

Graphical abstract

Keywords: S1PR1, Soft-drug, Plasma stability, Psoriasis, Topical
Highlights
-
•
Identification of soft drug S1PR agonists.
-
•
Development of a process for the evaluation of topical esterase soft drugs.
-
•
Study skin and plasma esterase SAR.
Abstract
The oral S1PR1 agonist ponesimod demonstrated substantial efficacy in a phase II clinical trial of psoriasis. Unfortunately, systemic side effects were observed, which included lymphopenia and transient bradycardia. We sought to develop a topical soft-drug S1PR1 agonist with an improved therapeutic index. By modifying ponesimod, we discovered an ester series of S1PR agonists. To increase metabolic instability in plasma we synthesised esters described as specific substrates for paraoxonase and butyrylcholinesterases, esterases present in human plasma.
Introduction
Psoriasis is a common chronic inflammatory skin disease that affects 2% of the population.1 52.3% of patients were dissatisfied with current treatments in a recent survey from the National Psoriasis Foundation in the US.1 Recently approved biological drugs targeting disease relevant receptors, such as secukinumab2 or ixekizumab3 for interleukin (IL)-17 and ustekinumab for IL-12/234 have brought great benefit to patients with severe symptoms of the disease. However, a need remains for safe, convenient, efficacious therapies for mild and moderate psoriasis.
Sphingosine-1-phosphate receptor (S1PR) agonists are of interest to the pharmaceutical industry, due to their potential to treat diseases of the immune system such as psoriasis and multiple sclerosis as well as cancer.5, 6 S1PR agonists, such as fingolimod and ponesimod (Fig. 1), initially activate sphingosine-1-phosphate receptors, but subsequently trigger receptor internalisation. This shuts down the sphingosine 1-phosphate signalling pathway, which then prevents the maturation and migration of lymphocytes.7 In 2010 fingolimod was approved for the treatment of relapsing/remitting multiple sclerosis and is the only S1PR1 agonist approved to date.8
Fig. 1.

Selected S1PR1 modulators.
In a phase 2 study in plaque psoriasis, a 40 mg oral dose of ponesimod, led to a 75% reduction in psoriasis area and severity index (PASI) score in 77% of the patients.9 This level of efficacy is competitive with other small molecules in development to treat psoriasis (apremilast10, tofacitinib11 and sotrastaurin12). Ponesimod was not however progressed into phase 3, possibly due to safety concerns. Oral use of pan-S1PR and S1PR1 selective modulators is associated with several severe side effects such as lymphopenia, bradycardia and dyspnoea, which limits their utility as therapies for non-life threatening chronic diseases.5
In mice, topical application of the S1PR agonist fingolimod led to a reduction in the number of Langerhans cells migrating to the lymph node.13 Work by Griffiths and co-workers has shown that Langerhans cell migration is higher in involved areas of patient’s skin than in non-involved areas of skin.14, 15 We hypothesised that local inhibition of Langerhans cell migration, through the effects of topical S1PR agonists, could restore a normal skin phenotype to patients with psoriasis.
Due to the potential of S1PR agonists to act as efficacious therapies, for the treatment of psoriasis, we decided to embark on developing a topical S1PR1 modulator using a fast-follower approach, inspired by ponesimod. Topical therapies apply drugs directly to the site of action and can lead to a reduction in the systemic exposure of a drug when compared to oral dosing. However, due to the chronic nature of psoriasis and the need to be able to treat large body surface areas (>10%), there is a danger that topical application could lead to biologically relevant systemic drug concentrations. To maximise the therapeutic index we decided to develop a soft drug.16, 17 Soft drugs are locally active, in this case in the skin, but then undergo rapid systemic metabolism, to metabolites, which are either inactive or rapidly cleared from the systemic circulation.18
Esters can be metabolised in liver by esterases and cytochrome P450 enzymes.19 However, most tissues are capable of metabolising an ester. Importantly, human blood contains a wide range of esterases and the rate of ester hydrolysis in blood can lead to ultra-rapid clearance,20 what we term a super-soft drug. Because S1PR agonists induce transient bradycardia, and as blood returning to circulation from skin passes through the heart before reaching the liver, we felt that a very rapid blood clearance mechanism could potentially discharge the cardiotoxicity risk. To select compounds which would be likely to demonstrate the desired pharmacokinetic profile, we targeted a human plasma stability half-life of <5 min. We chose to first develop benzoate ester analogues of ponesimod to determine if they could demonstrate potential as super-soft drugs. The desired compound would have S1PR pXC50 of >7 and a logP in the range of 2–4 which is believed to be optimal for skin penetration.21
Compounds 4a and 5a–d were synthesised as shown in Scheme 1. The (Z)-2-(propylimino)-3-(o-tolyl)thiazolidin-4-one core 2 was synthesised utilising a one-pot, two step reaction. Only the Z isomer was observed. N-propylamine was reacted with 1-isothiocyanato-2-methylbenzene 1 to give the resulting thiourea, which was condensed with 2-bromoacetyl bromide followed by addition of pyridine to furnish the desired thiazolidin-4-one 2. This core was then condensed with the appropriately substituted benzaldehyde 3a–b to furnish 4a and 5a. Finally, esterification or amidation of 4a gave the desired products. Based on the configurational analysis by X-ray, which characterised crystalline analogues of ponesimod as Z,Z22, all structurally related compounds 4a, 5a–d, 11, 12a–f and 15a, b were assigned to the Z,Z-isomer.
Scheme 1.

Reagents and conditions for preparation of compounds 4a and 5a–d: (a) n-PrNH2 (1 eq), CH2Cl2, rt (b) 2-bromoacetyl bromide (1 eq) and pyridine (2 eq), CH2Cl2, 0 °C to rt (two steps) (c) aldehyde (1 eq), NaOAc (2 eq), AcOH, 65 °C (d) alcohol (5 eq), HOAt (1.5 eq), EDC (1.5 eq), DIPEA (2.5 eq), CH2Cl2, rt. eq: equivalent; rt: room temperature; HOAt: 1-Hydroxy-7-azabenzotriazole; EDC: 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide; DIPEA: N,N-Diisopropylethylamine.
We selected human plasma stability as a model for human blood metabolism. Esters 5a–c and amide 5d demonstrated half-lives of >180 min in human plasma, far greater than the <5 min we were aiming for. S1PR1 activity was measured using a PathHunter β-Arrestin recruitment assay.23 All of the ester and amide compounds in Table 1 were active and had pIC50 values ranging from 6.5 to 7.0, with the exception of compound 5a where a chlorine ortho to the ester provided 100-fold improvement in activity compared to 5b. Gratifyingly, the parent carboxylic acid 4a, which is the desired metabolite of our esters/amides was less active, giving a pIC50 of <6 and meeting our targeted criteria for producing inactive metabolites.
Table 1.
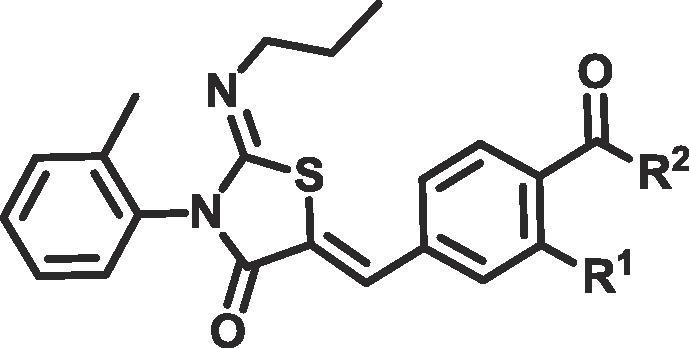
| Compound | R1 | R2 | Kinetic solubility (µM)a | CHI logDb | S1PR1 pIC50c | H Plasma Stability (half-life min)d |
|---|---|---|---|---|---|---|
| Ponesimod | – | – | 110 | 3.1 | 7.9 (0) | >180 |
| 5a | Cl | OMe | 39 | >4.4 | 8.6 (0.1) | >180 |
| 5b | H | OMe | 28 | >4.4 | 6.5 (0) | >180 |
| 4a | H | OH | 163 | 1.0 | <6 (0) | – |
| 5c | H | OCH2CH2OH | 110 | 3.4 | 6.9 (0) | >180 |
| 5d | H | NHCH2CH2OH | >250 | 2.3 | 7.0 (0) | >180 |
The aqueous solubility of the test compounds was measured using laser nephelometry.
Reverse-phase HPLC method to determine the chromatographic hydrophobicity index (CHI).
All pIC50s reported in this table correspond to n of 2, reported as their geometric mean. The range of pIC50 values is provided in brackets.
Incubated in human plasma at 37 °C.
Aqueous solubility is an important parameter for topically applied drugs. Increasing aqueous solubility, should directly increase skin penetration rates.24 Increasing aqueous solubility also increases the range of formulation options that can be used in pre-clinical development. We were pleased to see that introduction of a hydrogen bond donor at R2 in 5c led to a 4-fold increase in solubility relative to 5b. Changing the ester linker in 5c to an amide in 5d further improved solubility to >250 µM, probably due to improvements in lipophilicity (3.4 vs 2.3 respectively).
Encouraged by the solubility and activity data, we focused our efforts on optimising the metabolic profile of the esters. At this time, we identified that switching from the o-tolyl group used in ponesimod to an o-phenol 6 increased aqueous solubility 2-fold, presumably due to the reduction in lipophilicity (Fig. 2). As 6 was equipotent to ponesimod (pIC50 7.9) and to maximise compound solubility, subsequent compounds incorporated the o-phenol group.
Fig. 2.

Improvements in aqueous kinetic solubility from introduction of o-phenol group.
Compounds 11, 12a–c, 15a and 15b were synthesised using the route shown in Scheme 2. 12a–c were more soluble than ponesimod, probably due to significant improvements in CHI logD (Table 2). As expected a clear trend can be seen between low lipophilicity and improved solubility. Homologation of the ester group (15a, b) showed increased lipophilicity (3.2 and 3.5 respectively) and a commensurate reduction in solubility. However, as with 5a–d, amide 12a and esters 12b–c also showed a lack of metabolism in our human plasma stability assay (Table 2).
Scheme 2.

Reagents and conditions for preparation of compounds 11, 12a–f and 15a, b: (a) n-PrNH2 (1 eq), CH2Cl2, rt (b) 2-bromoacetyl bromide (1 eq) and pyridine (2 eq), CH2Cl2, 0 °C to rt (two steps) (c) aldehyde (1 eq), NaOAc (2 eq), AcOH, 85 °C (d) BBr3 (8 eq), CH2Cl2, −78 °C to rt (e) For 12a–c. amine (3 eq) or alcohol (5 eq), HOAt (1.5 eq), EDC (1.5 eq), DIPEA (2.5 eq), CH2Cl2, rt (f) For 12d–f. alcohol, alkyl chloride or alkyl tosylate (1.1–1.5 eq) K2CO3 (1.5 eq), NaI (0.1 eq), DMF, 70 °C (g) NEt3 (3 eq), EDC (1.1 eq), HOAt (1.1 eq), alcohol (1.5 eq) rt (h) BBr3 (5 eq), CH2Cl2, −78 °C to rt. eq: equivalent; rt: room temperature; HOAt: 1-Hydroxy-7-azabenzotriazole; EDC: 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide; DIPEA: N,N-Diisopropylethylamine; DMF: dimethyl formamide.
Table 2.
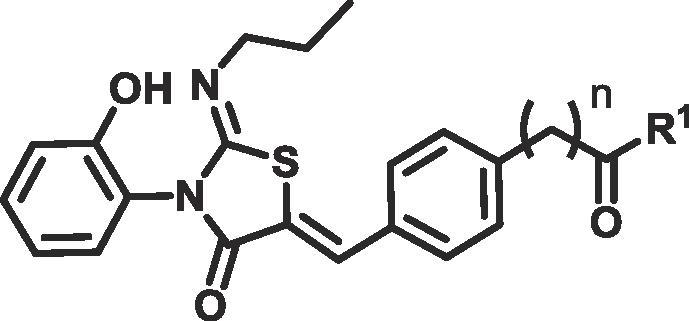
| Compound | n | R1 | Kinetic solubility (µM)a | CHI logDb | H Plasma Stability (half-life min)c |
|---|---|---|---|---|---|
| Ponesimod | – | – | 110 | 3.1 | >180 |
| 12a | 0 | NHCH2CONMe2 | 219 | 1.9 | >180 |
| 12b | 0 | OCH2CH2CH2OH | 183 | 2.3 | >180 |
| 12c | 0 | OCH2CH(±CH2OH)OH | >250 | 1.8 | >180 |
| 15a | 1 | OMe | 155 | 3.2 | >180 |
| 15b | 2 | OMe | 79 | 3.5 | >180 |
The aqueous solubility of the test compounds was measured using laser nephelometry.
Reverse-phase HPLC method to determine the chromatographic hydrophobicity index (CHI).
Incubated in human plasma at 37 °C.
As simple esters had shown themselves to be stable to human plasma, we turned our attention to ester functional groups which had been shown to be unstable in human plasma in the literature (half-life < 5min).20 The most successful studies focused on optimising metabolism by the esterases paraoxonase25 and butyrylcholinesterases26, which are mainly expressed in blood plasma.
We synthesised compounds 11 and 12d–f using the route shown in Scheme 2, which all contain ester groups that are unstable in human plasma when attached to other scaffolds. Although 12d or 12e did not show sufficient clearance in human plasma we were pleased to identify compound 12f containing a (2-oxo-1,3-dioxolan-4-yl)methyl ester which is rapidly degraded in human plasma (Table 3). The half-life of this ester in human plasma was 9 min and metabolite identification confirmed the conversion to the inactive parent acid 11 (Fig. 3).
Table 3.
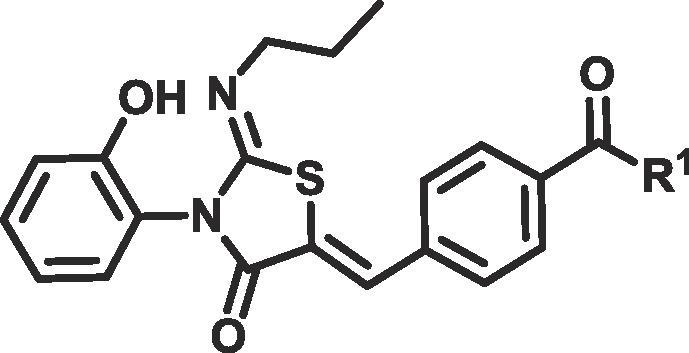
| Compound | R1 | H Plasma Stability (half-life min)a | Kinetic solubility (µM)b | CHI logDc | H Skin S9 (half-life min)d | S1PR1 pIC50e |
|---|---|---|---|---|---|---|
| 11 | OH | >180 | – | – | – | <6 (0) |
| 12d | 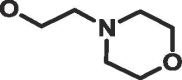 |
180 | 117 | 3.0 | 152 | – |
| 12e | 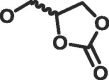 |
61 | 79 | 2.8 | 40 | – |
| 12f | 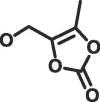 |
9 | 79 | 3.3 | 23 | <6 (0) |
Incubated in human plasma at 37 °C.
The aqueous solubility of the test compounds was measured using laser nephelometry.
Reverse-phase HPLC method to determine the chromatographic hydrophobicity index (CHI).
Stability measured in skin S9 over 180 mins in the presence of enzymatic cofactors.
All pIC50s reported in this table correspond to n of 2, reported as their geometric mean. The range of pIC50 values is provided in brackets.
Fig. 3.

Profile of compound 12f being hydrolysed to 11 in human plasma. 12f was incubated in human plasma at 37 °C. The experiment was conducted as an n of one to provide a qualitative analysis.
Unfortunately 12f was also rapidly metabolised in human skin S9 fraction with a half-life of just 23 min, far faster than the desirable half-life of >6 h that we target for once daily dosing. In fact, there are suggestions of a correlation between the relative stability of 12d–f in human plasma and skin S9 fraction. It may be that the esterases responsible for the metabolism of 12f are present in both human plasma and skin S9 fraction or that skin and plasma esterases have similar substrate affinities. Unfortunately, 12f was also inactive as a S1PR1 modulator (Table 3).
In summary, using a fast follower approach, we identified a series of ester S1PR1 modulators. Further optimisation led to improved solubility and identification of ester 12d that is plasma unstable. Unfortunately, the ester is inactive and is metabolised in human skin models. Further efforts will focus on identifying an active, skin stable compound that is rapidly metabolised systemically will be disclosed in due course.
Acknowledgement
The Centre for Dermatology and Genetic Medicine is supported by a Wellcome Trust Strategic Award (reference 098439/Z/12/Z).
Contributor Information
Mark Bell, Email: m.u.bell@dundee.ac.uk.
Andrew Woodland, Email: awoodland@dundee.ac.uk.
References
- 1.Parisi R., Symmons D.P., Griffiths C.E., Ashcroft D.M. J Invest Dermatol. 2013;133:377–385. doi: 10.1038/jid.2012.339. [DOI] [PubMed] [Google Scholar]
- 2.Langley R.G., Elewski B.E., Lebwohl M. N Engl J Med. 2014;371:326–338. doi: 10.1056/NEJMoa1314258. [DOI] [PubMed] [Google Scholar]
- 3.Gordon K.B., Blauvelt A., Papp K.A. Engl J Med. 2016;375:345–356. doi: 10.1056/NEJMoa1512711. [DOI] [PubMed] [Google Scholar]
- 4.Kimball A.B., Gordon K.B., Fakharzadeh S. Br J Dermatol. 2012;166:861–872. doi: 10.1111/j.1365-2133.2012.10901.x. [DOI] [PubMed] [Google Scholar]
- 5.Kunkel G.T., Maceyka M., Milstien S., Spiegel S. Nat Rev Drug Discov. 2013;12:688–702. doi: 10.1038/nrd4099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Huang W.C., Nagahashi M., Terracina K.P., Takabe K. Biomolecules. 2013;3 doi: 10.3390/biom3030408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Blaho V.A., Hla T. J Lipid Res. 2014;55:1596–1608. doi: 10.1194/jlr.R046300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kappos L., Radue E.W., O’Connor P. N Engl J Med. 2010;362:387–401. doi: 10.1056/NEJMoa0909494. [DOI] [PubMed] [Google Scholar]
- 9.Vaclavkova A., Chimenti S., Arenberger P. Lancet. 2014;384:2036–2045. doi: 10.1016/S0140-6736(14)60803-5. [DOI] [PubMed] [Google Scholar]
- 10.Papp K., Kristian Reich K., Leonardi C.L. J Am Acad Dermatol. 2015;73:37–49. doi: 10.1016/j.jaad.2015.03.049. [DOI] [PubMed] [Google Scholar]
- 11.Ports W.C., Khan S., Lan S. Br J Dermatol. 2013;169:137–145. doi: 10.1111/bjd.12266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Skvara H., Dawid M., Kleyn E. J Clin Invest. 2008;118:3151–3159. doi: 10.1172/JCI35636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Reines I., Kietzmann M., Mischke R. J Invest Dermatol. 2009;129:1954–1962. doi: 10.1038/jid.2008.454. [DOI] [PubMed] [Google Scholar]
- 14.Griffiths C.E., Dearman R.J., Cumberbatch M., Kimber I. Cytokine. 2005;32:67–70. doi: 10.1016/j.cyto.2005.07.011. [DOI] [PubMed] [Google Scholar]
- 15.Shaw F.L., Mellody K.T., Ogden S., Dearman R.J., Kimber I., Griffiths C.E. J Invest Dermatol. 2014;134:268–271. doi: 10.1038/jid.2013.289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bhardwaj Y.R., Pareek A., Jain V., Kishore D. Saudi Pharm J. 2014;22:290–302. doi: 10.1016/j.jsps.2013.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bodor N., Buchwald P. Pure Appl Chem. 2008;80 [Google Scholar]
- 18.Felding J., Sorensen M.D., Poulsen T.D. J Med Chem. 2014;57:5893–5903. doi: 10.1021/jm500378a. [DOI] [PubMed] [Google Scholar]
- 19.Roberts J.K., Moore C.D., Ward R.M., Yost G.S., Reilly C.A. J Pharmacol Exp Ther. 2013;345:308–316. doi: 10.1124/jpet.112.202556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Beaumont K., Webster R., Gardner I., Dack K. Curr Drug Metab. 2003;4:461–485. doi: 10.2174/1389200033489253. [DOI] [PubMed] [Google Scholar]
- 21.Singh P., Roberts M.S. J Pharmacol Exp Ther. 1994;268:144–151. Skin permeability and local tissue concentrations of nonsteroidal anti-inflammatory drugs after topical application. [PubMed] [Google Scholar]
- 22.Bolli M.H., Abele S., Binkert C. J Med Chem. 2010;53:4198–4211. doi: 10.1021/jm100181s. [DOI] [PubMed] [Google Scholar]
- 23.The PathHunter® CHO‐K1 EDG1 β‐Arrestin Cell Line (Discoverx) was used to assess agonist activity of the compounds. Briefly, compound at 5× desired concentration was added to cells and incubated at 37 °C for 90 min. Assay signal was generated through a single addition of PathHunter Detection reagent cocktail, followed by a one hour incubation at room temperature. Microplates were read following signal generation with a PerkinElmer Envision instrument for chemiluminescent signal detection. Percentage activity was calculated using the following formula: % Activity = 100% × (mean RLU of test sample − mean RLU of vehicle control)/(mean MAX control ligand − mean RLU of vehicle control).
- 24.Mitragotri S., Anissimov Y.G., Bunge A.L. Int J Pharm. 2011;418:115–129. doi: 10.1016/j.ijpharm.2011.02.023. [DOI] [PubMed] [Google Scholar]
- 25.Biggadike K., Angell R.M., Burgess C.M. J Med Chem. 2000;43:19–21. doi: 10.1021/jm990436t. [DOI] [PubMed] [Google Scholar]
- 26.Mørk N., Bundgaard H. Pharm Res. 1992;9:492–496. doi: 10.1023/a:1015888212948. [DOI] [PubMed] [Google Scholar]