translational modification of GSK-3β in cell lines, primary cardiomyocytes, and hearts from a guinea pig model of heart failure (S. Wang et al., 2018). This study showed that S-nitrosylation inhibits the phosphorylation of cytosolic GSK-3 substrates such as glycogen synthase and appears to alter the substrate specificity of GSK-3 for nuclear substrates, reducing phosphorylation of some targets and increasing the phosphorylation of others. Furthermore, S-nitrosylated GSK-3β is enriched in the nucleus and phosphoproteomic analysis of nuclear proteins identified multiple splicing factors, including SF3B1, SCAF1, SRSF2, SRSF4, SRSF11, NONO (Non-POU domain-containing octamer-binding protein), CSTF2 (cleavage stimulation factor 2), and CDC40 (Pre-mRNA-processing factor 17), as nitrosylation-dependent nuclear GSK-3 substrates; additional splicing factors were phosphorylated in a GSK-3 dependent manner that was independent of nitrosylation. Similar to the findings of Shinde et al, the majority of nuclear GSK-3 substrates lacked the S/TXXXS/T consensus; however, phosphorylated serines or threonines were frequently adjacent to proline, consistent with the known enrichment of proline adjacent to GSK-3 phosphorylation sites. Furthermore, tandem affinity purification (TAP-tag) of GSK-3 followed by mass spectrometry of the immune complex demonstrated that GSK-3 interacts with the spliceosome C complex (S. Wang et al., 2018). These findings are consistent with the prior studies in mESCs, human T-cells, and mouse neurons and together provide strong support for a broad role for GSK-3 in modifying splicing factors and other proteins involved in RNA biosynthesis and processing.
SR domain proteins
A major class of splicing proteins identified in the above phosphoproteomic analyses were SR proteins. SR proteins are highly conserved trans-acting mRNA binding proteins that play critical roles in splicing typically by binding to splicing enhancer elements in a sequence and context specific manner to modulate splicing (Busch & Hertel, 2012; Shepard & Hertel, 2009). The arginine-serine (RS) domains of SR proteins consist of multiple consecutive RS dipeptide repeats that undergo extensive phosphorylation by multiple kinases, including members of the SR protein kinase (SRPK) family (SRPK1 and SRPK2), hPRP4, topoisomerase I and the CDC2-like kinase (CLK) family (CLK1 to CLK4) (Giannakouros, Nikolakaki, Mylonis, & Georgatsou, 2011; Stamm, 2008; Zhou & Fu, 2013). GSK-3 phosphorylates multiple SR family protein splicing factor members within the RS motifs (Figure 3)(Shinde et al., 2017), which could modulate RNA binding, protein-protein interactions, and/or subcellular localization of RS proteins (Prasad, Colwill, Pawson, & Manley, 1999). For example, phosphorylation of SRSF1 (SF2/ASF) increases its binding to U1 70-kDa snRNP and regulates SRSF1 shuttling to the spliceosome from nuclear speckles (a subnuclear compartment enriched in small ribonucleoprotein particles and splicing factors) (Misteli et al., 1998; Stamm, 2008; Xiao & Manley, 1997, 1998).
GSK3 phosphorylates RBM8A (Y14), a component of the exon-junction complex involved in both splicing and nonsense mediated mRNA decay, at serines 166 and 168, which lie within adjacent RS motifs. Phosphorylation of these residues inhibits RBM8A interaction with other exon-junction complex components (Hsu et al., 2005; Ishigaki, Nakamura, Tatsuno, Ma, & Tomosugi, 2015), although these interactions have not yet been shown to be regulated specifically by GSK-3. SRSF10 is dephosphorylated in response to heat shock and plays an essential role in cell survival under stress conditions by inhibiting the splicing machinery (Shin, Feng, & Manley, 2004). Phosphorylation of the SR motifs also modulates the interaction of SR proteins with other splicing factors, as well as with pre-mRNAs during spliceosome assembly (Xiao & Manley, 1998).
GSK-3 dependent phosphorylation of other RNA binding proteins
In addition to spliceosomal proteins, GSK-3 regulates the phosphorylation of multiple RNA binding proteins that have been implicated in splicing as well as additional RNA processing functions. Most prominent among the proteins identified in the phosphoproteomic studies are NPM1 and PHF6, which were phosphorylated in a GSK-3 dependent manner and demonstrated in vitro phosphorylation by recombinant GSK-3β (Shinde et al., 2017). NPM1 is a nucleolar protein with diverse functions in the cell, including ribosome biogenesis, chromatin remodeling, and mRNA processing, and has been reported to function as a negative regulator of alternative splicing (Box et al., 2016; Tarapore et al., 2006). NPM1 post-translational modifications regulate both subcellular localization and function (Murano, Okuwaki, Hisaoka, & Nagata, 2008; Shandilya et al., 2014). NPM1 phosphorylation at serine 225 in mouse ESCs (ser-227 in human NPM1) is blocked by Gsk3 DKO and GSK-3ß phosphorylates human NPM1 in vitro at serine 222 (Shinde et al., 2017). PHF6 regulates chromatin architecture, suppresses ribosomal RNA synthesis and, when overexpressed, interacts with splicing factors and components of the NuRD complex (Soto-Feliciano et al., 2017; Todd, Ivanochko, & Picketts, 2015; Todd & Picketts, 2012; J. Wang et al., 2013). PHF6 is highly phosphorylated at serines 154 and 155 in a GSK3 dependent manner (Shinde et al., 2017). As with NPM1 and the SR proteins, the functional significance of PHF6 phosphorylation by GSK-3 has not yet been addressed, but these serines are adjacent to a nuclear localization signal and their phosphorylation may therefore regulate the subcellular localization of PHF6.
GSK-3 can also phosphorylate the AU-rich element binding protein p40AUF1 to regulate mRNA conformation in vitro. Some growth regulating mRNAs contain AU-rich elements in the 3’-untranslated regions that destabilize these mRNAs. p40AUF1 has an SXXXS motif that can be phosphorylated by GSK-3 in vitro. Phosphorylation of ARE-bound p40AUF1 causes remodeling of the protein-ARE complex and increases susceptibility to degradation. Conversely, when p40AUF1 is dephosphorylated the ARE/protein complex assumes a condensed conformation that correlates with increased stability. Hence GSK-3 may regulate mRNA turnover by remodeling the ARE-ribonucleoprotein complex (Wilson et al., 2003). GSK-3 also phosphorylates and destabilizes the RNA demethylase FTO (fat mass and obesity-associated protein); inhibition of GSK-3 causes accumulation of FTO which reduces overall levels of methylated (N6-methyladenosine or m6A) mRNAs (Faulds et al., 2018). In turn, m6A may regulate mRNA splicing, export, stability, or translation (Covelo-Molares, Bartosovic, & Vanacova, 2018).
It is important to note that the cell-based studies described above cannot distinguish whether the identified proteins are phosphorylated directly by GSK-3 or indirectly by a GSK-3 regulated protein kinase. For example, inhibition of GSK-3 activates the mechanistic target of rapamycin complex 1 (mTORC1) (Inoki et al., 2006), which was recently shown to activate the SR protein kinase SRPK2 and regulate splicing of lipogenic pre-mRNAs (G. Lee et al., 2017). Conversely, inhibition of GSK-3β causes alternative splicing of RAC1 pre-mRNA in colorectal cancer cells indirectly through reduction of SRPK1 levels and reduced phosphorylation of SRSF1 (Goncalves et al., 2014). GSK-3 inhibition could therefore modulate splicing indirectly through the regulation of SRPK1, SRPK2, or other downstream kinases. Although in vitro phosphorylation experiments (Heyd & Lynch, 2010; Shinde et al., 2017) show that GSK-3 can directly phosphorylate SRSF2, SRSF9, NPM1, PHF6, RBM8A, and PSF, this supports but does not prove that the phosphorylation occurs directly in vivo. Similarly, functional experiments involving mutation of phosphorylation sites within putative substrates may support a role for a given serine or threonine, but do not generally identify the protein kinase or prove that phosphorylation per se regulated the observed function. Thus, validation of a direct in vivo phosphorylation by a given protein kinase is especially challenging and is usually inferred from multiple, orthogonal approaches.
GSK-3 regulates splicing
Several groups using varied approaches have established that GSK-3 phosphorylates factors involved in pre-mRNA splicing and more generally in mRNA processing. However, with the exception of PSF regulation of CD45 splicing, how these phosphorylation events affect splicing factor function is yet to be investigated in most cases. Addressing the functional impact of GSK-3 on splicing, high throughput sequencing analyses have suggested a broad role for GSK-3 in the regulation of alternative splicing (Martinez et al., 2015; Shinde et al., 2017). To investigate the effect of complete genetic loss of both Gsk3 isoforms on global splicing, wild-type and Gsk3 double knockout (DKO) mouse embryonic stem cells (mESCs) were subjected to high depth RNA-sequencing followed by analysis through the MAJIQ (Modeling Alternative Junction Inclusion Quantification) algorithm (Vaquero-Garcia et al., 2016) to identify local splice variants (LSVs) and to measure the relative LSV abundance (change in percent spliced in (dPSI)) in DKO compared to wild-type (Martinez et al., 2015; Shinde et al., 2017). This identified 194 significant splicing differences (dPSI >20) in 188 genes, including genes involved in the regulation of DNA methylation such as TET2, which is also commonly mutated in leukemia and myelodysplasia. When stringency was reduced to dPSI > 10, 620 splicing events were altered in Gsk3 DKO compared to wild-type mESCs. The most common changes observed were cassette exons, intron retention, and alternative first exon use. Additionally, human T-cells were treated with the GSK-3 inhibitor SB216763 and then a high throughput method termed RNA-mediated oligonucleotide annealing, selection, and ligation coupled with sequencing (RASL-seq) was used to identify multiple LSVs that were sensitive to GSK-3 inhibition (Martinez et al., 2015; Shinde et al., 2017). Similarly, T-cell derived Jurkat cells treated with the GSK-3 inhibitor demonstrated splicing variants in >200 pre-mRNAs, including genes that regulate apoptosis, splicing, and, intriguingly, RUNX1 and ASXL1, which are associated with human acute myeloid leukemia and carcinomas, as described below (Kristen Lynch, personal communication). Taken together, these high throughput analyses of local splicing variants demonstrate a broad role for GSK-3 in context specific alternative splicing of multiple pre-mRNAs.
Wnt and RTK pathways may regulate splicing through inhibition of GSK-3
Wnt, cytokine, and receptor tyrosine kinase (RTK) pathways inhibit GSK-3 to activate downstream signaling (Figure 1), albeit through biochemically distinct mechanisms. Therefore, activation of these pathways might be predicted to alter splicing in part through inhibition of GSK-3. This mode of signal dependent regulation was demonstrated for TCR activation in human T-cells, as described for alternative splicing of CD45. In addition, both insulin and Wnt signaling alter splicing of 149 (insulin) and 85 (Wnt/wingless) genes in Drosophila S2 cells (Hartmann et al., 2009). The involvement of GSK-3 was not investigated in that study, but these pathways could, in principle, regulate alternative splicing through inhibition of GSK-3. Although the sets of alternatively spliced mRNAs were distinct of the two pathways, this could reflect the distinct mechanisms of GSK-3 inhibition or that each pathway signals through multiple effectors in addition to GSK-3. However, the regulation of splicing by these pathways is likely to be complex and involve multiple mechanisms. For example, the Wnt pathway also activates the transcription of SRSF3 which in turn modulates the splicing of CD44 mRNA (Bordonaro, 2013; Goncalves, Matos, & Jordan, 2008).
SPLICING MUTATIONS IN CANCER
Leukemia
Although alternative splicing provides cells with a means to diversify the proteome, pathological disruption of splicing factors may also promote the initiation and/or maintenance of cancer. The importance of differential splicing in normal and malignant hematopoiesis is supported by evidence showing distinct patterns of alternative splicing at each step of the hematopoietic hierarchy (L. Chen et al., 2014; Edwards et al., 2016; Inoue, Bradley, & Abdel-Wahab, 2016; Pimentel et al., 2016; J. J. Wong et al., 2013). The observations from the Cancer Genome Atlas (TCGA) that 14% of AML patients have mutations in spliceosomal factors focused attention on the potential oncogenic roles of splicing factors (Cancer Genome Atlas Research, 2013; Kandoth et al., 2013). Splicing factors are also frequently mutated in myelodysplastic syndromes (MDS), a group of clonal disorders of hematopoiesis characterized by hypercellular bone marrow, morphological dysplasia, ineffective hematopoiesis leading to anemia, thrombocytopenia, and/or neutropenia, and a high risk of progression to AML. The role of splicing and splicing factor mutations in cancer is covered in more detail in several recent, comprehensive reviews (Dvinge et al., 2016; Goncalves, Pereira, & Jordan, 2017; A. C. H. Wong, Rasko, & Wong, 2018), and here we will focus on splicing factors that are phosphorylated by GSK-3 and that may play a role in leukemogenesis associated with disruption of GSK-3 activity. These include SF3B1, SRSF2, PSF, NPM1, PHF6, and other factors identified in the phosphoproteomic analyses described above.
Although GSK3 mutations have not been identified in TCGA analyses of acute leukemias or solid tumors, GSK-3 is both a target and a negative regulator of pathways that are commonly activated by somatic mutations in malignancies, including RTK/PI3K/AKT and Wnt signaling. Thus, upstream pathway stimulation inhibits GSK-3 and activates signaling downstream of GSK-3. The rarity of GSK3 mutations in cancer may be explained by the presence of two, mostly redundant GSK3 genes, GSK3A and GSK3B, so that oncogenic mutations would require mutation of 3 to 4 separate loci. Nevertheless, a strong argument for GSK-3 regulated pathways in oncogenic transformation can be made for colorectal carcinoma (e.g. Wnt signaling) and leukemias (e.g. RTK signaling) based on known mutations and data from animal models.
RTK signaling is activated in a high frequency of AML cases, for example due to mutations in FLT3 or c-KIT (Cancer Genome Atlas Research, 2013). RTKs activate multiple cytosolic effectors, including phosphatidylinositol-3 kinase (PI3K) and AKT (Q. Xu, Simpson, Scialla, Bagg, & Carroll, 2003), which in turn phosphorylates and inhibits GSK-3 (Cross, Alessi, Cohen, Andjelkovich, & Hemmings, 1995) and enhances survival signaling (Beurel et al., 2015; Beurel & Jope, 2006). Hematopoietic cytokines, including thrombopoietin and erythropoietic, can also activate AKT and inhibit GSK-3 (Soda, Willert, Kaushansky, Geddis, & Ibarra, 2008; Somervaille, Linch, & Khwaja, 2001). Although a role for AKT regulation of GSK-3 in AML has not been established, AKT is constitutively active in primary AML cells from a majority of patients (Martelli et al., 2011; Q. Xu et al., 2003). The activated mutant of FLT3, FLT3-ITD, may also inhibit GSK-3 through a non-canonical mechanism to activate ß-catenin in primary AML cells (Jiang & Griffin, 2010; Tickenbrock et al., 2005).
A role for Wnt signaling in leukemia is supported by evidence from patient samples and experimental models of AML (Morgan et al., 2013; Simon, Grandage, Linch, & Khwaja, 2005; Y. Wang et al., 2010; Ysebaert et al., 2006), chronic myeloid leukemia (CML) (Abrahamsson et al., 2009; Heidel et al., 2012; Jamieson et al., 2004), and mixed lineage leukemia (MLL) (Yeung et al., 2010);(reviewed in (McCubrey et al., 2014; Mikesch, Steffen, Berdel, Serve, & Muller-Tidow, 2007; Staal & Clevers, 2005)). ß-catenin protein levels are elevated in human AML (Morgan et al., 2013; Simon et al., 2005), and a Wnt/ß-catenin transcription reporter showed increased activity in AML patient samples (Simon et al., 2005). ß-catenin protein expression also correlates with increased colony formation and serial replating in human AML and is associated with poor overall survival (Ysebaert et al., 2006). ß-catenin is also required for leukemogenesis induced in mice by overexpression of HoxA9/Meis1a or MLL-AF9 (Y. Wang et al., 2010).
Adenomatous polyposis coli (APC) is a tumor suppressor and core component of the Wnt pathway that enhances GSK-3 activity (Valvezan et al., 2012) to inhibit Wnt signaling. Loss of APC, as observed in the majority of colorectal cancers, inhibits GSK-3 and constitutively activates downstream Wnt/ß-catenin signaling (Polakis, 2000). The APC gene lies within a region of chromosome 5q that is frequently deleted in MDS and in AML and haploinsufficiency for Apc in mice causes an MDS/myeloproliferative phenotype (Lane et al., 2010; J. Wang, Fernald, Anastasi, Le Beau, & Qian, 2010), consistent with a role for GSK-3 inhibition in myeloid neoplasms. Interestingly, casein kinase 1α cooperates with GSK-3 to suppress ß-catenin; the CK1a gene (CSKN1A1) also lies within the 5q commonly deleted region, and CSKN1A1 haploinsufficiency has been strongly implicated in the pathogenesis of 5q- associated MDS (Kronke et al., 2015; Schneider et al., 2014). A direct role for GSK-3 in CML is also supported by the observation of a splice deletion in GSK3B mRNA specifically within granulocyte macrophage progenitors in a series of patients in blast crisis (Abrahamsson et al., 2009; Jamieson et al., 2004). Finally, the GSK-3 interacting protein and inhibitor GSKIP is present in a 700bp duplication within chromosome 14q in four families with inherited myeloproliferative disorder and predisposition to AML. Overexpression of GSKIP, together with ATG2B, in iPSC derived hematopoietic precursor cells and in primary cells from affected patients correlated with enhanced differentiation of hematopoietic progenitor cells, consistent with the hypothesis that inhibition of GSK-3 promotes myeloid neoplasia (Saliba et al., 2015), although it remains possible that other genes within the duplicated region contribute to this familial MPN.
Primary AML blasts from patients are difficult to maintain in culture, and their ex vivo viability can be substantially improved by adding a GSK-3 inhibitor or knocking down Gsk3a/b expression (Bhavanasi et al., 2017). Furthermore, combined inhibition of GSK-3 and mTORC1 maintains cells capable of initiating leukemia in mouse xenografts (“leukemia initiating cells”). These observations are consistent with the hypothesis that the survival of AML cells in vivo requires endogenous signals that act through pathways that inhibit GSK-3, such as RTK, cytokine, or Wnt signaling; poor ex vivo survival would then be due in part to the lack of these signals, and GSK-3 inhibitors would then mimic the endogenous signals to improve AML survival ex vivo (Bhavanasi et al., 2017). The downstream effectors of GSK-3 in this context are not known, but splicing factors, NPM1, and PHF6 are plausible candidates.
Additional evidence for a direct role for GSK-3 in myelodysplasia and leukemia comes from work with GSK-3 inhibitors and Gsk3 knockouts in mice (Guezguez et al., 2016; Huang et al., 2009; McCubrey et al., 2014). Pharmacological inhibition or RNAi-mediated knockdown of Gsk3 in mouse bone marrow causes a transient myeloproliferative state (Focosi, Azzara, Kast, Carulli, & Petrini, 2009; Huang et al., 2009), similar to Pten knockout in hematopoietic cells(Banerji et al., 2012; Yilmaz et al., 2006; J. Zhang et al., 2006), and complete knockout of Gsk3a and Gsk3b in mouse bone marrow causes a more severe myeloproliferative and dysplastic phenotype with marked increase in mature and immature granulocytes and dysplasia, as well as an increase in myeloid blasts that was suggestive of AML (Guezguez et al., 2016). The mechanism downstream of GSK-3 appears to be in part through enhanced Wnt/ß-catenin but additional downstream effectors of GSK-3 likely contribute to this dramatic phenotype. An intriguing possibility is that loss of Gsk3 in mouse hematopoietic cells alters splicing factor phosphorylation and function, leading to myelodysplasia in mice that mimics human MDS and AML associated with mutations in RNA splicing/processing factors. Furthermore, activation of pathways that inhibit GSK-3, such as FLT3 and c-Kit, or pharmacological inhibition of GSK-3, may similarly enhance AML cell viability by regulating pre-mRNA splicing or other aspects of RNA processing through GSK-3 inhibition.
It is therefore intriguing that GSK-3 regulates the phosphorylation of splicing factors that are recurrently mutated in MDS and AML, including SF3B1 and SRSF2, as well as other factors implicated in RNA biosynthesis that are frequently mutated in AML and other malignancies, including NPM1 and PHF6.
Mutations in SF3B1 and SRSF2, which encode proteins involved in 3’-splice site recognition during pre-mRNA processing, are the most common class of mutations in patients with MDS and occur across the entire spectrum of myeloid malignancies, including in 10–25% of patients with AML and in a higher proportion of patients with AML transformed from MDS (Cancer Genome Atlas Research, 2013; Gelsi-Boyer et al., 2009; Inoue et al., 2016; Papaemmanuil et al., 2011; Yoshida et al., 2011). These mutations occur early in the evolution of MDS, based on clonal architecture, consistent with driver mutations (Dvinge et al., 2016; Kandoth et al., 2013; Lindsley et al., 2015; Mian et al., 2015; Papaemmanuil et al., 2011).
Mutations in SF3B1 are common in a form of MDS termed refractory anemia with ringed sideroblasts (RARS) and also occur in other myeloid malignancies, as well as uveal melanoma, bladder, breast, and pancreatic cancers. Hotspots for recurrent mutations in SF3B1 in myeloid neoplasms as well as breast and pancreatic cancers include codons for V600, K666, and other sites that vary in frequency depending on the type of malignancy, with R625 more commonly mutated, for example, in uveal melanoma and bladder cancers. These missense mutations are exclusively heterozygous, suggesting a dominant effect rather than loss of function (Dvinge et al., 2016; Papaemmanuil et al., 2011). As SF3B1 is a highly conserved, integral component of U2 snRNPs, these mutations could have far reaching global effects on splicing. It is not yet clear how the recurrent mutations in SF3B1 affect splicing but may involve disruption of SF3B1 interaction with pre-mRNAs (Jenkins & Kielkopf, 2017). SF3B1 phosphorylation is coupled with splicing catalysis and occurs at multiple sites throughout the molecule (C. Wang et al., 1998). Thus, GSK-3 or other protein kinases might regulate SF3B1 function by modulating protein-RNA interaction as well as protein-protein interactions (Boudrez, Beullens, Waelkens, Stalmans, & Bollen, 2002) or subcellular localization (Eto, Sonoda, & Abe, 2011).
SRSF2 mutations are present in up to 47% of cases of the myelodysplastic/myeloproliferative disorder chronic myelomonocytic leukemia (CMML) (Meggendorfer et al., 2012) and also occur at lower frequency in RARS, other forms of MDS, and AML. SRSF2 is recurrently mutated at proline-95, which alters the specificity of SRSF2 binding to ESEs, changing the splicing of hundreds of mRNAs. Interestingly, SRSF2P95 alters the splicing pattern of EZH2, which is also frequently mutated in MDS, to generate a poison exon containing a premature termination codon that in turn leads to nonsense mediated decay of EZH2 mRNA (Kim et al., 2015). Furthermore, knock-in of SRSF2P95H in mouse hematopoietic cells drives hyperproliferation of hematopoietic precursor cells, anemia, leukopenia, and dysplasia, mimicking human MDS (Kim et al., 2015). SRSF2 is known to be phosphorylated by multiple kinases, but it is not known whether inhibition of GSK-3 mediated SRSF2 phosphorylation would mimic the specific effects of the SRSF2P95 mutation.
NPM1, a nucleolar protein with multiple proposed functions including roles in ribosome biogenesis and splicing, is mutated or rearranged in 35% of AML patients (Falini et al., 2005). NPM is highly phosphorylated by multiple protein kinases, including GSK-3. NPM1 phosphorylation in general is required for centrosome duplication, promotes NPM1 localization to nuclear speckles (Tarapore et al., 2006), and regulates NPM1 binding to RNA (Okuwaki, Tsujimoto, & Nagata, 2002). Oncogenic mutations in NPM1 disrupt the C-terminal domain resulting in sequestration of NPM1 protein in the cytoplasm, effectively inhibiting nuclear functions of NPM1. Thus, while the specific role of phosphorylation by GSK-3 is not known, loss of GSK-3 phosphorylation could affect subcellular localization or otherwise interfere with any of the nuclear functions of NPM1.
PHF6 is mutated in 3% of AML (Cancer Genome Atlas Research, 2013). Germline mutations in PHF6 causes the X-linked neurodevelopmental disorder Borjeson-Forssman-Lehmann syndrome (Lower et al., 2002), whereas somatic mutations of PHF6 were first identified in human T-ALL and myeloid leukemias, suggesting a role of PHF6 as tumor suppressor gene (Todd et al., 2015; Van Vlierberghe et al., 2010; Van Vlierberghe et al., 2011). On the other hand, knockdown of PHF6 impairs growth of BCR-ABL1+ B-ALL cells in vivo (Meacham et al., 2015), raising the possibility of context dependent roles for PHF6 as both tumor suppressor and oncogene in hematopoietic neoplasms.
RBM8A (Y14) is a core component of the exon junction complex that also regulates pre-mRNA splicing (Chuang, Lee, & Tarn, 2015; Michelle et al., 2012). Heterozygous loss of RBM8A is associated with the rare hematopoietic disorder thrombocytopenia with absent radius (TAR) syndrome. RBM8A mutations have not been associated with AML or MDS, but may affect cell proliferation or viability of hematopoietic cells through the alternative splicing of pre-mRNAs encoding p53, Bcl-x, Bim, and other regulators of apoptosis. Depletion of RMB8A increases the expression of the proapoptotic Bcl-x splice variant Bcl-x(S) (Michelle et al., 2012). Phosphorylation of RBM8A promotes dissociation from the exon junction complex and may therefore mimic RBM8A depletion to promote apoptosis. If GSK-3 phosphorylation of RBM8A similarly impairs its interaction with the exon junction complex, this could suggest a mechanism for the known propapoptic function of GSK-3 (Beurel et al., 2015; Bijur, De Sarno, & Jope, 2000; Pap & Cooper, 1998), and conversely, explain enhanced cell survival commonly observed with GSK-3 inhibitors (Nonaka, Hough, & Chuang, 1998).
How might inhibition of GSK-3 dependent splicing factor phosphorylation contribute to leukemogenesis? As discussed above, endogenous signaling through pathological activation of RTK or Wnt signaling could alter splicing factor function and affect global splicing patterns or splicing of specific genes known to drive leukemia when mutated. Based on RASL-seq analysis, inhibition of GSK-3 in human T-cell derived Jurkat cells disrupts the splicing of RUNX1, ASXL1, CD47, and other pre-RNAs. RUNX1 (also known as AML1) is a transcription factor crucial for hematopoietic cell development and a frequent target of translocations and mutations in MDS, AML, and ALL (Lam & Zhang, 2012). Inhibition of GSK-3 in Jurkat cells caused alternative splicing of RUNX1 to generate RUNX1a, a dominant negative form of RUNX1 (Komeno et al., 2014; Ran et al., 2013; Tanaka et al., 1995), in 25% of transcripts. GSK-3 inhibition also induced alternative splicing of ASXL1, a polycomb group gene frequently mutated in MDS and AML, to generate a splice form with an early premature termination codon in 80% of transcripts. Although these RASL-seq data have not yet been validated in primary cells, ASXL1 was also found to be alternatively spliced in hematopoietic stem and progenitor cells of MDS patients with mutations in SF3B1 (Dolatshad et al., 2015). Additionally, knockout of Gsk3 in mESCs caused alternative splicing of TET2, a DNA demethylating enzyme frequently mutated in MDS and AML (Shinde et al., 2017). These observations with RUNX1, ASXL1, and TET2 are based on RASL-seq or RNA-seq data and are yet to be confirmed in primary cells, but raise interesting questions about the role of alternative splicing in response to GSK-3 inhibition by small molecule inhibitors, genetic loss of function, or endogenous signals that normally signal by inhibiting GSK-3.
Alternative splicing as a therapeutic target in cancer
Targeting splicing may have therapeutic use in cancers that are driven by splicing factor mutations and/or pathologic splicing of specific oncogenic mRNAs. However, despite recurrent mutations in core splicing factors, transcriptome analyses in SF3B1, SRSF2, and U2AF1 mutated MDS have not revealed a unifying pattern of alternative splicing (Qiu et al., 2016). Other explanations could be that oncogenic mRNAs are mis-spliced a manner specific to each mutation, that integration of global changes in splicing enhance malignant transformation in a way that is not evident from bioinformatic analysis, or that the splicing factors serve additional functions apart from splicing per se, such as regulation of DNA damage or chromatin architecture.
Recurrent mutations in SF3B1 and SRSF2 (as well as U2AF1) are exclusively heterozygous missense mutations, suggesting that cells are extremely sensitive to perturbations in core splicing factor function and that maintaining a wild-type copy is essential for viability. Thus, pharmacological inhibition of these factors may be lethal for heterozygous cells and, at an optimal level of inhibition, spare wild-type cells (Yoshida et al., 2011). In support of this hypothesis, leukemias with spliceosomal gene mutations are preferentially susceptible to additional splicing perturbations in vivo compared to leukemias without such mutations (Cancer Genome Atlas Research, 2013). Furthermore murine myeloid leukemias treated with the spliceosome inhibitor E7107 demonstrate preferential cell death of leukemia cells bearing the Srsf2P95/+ mutation compared to isogenic leukemias with wild-type Srsf2 (S. C. Lee et al., 2016). Similar synthetic-lethal interactions between mutated U2AF1 and exposure to spliceosome inhibitors have also been reported (Shirai et al., 2015). These results highlight the possibility that manipulation of splicing might provide therapeutic benefit in leukemia.
Although the precise physiologic function of phosphorylation of most SR proteins is not well defined, inhibition of phosphorylation of SR proteins has emerged as a potential means to modulate splicing by altering the function and localization of splicing factors. A variety of screens have been performed to identify compounds that inhibit splicing factor protein kinases such as SRPK and/or CLK. Conversely, factors that inhibit GSK-3 improve AML cell viability in culture (Bhavanasi et al., 2017), and thus agents that increase GSK-3 activity could, in principle, impair AML cell survival in vivo by disrupting splicing factor function or by mimicking mutations in NPM1 and PHF6. However, further studies are needed to understand the potential therapeutic relevance of phosphorylation inhibitors and whether any genetic or histologic cancer subtypes are susceptible to these compounds (Araki et al., 2015; Muraki et al., 2004).
Conclusion
CONCLUSIONS AND FUTURE DIRECTIONS
Focused examination of signal regulated splicing as well as unbiased proteome-wide analyses have revealed that multiple RNA processing proteins are phosphorylated in a GSK-3 dependent manner, with biochemical evidence for direct phosphorylation by GSK-3 in several of these. Functional data support global regulation of mRNA splicing by GSK-3 based on Gsk3 knockout and inhibitor studies in diverse cell types. A detailed mechanism has been worked out for one of these examples, the TCR-dependent inhibition of GSK-3 mediated phosphorylation of PSF in the regulation of CD45 splicing in human T-cells, whereas mechanistic understanding of the impact of GSK-3 on splicing factor function in other contexts remains to be worked out in detail. Nevertheless, these findings point to RNA processing factors as new effectors of GSK-3 dependent signaling pathways.
The observations that: 1) GSK-3 regulated factors, including SF3B1, SRSF2, NPM1, and PHF6, are frequently mutated in AML and MDS, 2) mutational activation of pathways that inhibit GSK-3 occurs frequently in AML, and 3) inhibition of GSK-3 in normal hematopoietic cells causes a myeloproliferative/myelodysplastic neoplasm, raise the intriguing possibility that GSK-3 plays a central role in leukemia pathogenesis through the regulation of RNA splicing/processing enzymes. Future research will need to define in detail the molecular and functional impact of GSK-3 phosphorylation of these factors and establish a mechanistic link between GSK-3, RNA processing, and leukemia pathogenesis, but this is an attractive and testable hypothesis that will be an exciting area for future study.
Acknowledgments
The authors thank Kristen Lynch, Yoseph Barash, and Martin Carroll for helpful discussions. PSK was supported by grants from the National Institutes of Health (1R01MH100923 and 1R01GM115517), the Penn-CHOP Blood Center for Patient Care and Discovery at the University of Pennsylvania Perelman School of Medicine, and an ASH Bridge Award from the American Society of Hematology.
Contributor Information
Xiaolei Liu, Department of Medicine, Division of Hematology-Oncology, Perelman School of Medicine at the University of Pennsylvania, Philadelphia, PA, Xiaolei.Liu@pennmedicine.upenn.edu.
Peter S. Klein, Department of Medicine, Division of Hematology-Oncology, Perelman School of Medicine at the University of Pennsylvania, Philadelphia, PA, pklein@pennmedicine.upenn.edu.
References
- Abrahamsson AE, Geron I, Gotlib J, Dao KH, Barroga CF, Newton IG, … Jamieson CH (2009). Glycogen synthase kinase 3beta missplicing contributes to leukemia stem cell generation. Proc Natl Acad Sci U S A, 106(10), 3925–3929. doi: 10.1073/pnas.0900189106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alt JR, Cleveland JL, Hannink M, & Diehl JA (2000). Phosphorylation-dependent regulation of cyclin D1 nuclear export and cyclin D1-dependent cellular transformation. Genes Dev, 14(24), 3102–3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araki S, Dairiki R, Nakayama Y, Murai A, Miyashita R, Iwatani M, … Nakanishi O (2015). Inhibitors of CLK protein kinases suppress cell growth and induce apoptosis by modulating pre-mRNA splicing. PLoS One, 10(1), e0116929. doi: 10.1371/journal.pone.0116929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azoulay-Alfaguter I, Yaffe Y, Licht-Murava A, Urbanska M, Jaworski J, Pietrokovski S, … Eldar-Finkelman H (2011). Distinct molecular regulation of glycogen synthase kinase-3alpha isozyme controlled by its N-terminal region: functional role in calcium/calpain signaling. J Biol Chem, 286(15), 13470–13480. doi: 10.1074/jbc.M110.127969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerji V, Frumm SM, Ross KN, Li LS, Schinzel AC, Hahn CK, … Stegmaier K (2012). The intersection of genetic and chemical genomic screens identifies GSK-3alpha as a target in human acute myeloid leukemia. J Clin Invest, 122(3), 935–947. doi: 10.1172/JCI46465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrell WB, Szabo-Rogers HL, & Liu KJ (2012). Novel reporter alleles of GSK-3alpha and GSK-3beta. PLoS One, 7(11), e50422. doi: 10.1371/journal.pone.0050422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beals CR, Sheridan CM, Turck CW, Gardner P, & Crabtree GR (1997). Nuclear export of NF-ATc enhanced by glycogen synthase kinase-3. Science, 275(5308), 1930–1934. [DOI] [PubMed] [Google Scholar]
- Beurel E, Grieco SF, & Jope RS (2015). Glycogen synthase kinase-3 (GSK3): regulation, actions, and diseases. Pharmacol Ther, 148, 114–131. doi: 10.1016/j.pharmthera.2014.11.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beurel E, & Jope RS (2006). The paradoxical pro- and anti-apoptotic actions of GSK3 in the intrinsic and extrinsic apoptosis signaling pathways. Prog Neurobiol, 79(4), 173–189. doi: 10.1016/j.pneurobio.2006.07.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhavanasi D, Wen KW, Liu X, Vergez F, Danet-Desnoyers G, Carroll M, … Klein PS (2017). Signaling mechanisms that regulate ex vivo survival of human acute myeloid leukemia initiating cells. Blood Cancer J, 7(12), 636. doi: 10.1038/s41408-017-0003-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bijur GN, De Sarno P, & Jope RS (2000). Glycogen synthase kinase-3beta facilitates staurosporine- and heat shock-induced apoptosis. Protection by lithium. J Biol Chem, 275(11), 7583–7590. [DOI] [PubMed] [Google Scholar]
- Bijur GN, & Jope RS (2001). Proapoptotic stimuli induce nuclear accumulation of glycogen synthase kinase-3 beta. J Biol Chem, 276(40), 37436–37442. doi: 10.1074/jbc.M105725200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaustein M, Pelisch F, Tanos T, Munoz MJ, Wengier D, Quadrana L, … Srebrow A (2005). Concerted regulation of nuclear and cytoplasmic activities of SR proteins by AKT. Nat Struct Mol Biol, 12(12), 1037–1044. doi: 10.1038/nsmb1020 [DOI] [PubMed] [Google Scholar]
- Bordonaro M (2013). Crosstalk between Wnt Signaling and RNA Processing in Colorectal Cancer. J Cancer, 4(2), 96–103. doi: 10.7150/jca.5470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudrez A, Beullens M, Waelkens E, Stalmans W, & Bollen M (2002). Phosphorylation-dependent interaction between the splicing factors SAP155 and NIPP1. J Biol Chem, 277(35), 31834–31841. doi: 10.1074/jbc.M204427200 [DOI] [PubMed] [Google Scholar]
- Box JK, Paquet N, Adams MN, Boucher D, Bolderson E, O’Byrne KJ, & Richard DJ (2016). Nucleophosmin: from structure and function to disease development. BMC Mol Biol, 17(1), 19. doi: 10.1186/s12867-016-0073-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busch A, & Hertel KJ (2012). Evolution of SR protein and hnRNP splicing regulatory factors. Wiley Interdiscip Rev RNA, 3(1), 1–12. doi: 10.1002/wrna.100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research, N. (2013). Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med, 368(22), 2059–2074. doi: 10.1056/NEJMoa1301689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Kostadima M, Martens JH, Canu G, Garcia SP, Turro E, … Rendon A (2014). Transcriptional diversity during lineage commitment of human blood progenitors. Science, 345(6204), 1251033. doi: 10.1126/science.1251033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Wang R, Liu X, Wu Y, Zhou T, Yang Y, … Ying QL (2017). A Chemical-Genetic Approach Reveals the Distinct Roles of GSK3alpha and GSK3beta in Regulating Embryonic Stem Cell Fate. Dev Cell, 43(5), 563–576 e564. doi: 10.1016/j.devcel.2017.11.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang TW, Lee KM, & Tarn WY (2015). Function and pathological implications of exon junction complex factor Y14. Biomolecules, 5(2), 343–355. doi: 10.3390/biom5020343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Covelo-Molares H, Bartosovic M, & Vanacova S (2018). RNA methylation in nuclear pre-mRNA processing. Wiley Interdiscip Rev RNA, e1489. doi: 10.1002/wrna.1489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross D, Alessi D, Cohen P, Andjelkovich M, & Hemmings B (1995). Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature, 378, 785–789. [DOI] [PubMed] [Google Scholar]
- Ding VW, Chen RH, & McCormick F (2000). Differential regulation of glycogen synthase kinase 3beta by insulin and Wnt signaling. J Biol Chem, 275(42), 32475–32481. [DOI] [PubMed] [Google Scholar]
- Doble BW, Patel S, Wood GA, Kockeritz LK, & Woodgett JR (2007). Functional redundancy of GSK-3alpha and GSK-3beta in Wnt/beta-catenin signaling shown by using an allelic series of embryonic stem cell lines. Dev Cell, 12(6), 957–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doble BW, & Woodgett JR (2003). GSK-3: tricks of the trade for a multi-tasking kinase. J Cell Sci, 116(Pt 7), 1175–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolatshad H, Pellagatti A, Fernandez-Mercado M, Yip BH, Malcovati L, Attwood M, … Boultwood J (2015). Disruption of SF3B1 results in deregulated expression and splicing of key genes and pathways in myelodysplastic syndrome hematopoietic stem and progenitor cells. Leukemia, 29(5), 1092–1103. doi: 10.1038/leu.2014.331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dvinge H, Kim E, Abdel-Wahab O, & Bradley RK (2016). RNA splicing factors as oncoproteins and tumour suppressors. Nat Rev Cancer, 16(7), 413–430. doi: 10.1038/nrc.2016.51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edmond V, Moysan E, Khochbin S, Matthias P, Brambilla C, Brambilla E, … Eymin B (2011). Acetylation and phosphorylation of SRSF2 control cell fate decision in response to cisplatin. EMBO J, 30(3), 510–523. doi: 10.1038/emboj.2010.333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards CR, Ritchie W, Wong JJ, Schmitz U, Middleton R, An X, … Blobel GA (2016). A dynamic intron retention program in the mammalian megakaryocyte and erythrocyte lineages. Blood. doi: 10.1182/blood-2016-01-692764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eto K, Sonoda Y, & Abe S (2011). The kinase DYRKIA regulates pre-mRNA splicing in spermatogonia and proliferation of spermatogonia and Sertoli cells by phosphorylating a spliceosomal component, SAP155, in postnatal murine testes. Mol Cell Biochem, 355(1–2), 217–222. doi: 10.1007/s11010-011-0857-7 [DOI] [PubMed] [Google Scholar]
- Falini B, Mecucci C, Tiacci E, Alcalay M, Rosati R, Pasqualucci L, … Party GALW (2005). Cytoplasmic nucleophosmin in acute myelogenous leukemia with a normal karyotype. N Engl J Med, 352(3), 254–266. doi: 10.1056/NEJMoa041974 [DOI] [PubMed] [Google Scholar]
- Faulds KJ, Egelston JN, Sedivy LJ, Mitchell MK, Garimella S, Kozlowski H, … Phiel CJ (2018). Glycogen synthase kinase-3 (Gsk-3) activity regulates mRNA methylation in mouse embryonic stem cells. J Biol Chem. doi: 10.1074/jbc.RA117.001298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiol CJ, Mahrenholz AM, Wang Y, Roeske RW, & Roach PJ (1987). Formation of protein kinase recognition sites by covalent modification of the substrate. Molecular mechanism for the synergistic action of casein kinase II and glycogen synthase kinase 3. J Biol Chem, 262(29), 14042–14048. [PubMed] [Google Scholar]
- Focosi D, Azzara A, Kast RE, Carulli G, & Petrini M (2009). Lithium and hematology: established and proposed uses. Journal of Leukocyte Biology, 85(1), 20–28. [DOI] [PubMed] [Google Scholar]
- Frame S, Cohen P, & Biondi RM (2001). A common phosphate binding site explains the unique substrate specificity of GSK3 and its inactivation by phosphorylation. Mol Cell, 7(6), 1321–1327. [DOI] [PubMed] [Google Scholar]
- Fu XD, & Ares M Jr. (2014). Context-dependent control of alternative splicing by RNA-binding proteins. Nat Rev Genet, 15(10), 689–701. doi: 10.1038/nrg3778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelsi-Boyer V, Trouplin V, Adelaide J, Bonansea J, Cervera N, Carbuccia N, … Birnbaum D (2009). Mutations of polycomb-associated gene ASXL1 in myelodysplastic syndromes and chronic myelomonocytic leukaemia. Br J Haematol, 145(6), 788–800. doi: 10.1111/j.1365-2141.2009.07697.x [DOI] [PubMed] [Google Scholar]
- Giannakouros T, Nikolakaki E, Mylonis I, & Georgatsou E (2011). Serine-arginine protein kinases: a small protein kinase family with a large cellular presence. FEBS J, 278(4), 570–586. doi: 10.1111/j.1742-4658.2010.07987.x [DOI] [PubMed] [Google Scholar]
- Goncalves V, Henriques AF, Pereira JF, Neves Costa A, Moyer MP, Moita LF, … Jordan P (2014). Phosphorylation of SRSF1 by SRPK1 regulates alternative splicing of tumor-related Rac1b in colorectal cells. RNA, 20(4), 474–482. doi: 10.1261/rna.041376.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goncalves V, Matos P, & Jordan P (2008). The beta-catenin/TCF4 pathway modifies alternative splicing through modulation of SRp20 expression. RNA, 14(12), 2538–2549. doi: 10.1261/rna.1253408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goncalves V, Pereira JFS, & Jordan P (2017). Signaling Pathways Driving Aberrant Splicing in Cancer Cells. Genes (Basel), 9(1). doi: 10.3390/genes9010009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guezguez B, Almakadi M, Benoit YD, Shapovalova Z, Rahmig S, Fiebig-Comyn A, … Bhatia M (2016). GSK3 Deficiencies in Hematopoietic Stem Cells Initiate Pre-neoplastic State that Is Predictive of Clinical Outcomes of Human Acute Leukemia. Cancer Cell, 29(1), 61–74. doi: 10.1016/j.ccell.2015.11.012 [DOI] [PubMed] [Google Scholar]
- Hartmann B, Castelo R, Blanchette M, Boue S, Rio DC, & Valcarcel J (2009). Global analysis of alternative splicing regulation by insulin and wingless signaling in Drosophila cells. Genome Biol, 10(1), R11. doi: 10.1186/gb-2009-10-1-r11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heidel FH, Bullinger L, Feng Z, Wang Z, Neff TA, Stein L, … Armstrong SA (2012). Genetic and pharmacologic inhibition of beta-catenin targets imatinib-resistant leukemia stem cells in CML. Cell Stem Cell, 10(4), 412–424. doi: 10.1016/j.stem.2012.02.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermiston ML, Xu Z, Majeti R, & Weiss A (2002). Reciprocal regulation of lymphocyte activation by tyrosine kinases and phosphatases. J Clin Invest, 109(1), 9–14. doi: 10.1172/JCI14794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez F, Perez M, Lucas JJ, Mata AM, Bhat R, & Avila J (2004). Glycogen synthase kinase-3 plays a crucial role in tau exon 10 splicing and intranuclear distribution of SC35. Implications for Alzheimer’s disease. J Biol Chem, 279(5), 3801–3806. doi: 10.1074/jbc.M311512200 [DOI] [PubMed] [Google Scholar]
- Heyd F, & Lynch KW (2010). Phosphorylation-dependent regulation of PSF by GSK3 controls CD45 alternative splicing. Mol Cell, 40(1), 126–137. doi: 10.1016/j.molcel.2010.09.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heyd F, & Lynch KW (2011). Degrade, move, regroup: signaling control of splicing proteins. Trends Biochem Sci, 36(8), 397–404. doi: 10.1016/j.tibs.2011.04.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoeflich KP, Luo J, Rubie EA, Tsao MS, Jin O, & Woodgett JR (2000). Requirement for glycogen synthase kinase-3beta in cell survival and NF- kappaB activation. Nature, 406(6791), 86–90. [DOI] [PubMed] [Google Scholar]
- Hsu L-W, Hsu M, Li C, Chuang TW, Lin RI, & Tarn WY (2005). Phosphorylation of Y14 modulates its interaction with proteins involved in mRNA metabolism and influences its methylation. J Biol Chem, 280(41), 34507–34512. doi: 10.1074/jbc.M507658200 [DOI] [PubMed] [Google Scholar]
- Huang J, Zhang Y, Bersenev A, O’Brien WT, Tong W, Emerson SG, & Klein PS (2009). Pivotal role for glycogen synthase kinase-3 in hematopoietic stem cell homeostasis in mice. J Clin Invest, 119(12), 3519–3529. doi:40572 [pii] 10.1172/JCI40572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoki K, Ouyang H, Zhu T, Lindvall C, Wang Y, Zhang X, … Guan KL (2006). TSC2 integrates Wnt and energy signals via a coordinated phosphorylation by AMPK and GSK3 to regulate cell growth. Cell, 126, 955–968. [DOI] [PubMed] [Google Scholar]
- Inoue D, Bradley RK, & Abdel-Wahab O (2016). Spliceosomal gene mutations in myelodysplasia: molecular links to clonal abnormalities of hematopoiesis. Genes Dev, 30(9), 989–1001. doi: 10.1101/gad.278424.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishigaki Y, Nakamura Y, Tatsuno T, Ma S, & Tomosugi N (2015). Phosphorylation status of human RNA-binding protein 8A in cells and its inhibitory regulation by Magoh. Exp Biol Med (Maywood), 240(4), 438–445. doi: 10.1177/1535370214556945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamieson CH, Ailles LE, Dylla SJ, Muijtjens M, Jones C, Zehnder JL, … Weissman IL (2004). Granulocyte-macrophage progenitors as candidate leukemic stem cells in blast-crisis CML. N Engl J Med, 351(7), 657–667. doi: 10.1056/NEJMoa040258 [DOI] [PubMed] [Google Scholar]
- Jenkins JL, & Kielkopf CL (2017). Splicing Factor Mutations in Myelodysplasias: Insights from Spliceosome Structures. Trends Genet, 33(5), 336–348. doi: 10.1016/j.tig.2017.03.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang J, & Griffin JD (2010). Wnt/beta-catenin Pathway Modulates the Sensitivity of the Mutant FLT3 Receptor Kinase Inhibitors in a GSK-3beta Dependent Manner. Genes Cancer, 1(2), 164–176. doi: 10.1177/1947601910362446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaidanovich-Beilin O, & Woodgett JR (2011). GSK-3: Functional Insights from Cell Biology and Animal Models. Front Mol Neurosci, 4, 40. doi: 10.3389/fnmol.2011.00040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandoth C, McLellan MD, Vandin F, Ye K, Niu B, Lu C, … Ding L (2013). Mutational landscape and significance across 12 major cancer types. Nature, 502(7471), 333–339. doi: 10.1038/nature12634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katzenberger RJ, Marengo MS, & Wassarman DA (2009). Control of alternative splicing by signal-dependent degradation of splicing-regulatory proteins. J Biol Chem, 284(16), 10737–10746. doi: 10.1074/jbc.M809506200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim E, Ilagan JO, Liang Y, Daubner GM, Lee SC, Ramakrishnan A, … Abdel-Wahab O (2015). SRSF2 Mutations Contribute to Myelodysplasia by Mutant-Specific Effects on Exon Recognition. Cancer Cell, 27(5), 617–630. doi: 10.1016/j.ccell.2015.04.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komeno Y, Yan M, Matsuura S, Lam K, Lo MC, Huang YJ, … Zhang DE (2014). Runx1 exon 6-related alternative splicing isoforms differentially regulate hematopoiesis in mice. Blood, 123(24), 3760–3769. doi: 10.1182/blood-2013-08-521252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kronke J, Fink EC, Hollenbach PW, MacBeth KJ, Hurst SN, Udeshi ND, … Ebert BL (2015). Lenalidomide induces ubiquitination and degradation of CK1alpha in del(5q) MDS. Nature, 523(7559), 183–188. doi: 10.1038/nature14610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam K, & Zhang DE (2012). RUNX1 and RUNX1-ETO: roles in hematopoiesis and leukemogenesis. Front Biosci (Landmark Ed), 17, 1120–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane SW, Sykes SM, Al-Shahrour F, Shterental S, Paktinat M, Lo Celso C, … Gilliland DG (2010). The Apc(min) mouse has altered hematopoietic stem cell function and provides a model for MPD/MDS. Blood, 115(17), 3489–3497. doi:blood-2009-11-251728 [pii]; 10.1182/blood-2009-11-251728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee G, Zheng Y, Cho S, Jang C, England C, Dempsey JM, … Blenis J (2017). Post-transcriptional Regulation of De Novo Lipogenesis by mTORC1-S6K1-SRPK2 Signaling. Cell, 171(7), 1545–1558 e1518. doi: 10.1016/j.cell.2017.10.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SC, Dvinge H, Kim E, Cho H, Micol JB, Chung YR, … Abdel-Wahab O (2016). Modulation of splicing catalysis for therapeutic targeting of leukemia with mutations in genes encoding spliceosomal proteins. Nat Med, 22(6), 672–678. doi: 10.1038/nm.4097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin S, & Fu XD (2007). SR proteins and related factors in alternative splicing. Adv Exp Med Biol, 623, 107–122. [DOI] [PubMed] [Google Scholar]
- Lindsley RC, Mar BG, Mazzola E, Grauman PV, Shareef S, Allen SL, … Ebert BL (2015). Acute myeloid leukemia ontogeny is defined by distinct somatic mutations. Blood, 125(9), 1367–1376. doi: 10.1182/blood-2014-11-610543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu KJ, Arron JR, Stankunas K, Crabtree GR, & Longaker MT (2007). Chemical rescue of cleft palate and midline defects in conditional GSK-3beta mice. Nature, 446(7131), 79–82. doi: 10.1038/nature05557 [DOI] [PubMed] [Google Scholar]
- Lower KM, Turner G, Kerr BA, Mathews KD, Shaw MA, Gedeon AK, … Gecz J (2002). Mutations in PHF6 are associated with Borjeson-Forssman-Lehmann syndrome. Nat Genet, 32(4), 661–665. doi: 10.1038/ng1040 [DOI] [PubMed] [Google Scholar]
- Lynch KW (2007). Regulation of alternative splicing by signal transduction pathways. Adv Exp Med Biol, 623, 161–174. [DOI] [PubMed] [Google Scholar]
- Lynch KW, & Weiss A (2000). A model system for activation-induced alternative splicing of CD45 pre-mRNA in T cells implicates protein kinase C and Ras. Mol Cell Biol, 20(1), 70–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martelli AM, Evangelisti C, Chappell W, Abrams SL, Basecke J, Stivala F, … McCubrey JA (2011). Targeting the translational apparatus to improve leukemia therapy: roles of the PI3K/PTEN/Akt/mTOR pathway. Leukemia, 25(7), 1064–1079. doi: 10.1038/leu.2011.46 [DOI] [PubMed] [Google Scholar]
- Martinez NM, Agosto L, Qiu J, Mallory MJ, Gazzara MR, Barash Y, … Lynch KW (2015). Widespread JNK-dependent alternative splicing induces a positive feedback loop through CELF2-mediated regulation of MKK7 during T-cell activation. Genes Dev, 29(19), 2054–2066. doi: 10.1101/gad.267245.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez NM, & Lynch KW (2013). Control of alternative splicing in immune responses: many regulators, many predictions, much still to learn. Immunol Rev, 253(1), 216–236. doi: 10.1111/imr.12047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez NM, Pan Q, Cole BS, Yarosh CA, Babcock GA, Heyd F, … Lynch KW (2012). Alternative splicing networks regulated by signaling in human T cells. RNA, 18(5), 1029–1040. doi: 10.1261/rna.032243.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCubrey JA, Steelman LS, Bertrand FE, Davis NM, Abrams SL, Montalto G, … Martelli AM (2014). Multifaceted roles of GSK-3 and Wnt/beta-catenin in hematopoiesis and leukemogenesis: opportunities for therapeutic intervention. Leukemia, 28(1), 15–33. doi: 10.1038/leu.2013.184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McManus EJ, Sakamoto K, Armit LJ, Ronaldson L, Shpiro N, Marquez R, & Alessi DR (2005). Role that phosphorylation of GSK3 plays in insulin and Wnt signalling defined by knockin analysis. EMBO J, 24(8), 1571–1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meacham CE, Lawton LN, Soto-Feliciano YM, Pritchard JR, Joughin BA, Ehrenberger T, … Hemann MT (2015). A genome-scale in vivo loss-of-function screen identifies Phf6 as a lineage-specific regulator of leukemia cell growth. Genes Dev, 29(5), 483–488. doi: 10.1101/gad.254151.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meggendorfer M, Roller A, Haferlach T, Eder C, Dicker F, Grossmann V, … Schnittger S (2012). SRSF2 mutations in 275 cases with chronic myelomonocytic leukemia (CMML). Blood, 120(15), 3080–3088. doi: 10.1182/blood-2012-01-404863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mian SA, Rouault-Pierre K, Smith AE, Seidl T, Pizzitola I, Kizilors A, … Mufti GJ (2015). SF3B1 mutant MDS-initiating cells may arise from the haematopoietic stem cell compartment. Nat Commun, 6, 10004. doi: 10.1038/ncomms10004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michelle L, Cloutier A, Toutant J, Shkreta L, Thibault P, Durand M, … Chabot B (2012). Proteins associated with the exon junction complex also control the alternative splicing of apoptotic regulators. Mol Cell Biol, 32(5), 954–967. doi: 10.1128/MCB.06130-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikesch JH, Steffen B, Berdel WE, Serve H, & Muller-Tidow C (2007). The emerging role of Wnt signaling in the pathogenesis of acute myeloid leukemia. Leukemia, 21(8), 1638–1647. doi: 10.1038/sj.leu.2404732 [DOI] [PubMed] [Google Scholar]
- Misteli T, Caceres JF, Clement JQ, Krainer AR, Wilkinson MF, & Spector DL (1998). Serine phosphorylation of SR proteins is required for their recruitment to sites of transcription in vivo. J Cell Biol, 143(2), 297–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan RG, Pearn L, Liddiard K, Pumford SL, Burnett AK, Tonks A, & Darley RL (2013). gamma-Catenin is overexpressed in acute myeloid leukemia and promotes the stabilization and nuclear localization of beta-catenin. Leukemia, 27(2), 336–343. doi: 10.1038/leu.2012.221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muraki M, Ohkawara B, Hosoya T, Onogi H, Koizumi J, Koizumi T, … Hagiwara M (2004). Manipulation of alternative splicing by a newly developed inhibitor of Clks. J Biol Chem, 279(23), 24246–24254. doi: 10.1074/jbc.M314298200 [DOI] [PubMed] [Google Scholar]
- Murano K, Okuwaki M, Hisaoka M, & Nagata K (2008). Transcription regulation of the rRNA gene by a multifunctional nucleolar protein, B23/nucleophosmin, through its histone chaperone activity. Mol Cell Biol, 28(10), 3114–3126. doi: 10.1128/MCB.02078-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers RL, Yousefi M, Lengner CJ, & Klein PS (2018). Wnt Signaling in Intestinal Stem Cells and Cancer. Encyclopedia of Cancer, in press. [Google Scholar]
- Nonaka S, Hough CJ, & Chuang DM (1998). Chronic lithium treatment robustly protects neurons in the central nervous system against excitotoxicity by inhibiting N-methyl-D- aspartate receptor-mediated calcium influx. Proc Natl Acad Sci U S A, 95(5), 2642–2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okuwaki M, Tsujimoto M, & Nagata K (2002). The RNA binding activity of a ribosome biogenesis factor, nucleophosmin/B23, is modulated by phosphorylation with a cell cycle-dependent kinase and by association with its subtype. Mol Biol Cell, 13(6), 2016–2030. doi: 10.1091/mbc.02-03-0036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan Q, Shai O, Lee LJ, Frey BJ, & Blencowe BJ (2008). Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat Genet, 40(12), 1413–1415. doi: 10.1038/ng.259 [DOI] [PubMed] [Google Scholar]
- Pap M, & Cooper GM (1998). Role of glycogen synthase kinase-3 in the phosphatidylinositol 3- Kinase/Akt cell survival pathway. J Biol Chem, 273(32), 19929–19932. [DOI] [PubMed] [Google Scholar]
- Papaemmanuil E, Cazzola M, Boultwood J, Malcovati L, Vyas P, Bowen D, … Chronic Myeloid Disorders Working Group of the International Cancer Genome, C. (2011). Somatic SF3B1 mutation in myelodysplasia with ring sideroblasts. N Engl J Med, 365(15), 1384–1395. doi: 10.1056/NEJMoa1103283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piao S, Lee SH, Kim H, Yum S, Stamos JL, Xu Y, … Ha NC (2008). Direct inhibition of GSK3beta by the phosphorylated cytoplasmic domain of LRP6 in Wnt/beta-catenin signaling. PLoS One, 3(12), e4046. doi: 10.1371/journal.pone.0004046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picton C, Woodgett J, Hemmings B, & Cohen P (1982). Multisite phosphorylation of glycogen synthase from rabbit skeletal muscle. Phosphorylation of site 5 by glycogen synthase kinase-5 (casein kinase-II) is a prerequisite for phosphorylation of sites 3 by glycogen synthase kinase-3. FEBS Lett, 150(1), 191–196. [DOI] [PubMed] [Google Scholar]
- Pimentel H, Parra M, Gee SL, Mohandas N, Pachter L, & Conboy JG (2016). A dynamic intron retention program enriched in RNA processing genes regulates gene expression during terminal erythropoiesis. Nucleic Acids Res, 44(2), 838–851. doi: 10.1093/nar/gkv1168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polakis P (2000). Wnt signaling and cancer. Genes Dev, 14(15), 1837–1851. [PubMed] [Google Scholar]
- Poorkaj P, Bird TD, Wijsman E, Nemens E, Garruto RM, Anderson L, … Schellenberg GD (1998). Tau is a candidate gene for chromosome 17 frontotemporal dementia. Ann Neurol, 43(6), 815–825. [DOI] [PubMed] [Google Scholar]
- Prasad J, Colwill K, Pawson T, & Manley JL (1999). The protein kinase Clk/Sty directly modulates SR protein activity: both hyper- and hypophosphorylation inhibit splicing. Mol Cell Biol, 19(10), 6991–7000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu J, Zhou B, Thol F, Zhou Y, Chen L, Shao C, … Heuser M (2016). Distinct splicing signatures affect converged pathways in myelodysplastic syndrome patients carrying mutations in different splicing regulators. RNA, 22(10), 1535–1549. doi: 10.1261/rna.056101.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran D, Shia WJ, Lo MC, Fan JB, Knorr DA, Ferrell PI, … Zhang DE (2013). RUNX1a enhances hematopoietic lineage commitment from human embryonic stem cells and inducible pluripotent stem cells. Blood, 121(15), 2882–2890. doi: 10.1182/blood-2012-08-451641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saliba J, Saint-Martin C, Di Stefano A, Lenglet G, Marty C, Keren B, … Plo I (2015). Germline duplication of ATG2B and GSKIP predisposes to familial myeloid malignancies. Nat Genet, 47(10), 1131–1140. doi: 10.1038/ng.3380 [DOI] [PubMed] [Google Scholar]
- Schneider RK, Adema V, Heckl D, Jaras M, Mallo M, Lord AM, … Ebert BL (2014). Role of casein kinase 1A1 in the biology and targeted therapy of del(5q) MDS. Cancer Cell, 26(4), 509–520. doi: 10.1016/j.ccr.2014.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shandilya J, Senapati P, Dhanasekaran K, Bangalore SS, Kumar M, Kishore AH, … Kundu TK (2014). Phosphorylation of multifunctional nucleolar protein nucleophosmin (NPM1) by aurora kinase B is critical for mitotic progression. FEBS Lett, 588(14), 2198–2205. doi: 10.1016/j.febslet.2014.05.014 [DOI] [PubMed] [Google Scholar]
- Shav-Tal Y, & Zipori D (2002). PSF and p54(nrb)/NonO--multi-functional nuclear proteins. FEBS Lett, 531(2), 109–114. [DOI] [PubMed] [Google Scholar]
- Shepard PJ, & Hertel KJ (2009). The SR protein family. Genome Biol, 10(10), 242. doi: 10.1186/gb-2009-10-10-242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin C, Feng Y, & Manley JL (2004). Dephosphorylated SRp38 acts as a splicing repressor in response to heat shock. Nature, 427(6974), 553–558. doi: 10.1038/nature02288 [DOI] [PubMed] [Google Scholar]
- Shin C, & Manley JL (2004). Cell signalling and the control of pre-mRNA splicing. Nat Rev Mol Cell Biol, 5(9), 727–738. doi: 10.1038/nrm1467 [DOI] [PubMed] [Google Scholar]
- Shinde MY, Sidoli S, Kulej K, Mallory MJ, Radens CM, Reicherter AL, … Klein PS (2017). Phosphoproteomics reveals that glycogen synthase kinase-3 phosphorylates multiple splicing factors and is associated with alternative splicing. J Biol Chem, 292(44), 18240–18255. doi: 10.1074/jbc.M117.813527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirai CL, Tripathi M, Ley JN, Ndonwi M, White BS, Tapia R, … Walter MJ (2015). Preclinical Activity of Splicing Modulators in U2AF1 Mutant MDS/AML. Blood, 126(23), 1653. [Google Scholar]
- Shultz JC, Goehe RW, Wijesinghe DS, Murudkar C, Hawkins AJ, Shay JW, … Chalfant CE (2010). Alternative splicing of caspase 9 is modulated by the phosphoinositide 3-kinase/Akt pathway via phosphorylation of SRp30a. Cancer Res, 70(22), 9185–9196. doi: 10.1158/0008-5472.CAN-10-1545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon M, Grandage VL, Linch DC, & Khwaja A (2005). Constitutive activation of the Wnt/beta-catenin signalling pathway in acute myeloid leukaemia. Oncogene, 24(14), 2410–2420. doi: 10.1038/sj.onc.1208431 [DOI] [PubMed] [Google Scholar]
- Soda M, Willert K, Kaushansky K, Geddis AE, & Ibarra YM (2008). Inhibition of GSK-3beta promotes survival and proliferation of megakaryocytic cells through a beta-catenin-independent pathway. Cell Signal, 20(12), 2317–2323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somervaille TC, Linch DC, & Khwaja A (2001). Growth factor withdrawal from primary human erythroid progenitors induces apoptosis through a pathway involving glycogen synthase kinase-3 and Bax. Blood, 98(5), 1374–1381. [DOI] [PubMed] [Google Scholar]
- Soto-Feliciano YM, Bartlebaugh JME, Liu Y, Sanchez-Rivera FJ, Bhutkar A, Weintraub AS, … Hemann MT (2017). PHF6 regulates phenotypic plasticity through chromatin organization within lineage-specific genes. Genes Dev, 31(10), 973–989. doi: 10.1101/gad.295857.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spillantini MG, Murrell JR, Goedert M, Farlow MR, Klug A, & Ghetti B (1998). Mutation in the tau gene in familial multiple system tauopathy with presenile dementia. Proc Natl Acad Sci U S A, 95(13), 7737–7741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staal FJ, & Clevers HC (2005). WNT signalling and haematopoiesis: a WNT-WNT situation. Nat Rev Immunol, 5(1), 21–30. [DOI] [PubMed] [Google Scholar]
- Stamm S (2008). Regulation of alternative splicing by reversible protein phosphorylation. J Biol Chem, 283(3), 1223–1227. doi: 10.1074/jbc.R700034200 [DOI] [PubMed] [Google Scholar]
- Sutherland C (2011). What Are the bona fide GSK3 Substrates? Int J Alzheimers Dis, 2011, 505607. doi: 10.4061/2011/505607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka T, Tanaka K, Ogawa S, Kurokawa M, Mitani K, Nishida J, … Hirai H (1995). An acute myeloid leukemia gene, AML1, regulates hemopoietic myeloid cell differentiation and transcriptional activation antagonistically by two alternative spliced forms. EMBO J, 14(2), 341–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarapore P, Shinmura K, Suzuki H, Tokuyama Y, Kim SH, Mayeda A, & Fukasawa K (2006). Thr199 phosphorylation targets nucleophosmin to nuclear speckles and represses pre-mRNA processing. FEBS Lett, 580(2), 399–409. doi: 10.1016/j.febslet.2005.12.022 [DOI] [PubMed] [Google Scholar]
- Tickenbrock L, Schwable J, Wiedehage M, Steffen B, Sargin B, Choudhary C, … Serve H (2005). Flt3 tandem duplication mutations cooperate with Wnt signaling in leukemic signal transduction. Blood, 105(9), 3699–3706. doi: 10.1182/blood-2004-07-2924 [DOI] [PubMed] [Google Scholar]
- Todd MA, Ivanochko D, & Picketts DJ (2015). PHF6 Degrees of Separation: The Multifaceted Roles of a Chromatin Adaptor Protein. Genes (Basel), 6(2), 325–352. doi: 10.3390/genes6020325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todd MA, & Picketts DJ (2012). PHF6 interacts with the nucleosome remodeling and deacetylation (NuRD) complex. J Proteome Res, 11(8), 4326–4337. doi: 10.1021/pr3004369 [DOI] [PubMed] [Google Scholar]
- Tran H, & Polakis P (2012). Reversible modification of adenomatous polyposis coli (APC) with K63-linked polyubiquitin regulates the assembly and activity of the beta-catenin destruction complex. J Biol Chem, 287(34), 28552–28563. doi:M112.387878 [pii]; 10.1074/jbc.M112.387878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valvezan AJ, & Klein PS (2011). GSK-3 and Wnt Signaling in Neurogenesis and Bipolar Disorder. Front Mol Neurosci, 5, 1. doi: 10.3389/fnmol.2012.00001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valvezan AJ, Zhang F, Diehl JA, & Klein PS (2012). Adenomatous Polyposis Coli (APC) Regulates Multiple Signaling Pathways by Enhancing Glycogen Synthase Kinase-3 (GSK-3) Activity. J Biol Chem, 287(6), 3823–3832. doi:M111.323337 [pii]; 10.1074/jbc.M111.323337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Vlierberghe P, Palomero T, Khiabanian H, Van der Meulen J, Castillo M, Van Roy N, … Ferrando A (2010). PHF6 mutations in T-cell acute lymphoblastic leukemia. Nat Genet, 42(4), 338–342. doi: 10.1038/ng.542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Vlierberghe P, Patel J, Abdel-Wahab O, Lobry C, Hedvat CV, Balbin M, … Ferrando A (2011). PHF6 mutations in adult acute myeloid leukemia. Leukemia, 25(1), 130–134. doi: 10.1038/leu.2010.247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaquero-Garcia J, Barrera A, Gazzara MR, Gonzalez-Vallinas J, Lahens NF, Hogenesch JB, … Barash Y (2016). A new view of transcriptome complexity and regulation through the lens of local splicing variations. Elife, 5, e11752. doi: 10.7554/eLife.11752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahl MC, Will CL, & Luhrmann R (2009). The spliceosome: design principles of a dynamic RNP machine. Cell, 136(4), 701–718. doi: 10.1016/j.cell.2009.02.009 [DOI] [PubMed] [Google Scholar]
- Wang C, Chua K, Seghezzi W, Lees E, Gozani O, & Reed R (1998). Phosphorylation of spliceosomal protein SAP 155 coupled with splicing catalysis. Genes Dev, 12(10), 1409–1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang ET, Sandberg R, Luo S, Khrebtukova I, Zhang L, Mayr C, … Burge CB (2008). Alternative isoform regulation in human tissue transcriptomes. Nature, 456(7221), 470–476. doi: 10.1038/nature07509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Fernald AA, Anastasi J, Le Beau MM, & Qian Z (2010). Haploinsufficiency of Apc leads to ineffective hematopoiesis. Blood, 115(17), 3481–3488. doi: 10.1182/blood-2009-11-251835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Leung JW, Gong Z, Feng L, Shi X, & Chen J (2013). PHF6 regulates cell cycle progression by suppressing ribosomal RNA synthesis. J Biol Chem, 288(5), 3174–3183. doi: 10.1074/jbc.M112.414839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Venkatraman V, Crowgey EL, Liu T, Fu Z, Holewinski RJ, … Van Eyk JE (2018). Protein S-Nitrosylation Controls Glycogen Synthase Kinase 3beta Function Independent of its Phosphorylation State. Circ Res. doi: 10.1161/CIRCRESAHA.118.312789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Krivtsov AV, Sinha AU, North TE, Goessling W, Feng Z, … Armstrong SA (2010). The Wnt/beta-catenin pathway is required for the development of leukemia stem cells in AML. Science, 327(5973), 1650–1653. doi: 10.1126/science.1186624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- White ES, Sagana RL, Booth AJ, Yan M, Cornett AM, Bloomheart CA, … Muro AF (2010). Control of fibroblast fibronectin expression and alternative splicing via the PI3K/Akt/mTOR pathway. Exp Cell Res, 316(16), 2644–2653. doi: 10.1016/j.yexcr.2010.06.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson GM, Lu J, Sutphen K, Suarez Y, Sinha S, Brewer B, … Brewer G (2003). Phosphorylation of p40AUF1 regulates binding to A + U-rich mRNA-destabilizing elements and protein-induced changes in ribonucleoprotein structure. J Biol Chem, 278(35), 33039–33048. doi: 10.1074/jbc.M305775200 [DOI] [PubMed] [Google Scholar]
- Wong ACH, Rasko JEJ, & Wong JJ (2018). We skip to work: alternative splicing in normal and malignant myelopoiesis. Leukemia, 32(5), 1081–1093. doi: 10.1038/s41375-018-0021-4 [DOI] [PubMed] [Google Scholar]
- Wong JJ, Ritchie W, Ebner OA, Selbach M, Wong JW, Huang Y, … Rasko JE (2013). Orchestrated intron retention regulates normal granulocyte differentiation. Cell, 154(3), 583–595. doi: 10.1016/j.cell.2013.06.052 [DOI] [PubMed] [Google Scholar]
- Xiao SH, & Manley JL (1997). Phosphorylation of the ASF/SF2 RS domain affects both protein-protein and protein-RNA interactions and is necessary for splicing. Genes Dev, 11(3), 334–344. [DOI] [PubMed] [Google Scholar]
- Xiao SH, & Manley JL (1998). Phosphorylation-dephosphorylation differentially affects activities of splicing factor ASF/SF2. EMBO J, 17(21), 6359–6367. doi: 10.1093/emboj/17.21.6359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu C, Kim NG, & Gumbiner BM (2009). Regulation of protein stability by GSK3 mediated phosphorylation. Cell Cycle, 8(24), 4032–4039. doi: 10.4161/cc.8.24.10111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Q, Simpson SE, Scialla TJ, Bagg A, & Carroll M (2003). Survival of acute myeloid leukemia cells requires PI3 kinase activation. Blood, 102(3), 972–980. doi: 10.1182/blood-2002-11-3429 [DOI] [PubMed] [Google Scholar]
- Yeung J, Esposito MT, Gandillet A, Zeisig BB, Griessinger E, Bonnet D, & So CW (2010). beta-Catenin mediates the establishment and drug resistance of MLL leukemic stem cells. Cancer Cell, 18(6), 606–618. doi: 10.1016/j.ccr.2010.10.032 [DOI] [PubMed] [Google Scholar]
- Yilmaz OH, Valdez R, Theisen BK, Guo W, Ferguson DO, Wu H, & Morrison SJ (2006). Pten dependence distinguishes haematopoietic stem cells from leukaemia-initiating cells. Nature, 441(7092), 475–482. [DOI] [PubMed] [Google Scholar]
- Yin L, Wang J, Klein PS, & Lazar MA (2006). Nuclear receptor Rev-erbalpha is a critical lithium-sensitive component of the circadian clock. Science, 311(5763), 1002–1005. [DOI] [PubMed] [Google Scholar]
- Yoshida K, Sanada M, Shiraishi Y, Nowak D, Nagata Y, Yamamoto R, … Ogawa S (2011). Frequent pathway mutations of splicing machinery in myelodysplasia. Nature, 478(7367), 64–69. doi: 10.1038/nature10496 [DOI] [PubMed] [Google Scholar]
- Ysebaert L, Chicanne G, Demur C, De Toni F, Prade-Houdellier N, Ruidavets JB, … Racaud-Sultan C (2006). Expression of beta-catenin by acute myeloid leukemia cells predicts enhanced clonogenic capacities and poor prognosis. Leukemia, 20(7), 1211–1216. doi: 10.1038/sj.leu.2404239 [DOI] [PubMed] [Google Scholar]
- Zhang J, Grindley JC, Yin T, Jayasinghe S, He XC, Ross JT, … Li L (2006). PTEN maintains haematopoietic stem cells and acts in lineage choice and leukaemia prevention. Nature, 441(7092), 518–522. [DOI] [PubMed] [Google Scholar]
- Zhang W, DePaoli-Roach AA, & Roach PJ (1993). Mechanisms of multisite phosphorylation and inactivation of rabbit muscle glycogen synthase. Arch Biochem Biophys, 304(1), 219–225. [DOI] [PubMed] [Google Scholar]
- Zhou Z, & Fu XD (2013). Regulation of splicing by SR proteins and SR protein-specific kinases. Chromosoma, 122(3), 191–207. doi: 10.1007/s00412-013-0407-z [DOI] [PMC free article] [PubMed] [Google Scholar]