

Abstract
To date the only Neandertal genome that has been sequenced to high quality is from an individual found in Southern Siberia. We sequenced the genome of a female Neandertal from ~50 thousand years ago from Vindija Cave, Croatia to ~30-fold genomic coverage. She carried 1.6 differences per ten thousand base pairs between the two copies of her genome, fewer than present-day humans, suggesting that Neandertal populations were of small size. Our analyses indicate that she was more closely related to the Neandertals that mixed with the ancestors of present-day humans living outside of sub-Saharan Africa than the previously sequenced Neandertal from Siberia, allowing 10-20% more Neandertal DNA to be identified in present-day humans, including variants involved in LDL cholesterol levels, schizophrenia and other diseases.
One Sentence Summary:
A high-quality Neandertal genome from Croatia is substantially closer to the Neandertals that contributed DNA to present-day humans.
Main Text:
Neandertals are the closest evolutionary relatives identified to date of all present-day humans and therefore provide a unique perspective on human biology and history. In particular, comparisons of genome sequences from Neandertals with those of present-day humans have allowed genetic features unique to modern humans to be identified (1, 2) and have shown that Neandertals mixed with the ancestors of present-day people living outside sub-Saharan Africa (3). Many of the DNA sequences acquired by non-Africans from Neandertals were likely detrimental and were purged from the human genome via negative selection (4–8) but some appear to have been beneficial and were positively selected (9); among people today, alleles derived from Neandertals are associated with both susceptibility and resistance to diseases (7, 10–12)
However, our knowledge about the genetic variation among Neandertals is still limited. To date genome-wide DNA sequences of five Neandertals have been determined. One of these, the “Altai Neandertal”, found in Denisova Cave in the Altai Mountains in southern Siberia, the eastern-most known reach of the Neandertal range, yielded a high quality genome sequence (~50-fold genomic coverage) (2). In addition, a composite genome sequence from three Neandertal individuals has been generated from Vindija Cave in Croatia in southern Europe but is of low quality (~1.2-fold total coverage) (3), while a Neandertal genome from Mezmaiskaya Cave in the Caucasus (2) is of even lower quality (~0.5-fold coverage). In addition, chromosome 21 (13) and exome sequences (14) have been generated from a different individual from Vindija Cave and one from Sidron Cave in Spain. The lack of high-quality Neandertal genome sequences, especially from the center of their geographical range and from the time close to when they were estimated to have mixed with modern humans, limits our ability to reconstruct their history and the extent of their genetic contribution to present-day humans.
Neandertals lived in Vindija Cave in Croatia until relatively late in their history (3, 15). The cave has yielded Neandertal and animal bones, many of them too fragmentary to determine from their morphology from what species they derive. Importantly, DNA preservation in Vindija Cave is relatively good and allowed the determination of Pleistocene nuclear DNA from a cave bear (16), a Neandertal genome (3), exome and chromosome 21 sequences (13, 14).
To generate DNA suitable for deep sequencing, we extracted DNA (17) and generated DNA libraries (18) from 12 samples from Vindija 33.19, one of 19 bone fragments from Vindija Cave determined to be of Neandertal origin by mitochondrial (mt) DNA analyses (19). In addition, 567 milligram (mg) were removed for radiocarbon dating and yielded a date of greater than 45.5 thousand years before present (OxA 32,278). One of the DNA extracts, generated from 41 mg of bone material, contained more hominin DNA than the other extracts. We created additional libraries from this extract, but to maximize the number of molecules retrieved from the specimen we omitted the uracil-DNA-glycosylase (UDG) treatment (20, 21). A total of 24 billion DNA fragments were sequenced and approximately 10% of these could be mapped to the human genome. Their average length was 53 base pairs (bp) and they yielded 30-fold coverage of the approximately 1.8 billion bases of the genome to which such short fragments can be confidently mapped.
We estimated present-day human DNA contamination among the DNA fragments (20). First, using positions in the mtDNA where present-day humans differ from Neandertals we estimated an mtDNA contamination rate of 1.4-1.7%. Similarly, using positions in the autosomal genome where all present-day humans carry derived variants whereas all archaic genomes studied to date carry ancestral variants we estimated a nuclear contamination rate of 0.17-0.48%. Because the coverage of the X chromosome is similar to that of the autosomes we inferred that the Vindija 33.19 individual is a female, allowing us to use DNA fragments that map to the Y chromosome to estimate a male DNA contamination of 0.74% (between 0.70-0.78% for each of the nine sequencing libraries). Finally, using a likelihood method (2, 3) we estimated the autosomal contamination to 0.18-0.23%. We conclude that the nuclear DNA contamination rate among the DNA fragments sequenced is less than 1%. After genotyping this will result in contamination that is much lower than 1%.
Because ~76% of the DNA fragments were not UDG-treated, they carry C to T substitutions throughout their lengths. This causes standard genotyping software to generate false heterozygous calls. To overcome this we implemented snpAD, a genotyping software that incorporates a position-dependent error-profile to estimate the most likely genotype for each position in the genome. This results in genotypes of comparable quality to UDG-treated ancient DNA given our genomic coverage (20). The high-coverage of the Vindija genome also allowed for characterization of longer structural variants and segmental duplications (20).
To gauge whether the Vindija 33.19 bone might stem from a previously sequenced individual from Vindija Cave we compared heterozygous sites in the Vindija 33.19 genome to DNA fragments sequenced from the other bones. The three bones from which a low-coverage composite genome has been generated (Vindija 33.16, 33.25 and 33.26) do not share variants with Vindija 33.19 at a level compatible with deriving from the same individual. In contrast, over 99% of heterozygous sites in the chromosome 21 sequence from Vindija 33.15 (13) are shared with Vindija 33.19, indicating that they come from the same individual (20). Additionally, two of the other three bones may come from individuals that shared a maternal ancestor to Vindija 33.19 relatively recently in their family history because all carry identical mtDNAs.
In addition to the Altai Neandertal genome, a genome from a Denisovan, an Asian relative of Neandertals, has been sequenced to high coverage (~30-fold) from Denisova Cave. These two genomes are similar in that their heterozygosity is about one fifth of that of present-day Africans and about one third of that of present-day Eurasians. We estimated the heterozygosity of the Vindija 33.19 autosomal genome to 1.6×10−5; similar to the Altai Neandertal genome and slightly lower than the Denisovan genome (1.8 ×10−5) (Fig. 1A). Thus, low heterozygosity may be a feature typical of archaic hominins, suggesting that they lived in small and isolated populations with an effective population size of around 3,000 individuals (20). In addition to low over-all heterozygosity, the Altai Neandertal genome carried segments of many megabases (Mb) (>10 centimorgans (cM)) without any differences between its two chromosomes, indicating that the parents of that individual were related at the level of half-sibs (2). Such segments are almost totally absent in the Vindija genome (Fig. 1B), suggesting that the extreme inbreeding between the parents of the Altai Neandertal was not ubiquitous among Neandertals. We note, however, that the Vindija genome carries extended homozygous segments (>2.5cM) comparable to what is seen in some isolated Native American populations today (20).
Fig. 1. Heterozygosity and inbreeding in the Vindija Neandertal.
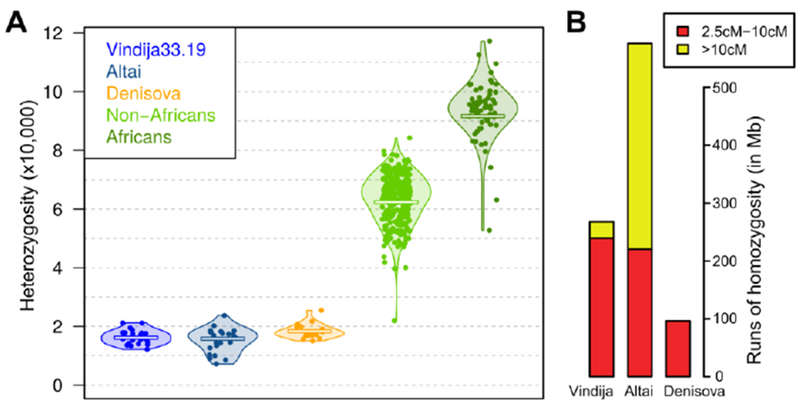
(A) Distribution of heterozygosity over all autosomes in the three archaic hominins, 12 Non-Africans and 3 Africans. Each dot represents the heterozygosity measured for one autosome. The center bar indicates the mean heterozygosity across the autosomal genome. (B) Genome covered by shorter (2.5–10cM, red) and longer (>10cM, yellow) runs of homozygosity in the three archaic hominins.
The high quality of the three archaic genome sequences allows their approximate ages to be estimated from the number of new nucleotide substitutions they carry relative to present-day humans when compared to the inferred ancestor shared with apes (1). Using this approach, we estimate that the Vindija 33.19 individual lived 52 thousand years ago (kya), the Altai Neandertal individual 122kya, and the Denisovan individual 72kya (Fig. 2) (20). Many factors make such absolute age estimates tentative. Among these are uncertainty in generation times and mutation rates. Nevertheless, these results indicate that the Altai Neandertal lived about twice as far back in time as the Vindija 33.19 Neandertal, while the Denisovan individual lived after the Altai but before the Vindija Neandertal.
Fig. 2. Approximate ages of specimens and population split times.
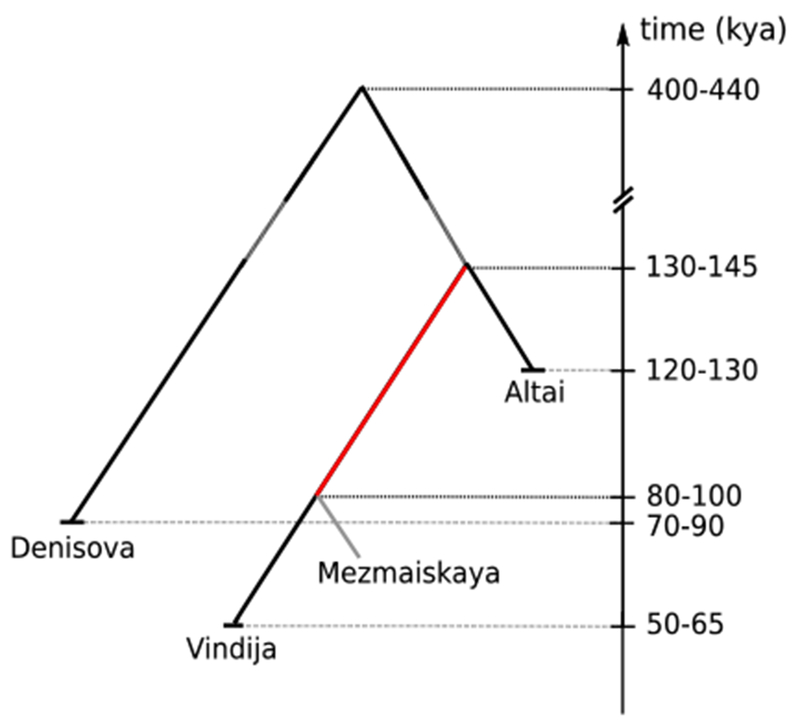
Age estimates for the genomes estimated from branch shortening, i.e. the absence of mutations in the archaic genomes, are indicated by dashed lines. Population split time estimates are indicated by dashed lines. The majority of Neandertal DNA in present-day people comes from a population that split from the branch indicated in red. All reported ages assume a human-chimpanzee divergence of 13 million years. Numbers show ranges over point estimates (split times), or ranges over different data filters (branch shortening).
We next estimated when ancestral populations that gave rise to the three archaic genomes and to modern humans split from each other based on the extent to which they share genetic variants (1–3, 20). The estimated population split time between the Vindija Neandertal and the Denisovan is 390-440kya and between the Vindija Neandertal and modern humans 520-630kya, in agreement with previous estimates using the Altai Neandertal (2). The split time between the Vindija and the Altai Neandertals is estimated to 130-145kya. To estimate the population split time to the Mezmaiskaya 1 Neandertal previously sequenced to 0.5-fold coverage, we prepared and sequenced libraries yielding an additional 1.4-fold coverage. Because the present-day human DNA contamination of these libraries is in the order of 2-3% (20), we estimated the population split time to the Vindija 33.19 individual with and without restricting the analysis of the Mezmaiskaya 1 individual to fragments that show evidence of deamination. The resulting split time estimates are 100kya for the deaminated fragments and 80kya for all fragments (Fig. 2).
It has been suggested that Denisovans received gene flow from a human lineage that diverged prior to the common ancestor of modern humans, Neandertals and Denisovans (2). In addition, it has been suggested that the ancestors of the Altai Neandertal received gene flow from early modern humans that may not have affected the ancestors of European Neandertals (13). In agreement with these studies, we find that the Denisovan genome carries fewer derived alleles that are fixed in Africans, and thus tend to be older, than the Altai Neandertal genome while the Altai genome carries more derived alleles that are of lower frequency in Africa, and thus younger, than the Denisovan genome (20). However, the Vindija and Altai genomes do not differ significantly in the sharing of derived alleles with Africans indicating that they may not differ with respect to their putative interactions with early modern humans (Fig. 3A & B). Thus, in contrast to earlier analyses of chromosome 21 data for the European Neandertals (13), analyses of the full genomes suggest that the putative early modern human gene flow into Neandertals occurred prior to the divergence of the populations ancestral to the Vindija and Altai Neandertals ~130-145 thousand years ago (Fig. 2). Coalescent simulations show that a model with only gene flow from a deeply diverged hominin into Denisovan ancestors explains the data better than one with only gene flow from early modern humans into Neandertal ancestors, but that a model involving both gene flows explains the data even better. It is likely that gene flow occurred between many or even most hominin groups in the late Pleistocene and that more such events will be detected as more ancient genomes of high quality become available.
Fig. 3. Allele sharing between archaic and modern humans.
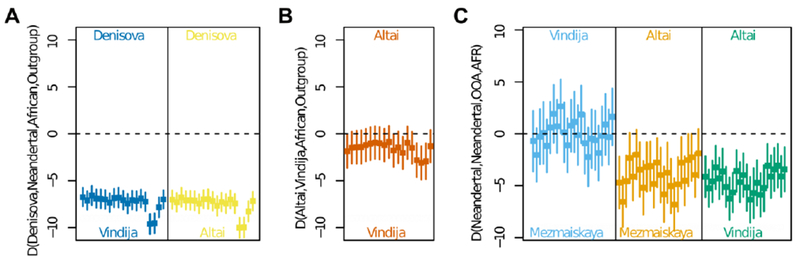
(A) Derived allele-sharing in percent of 19 African populations with the Altai and Denisovan, and Vindija and Denisovan genomes, respectively. (B) Sharing of derived alleles in each of the 19 African populations with the Vindija and Altai genomes. (C) Allele sharing of Neandertals with non-Africans and Africans. Points show derived allele sharing in percent for all pairwise comparisons between non-Africans (OAA: French, Sardinian, Han, Dai, Karitiana, Mixe, Australian, Papuan) and Africans (AFR: San, Mbuti, Yoruba). Mezmaiskaya 1 data were restricted to sequences showing evidence of deamination to reduce the influence of present-day human DNA contamination. Lines show two standard errors from the mean in all plots.
A proportion of the genomes of all present-day people whose roots are outside Africa derives from Neandertals (2, 3, 22). We tested if any of the three sequenced Neandertals falls closer to the lineage that contributed DNA to present-day non-Africans by asking if any of them shares more alleles with present-day non-Africans than the others (20, 23). The Vindija 33.19 and Mezmaiskaya 1 genomes share more alleles with non-Africans than the Altai Neandertal, and there is no indication that the former two genomes differ in the extent of their allele-sharing with present-day people (Fig. 3C). Using a likelihood approach we estimate the proportion of Neandertal DNA in present-day populations that is closer to the Vindija than the Altai genomes to be 99%−100% (20). Thus, the majority of Neandertal DNA in present-day populations appears to come from Neandertal populations that diverged from the Vindija and Mezmaiskaya 1 Neandertals prior to their divergence from each other some 80-100kya.
The two high-coverage Neandertal genomes allow us to estimate the proportion of the genomes of present-day people that derive from Neandertals with greater accuracy than was hitherto possible. We asked how many derived alleles non-Africans share with the Altai Neandertal relative to how many derived alleles the Vindija Neandertal shares with the Altai Neandertal - essentially asking how close non-Africans are to being 100% Neandertal (24). We find that non-African populations outside Oceania carry between 1.8-2.6% Neandertal DNA (Fig. 4A), higher than previous estimates of 1.5-2.1% (2). As described (25), East Asians carry somewhat more Neandertal DNA (2.3-2.6%) than people in Western Eurasia (1.8-2.4%).
Fig. 4. Estimates of fraction of Neandertal DNA for present-day populations.
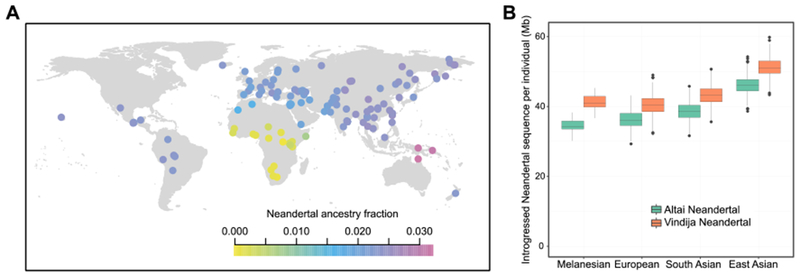
(A) Colors indicate Neandertal ancestry estimates (20). Oceanian populations show high estimates due to Denisovan ancestry that is difficult to distinguish from Neandertal ancestry. (B) Amount of Neandertal sequence in present-day Europeans, South Asians and East Asians (20).
We also identified (8) regions of Neandertal-ancestry in present-day Europeans and Asians using the Vindija and the Altai Neandertal genomes (20). The Vindija genome allows us to identify ~10% more Neandertal DNA sequences per individual than the Altai Neandertal genome (e.g. 40.4 Mb vs 36.3 Mb in Europeans) due to the closer relationship between the Vindija genome and the introgressing Neandertal populations. In Melanesians, the increased power to distinguish between Denisovan and Neandertal DNA sequences results in the identification of 20% more Neandertal DNA (Fig. 4B).
Many Neandertal variants associated with phenotypes and susceptibility to diseases have been identified in present-day non-Africans (6, 7, 10–12). The fact that the Vindija Neandertal genome is more closely related to the introgressing Neandertals allows ~15% more such variants to be identified (20). Among these are variants associated with plasma levels of LDL cholesterol (rs10490626) and vitamin D (rs6730714), eating disorders (rs74566133), visceral fat accumulation (rs2059397), rheumatoid arthritis (45475795), schizophrenia (rs16977195) and the response to antipsychotic drugs (rs1459148). This adds to mounting evidence that Neandertal ancestry influences disease risk in present-day humans, particularly with respect to neurological, psychiatric, immunological, and dermatological phenotypes (7).
Supplementary Material
Acknowledgments:
We thank J. Lenardić, and D. Brajković for expert assistance in the Institute for Quaternary Paleontology and Geology in Zagreb, B. Hoeber and A. Weihmann for DNA sequencing, and U. Stenzel for help with computational analyses. Q.F. is funded in part by National Key R&D Program of China (2016YFE0203700), CAS (XDB13000000, QYZDB-SS W-DQC003, XDPB05), NSFC (91731303,41672021, 41630102) and the Howard Hughes Medical Institute (grant no. 55008731), D.R. was supported by the U.S. National Science Foundation (grant BCS-1032255) and is an investigator of the Howard Hughes Medical Institute, E.E.E. was supported by the U.S. National Institutes of Health (NIH R01HG002385) and is an investigator of the Howard Hughes Medical Institute. This project was funded by the European Research Council (grant agreement no. 694707 to S.P.) and the Max Planck Foundation (grant 31-12LMP Pääbo to S.P.). P.K., M.H., C.H., S.N., T.M., Q.F. performed experiments. K.P., C.dF., S.G., F.M., M.H., B.V., L.S., P.H., St.P., Da.R., C.T., R.R., P.S., M.C., M.D., B.J.N., F.M.K., N.P., D.R., E.E.E., M.S., M.H.S., A.A., J.K., M.M. and S.P. analyzed data. P.R., Ž.K., I.G., L.V.G. and V.B.D. provided samples. K.P. and S.P. wrote and edited the manuscript with input from all authors. Sequencing data for Vindija 33.19 and Mezmaiskaya 1 are available in the European Nucleotide Archive under the study accession numbers PRJEB21157 and PRJEB21195, respectively, and the Vindija genome can be viewed at https://bioinf.eva.mpg.de/jbrowse.
Footnotes
Supplementary Materials
Supplementary text
Supplemental Figures
Supplemental Tables.
References (26-118)
References and Notes:
- 1.Meyer M et al. , A high-coverage genome sequence from an archaic Denisovan individual. Science 338, 222–226 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Prüfer K et al. , The complete genome sequence of a Neanderthal from the Altai Mountains. Nature 505, 43–49 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Green RE et al. , A draft sequence of the Neandertal genome. Science 328, 710–722 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Harris K, Nielsen R, The Genetic Cost of Neanderthal Introgression. Genetics 203, 881–891 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Juric I, Aeschbacher S, Coop G, The Strength of Selection against Neanderthal Introgression. PLoS Genet 12, e1006340 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sankararaman S et al. , The genomic landscape of Neanderthal ancestry in present-day humans. Nature 507, 354–357 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Simonti CN et al. , The phenotypic legacy of admixture between modern humans and Neandertals. Science 351, 737–741 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vernot B, Akey JM, Resurrecting Surviving Neandertal Lineages from Modern Human Genomes. Science 343, 1017–1021 (2014). [DOI] [PubMed] [Google Scholar]
- 9.Racimo F, Sankararaman S, Nielsen R, Huerta-Sanchez E, Evidence for archaic adaptive introgression in humans. Nat Rev Genet 16, 359–371 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sigma Type 2 Diabetes Consortium et al. , Sequence variants in SLC16A11 are a common risk factor for type 2 diabetes in Mexico. Nature 506, 97–101 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dannemann M, Andres AM, Kelso J, Introgression of Neandertal- and Denisovan-like Haplotypes Contributes to Adaptive Variation in Human Toll-like Receptors. Am J Hum Genet 98, 22–33 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Quach H et al. , Genetic Adaptation and Neandertal Admixture Shaped the Immune System of Human Populations. Cell 167, 643–656 e617 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kuhlwilm M et al. , Ancient gene flow from early modern humans into Eastern Neanderthals. Nature 530, 429–433 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Castellano S et al. , Patterns of coding variation in the complete exomes of three Neandertals. Proceedings of the National Academy of Sciences of the United States of America 111, 6666–6671 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Deviese T et al. , Direct dating of Neanderthal remains from the site of Vindija Cave and implications for the Middle to Upper Paleolithic transition. Proceedings of the National Academy of Sciences of the United States of America, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Greenwood AD, Paabo S, Nuclear insertion sequences of mitochondrial DNA predominate in hair but not in blood of elephants. Mol Ecol 8, 133–137 (1999). [DOI] [PubMed] [Google Scholar]
- 17.Dabney J et al. , Complete mitochondrial genome sequence of a Middle Pleistocene cave bear reconstructed from ultrashort DNA fragments. Proceedings of the National Academy of Sciences of the United States of America 110, 15758–15763 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gansauge MT, Meyer M, Single-stranded DNA library preparation for the sequencing of ancient or damaged DNA. Nat Protoc 8, 737–748 (2013). [DOI] [PubMed] [Google Scholar]
- 19.Gansauge MT, Meyer M, Selective enrichment of damaged DNA molecules for ancient genome sequencing. Genome Res 24, 1543–1549 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Materials and methods are available as supplementary materials.
- 21.Briggs AW et al. , Removal of deaminated cytosines and detection of in vivo methylation in ancient DNA. Nucleic Acids Res 38, e87 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mallick S et al. , The Simons Genome Diversity Project: 300 genomes from 142 diverse populations. Nature 538, 201–206 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Patterson N et al. , Ancient Admixture in Human History. Genetics 192, 1065–1093 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Reich D et al. , Denisova Admixture and the First Modern Human Dispersals into Southeast Asia and Oceania. The American Journal of Human Genetics 89, 516–528 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wall JD et al. , Higher Levels of Neanderthal Ancestry in East Asians than in Europeans. Genetics 194, 199–209 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Briggs AW et al. , Primer extension capture: targeted sequence retrieval from heavily degraded DNA sources. J Vis Exp, 1573 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rohland N, Hofreiter M, Comparison and optimization of ancient DNA extraction. Biotechniques 42, 343–352 (2007). [DOI] [PubMed] [Google Scholar]
- 28.Meyer M, Kircher M, Illumina sequencing library preparation for highly multiplexed target capture and sequencing. Cold Spring Harbor protocols 2010, pdb prot5448 (2010). [DOI] [PubMed] [Google Scholar]
- 29.Maricic T, Whitten M, Pääbo S, Multiplexed DNA Sequence Capture of Mitochondrial Genomes Using PCR Products. Plos One 5, (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Green RE et al. , A complete Neandertal mitochondrial genome sequence determined by high-throughput sequencing. Cell 134, 416–426 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Malez M, Ullrich H, Neuere paläanthropologische Untersuchungen am Material aus der Höhle Vindija (Kroatien, Jugoslawien). Palaeontologia Jugoslavica 29, 1–44 (1982). [Google Scholar]
- 32.Golovanova LV, Hoffecker JF, Kharitonov VM, Romanova GP, Mezmaiskaya cave: A Neanderthal occupation in the Northern Caucasus. Curr Anthropol 40, 77–86 (1999). [Google Scholar]
- 33.Skinner AR et al. , ESR dating at Mezmaiskaya Cave, Russia. Appl Radiat Isotopes 62, 219–224 (2005). [DOI] [PubMed] [Google Scholar]
- 34.Korlević P et al. , Reducing microbial and human contamination in DNA extractions from ancient bones and teeth. Biotechniques 59, 87–93 (2015). [DOI] [PubMed] [Google Scholar]
- 35.Dabney J, Meyer M, Length and GC-biases during sequencing library amplification: A comparison of various polymerase-buffer systems with ancient and modern DNA sequencing libraries. Biotechniques 52, 87–94 (2012). [DOI] [PubMed] [Google Scholar]
- 36.Gansauge MT et al. , Single-stranded DNA library preparation from highly degraded DNA using T4 DNA ligase. Nucleic Acids Res 45, e79 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kircher M, Sawyer S, Meyer M, Double indexing overcomes inaccuracies in multiplex sequencing on the Illumina platform. Nucleic Acids Res 40, e3 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kircher M, Stenzel U, Kelso J, Improved base calling for the Illumina Genome Analyzer using machine learning strategies. Genome Biol 10, R83 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Renaud G, Kircher M, Stenzel U, Kelso J, freeIbis: an efficient basecaller with calibrated quality scores for Illumina sequencers. Bioinformatics 29, 1208–1209 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Renaud G, Stenzel U, Kelso J, leeHom: adaptor trimming and merging for Illumina sequencing reads. Nucleic Acids Res 42, e141 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Renaud G, Stenzel U, Maricic T, Wiebe V, Kelso J, deML: robust demultiplexing of Illumina sequences using a likelihood-based approach. Bioinformatics 31, 770–772 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.1000 Genomes Phase 2 decoy sequences: ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/technical/reference/phase2_reference_assembly_sequence/hs37d5.fa.gz.
- 43.Li H, Durbin R, Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. https://bitbucket.org/ustenzel/network-aware-bwa.
- 45. https://bitbucket.org/ustenzel/biohazard-tools.
- 46.Fu Q et al. , Genome sequence of a 45,000-year-old modern human from western Siberia. Nature 514, 445–449 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lazaridis I et al. , Ancient human genomes suggest three ancestral populations for present-day Europeans. Nature 513, 409–413 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.McKenna A et al. , The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20, 1297–1303 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Varshney U, van de Sande JH, Specificities and kinetics of uracil excision from uracil-containing DNA oligomers by Escherichia coli uracil DNA glycosylase. Biochemistry 30, 4055–4061 (1991). [DOI] [PubMed] [Google Scholar]
- 50.The 1000 Genomes Project Consortium, A global reference for human genetic variation. Nature 526, 68–74 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nielsen R, Paul JS, Albrechtsen A, Song YS, Genotype and SNP calling from next-generation sequencing data. Nat Rev Genet 12, 443–451 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li R et al. , SNP detection for massively parallel whole-genome resequencing. Genome Res 19, 1124–1132 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Powell MJD, The BOBYQA algorithm for bound constrained optimization without derivatives. Technical Report DAMTP 2009/NA06, (2009). [Google Scholar]
- 54.Johnson SG, The NLopt nonlinear-optimization package, http://ab-initio.mit.edu/nlopt.
- 55.Benson G, Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res 27, 573–580 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rosenbloom KR et al. , The UCSC Genome Browser database: 2015 update. Nucleic Acids Res 43, D670–681 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Meyer M et al. , Nuclear DNA sequences from the Middle Pleistocene Sima de los Huesos hominins. Nature 531, 504–507 (2016). [DOI] [PubMed] [Google Scholar]
- 58.http://hgdownload.soe.ucsc.edu/goldenPath/hg19/database/simpleRepeat.txt.gz.
- 59.Hinch AG et al. , The landscape of recombination in African Americans. Nature 476, 170–175 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.DePristo MA et al. , A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet 43, 491–498 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bhatia G, Patterson N, Sankararaman S, Price AL, Estimating and interpreting FST: the impact of rare variants. Genome Res 23, 1514–1521 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Briggs AW et al. , Targeted retrieval and analysis of five Neandertal mtDNA genomes. Science 325, 318–321 (2009). [DOI] [PubMed] [Google Scholar]
- 63.Li H, Durbin R, Inference of human population history from individual whole-genome sequences. Nature 475, 493–496 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hudson RR, Generating samples under a Wright-Fisher neutral model of genetic variation. Bioinformatics 18, 337–338 (2002). [DOI] [PubMed] [Google Scholar]
- 65.Fenner JN, Cross-cultural estimation of the human generation interval for use in genetics-based population divergence studies. Am J Phys Anthropol 128, 415–423 (2005). [DOI] [PubMed] [Google Scholar]
- 66.Langergraber KE et al. , Generation times in wild chimpanzees and gorillas suggest earlier divergence times in great ape and human evolution. Proceedings of the National Academy of Sciences of the United States of America 109, 15716–15721 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Scally A, Durbin R, Revising the human mutation rate: implications for understanding human evolution. Nat Rev Genet 13, 745–753 (2012). [DOI] [PubMed] [Google Scholar]
- 68.Venn O et al. , Nonhuman genetics. Strong male bias drives germline mutation in chimpanzees. Science 344, 1272–1275 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Auton A et al. , A global reference for human genetic variation. Nature 526, 68–74 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Vernot B et al. , Excavating Neandertal and Denisovan DNA from the genomes of Melanesian individuals. Science, aad9416 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Reich D et al. , Genetic history of an archaic hominin group from Denisova Cave in Siberia. Nature 468, 1053–1060 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Durand EY, Patterson N, Reich D, Slatkin M, Testing for Ancient Admixture between Closely Related Populations. Mol Biol Evol 28, 2239–2252 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Busing FMTA, Meijer E, Van Der Leeden R, Delete-m jackknife for unequal m. Stat. Comput. 9, 3–8 (1999). [Google Scholar]
- 74.Qin P, Stoneking M, Denisovan Ancestry in East Eurasian and Native American Populations. Mol Biol Evol, msv141 (2015). [DOI] [PubMed] [Google Scholar]
- 75.Llorente MG et al. , Ancient Ethiopian genome reveals extensive Eurasian admixture throughout the African continent. Science 350, 820–822 (2015). [DOI] [PubMed] [Google Scholar]
- 76.Sánchez-Quinto F et al. , North African Populations Carry the Signature of Admixture with Neandertals. PLOS ONE 7, e47765 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Pickrell JK et al. , Ancient west Eurasian ancestry in southern and eastern Africa. Proceedings of the National Academy of Sciences of the United States of America 111, 2632–2637 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sankararaman S, Mallick S, Patterson N, Reich D, The Combined Landscape of Denisovan and Neanderthal Ancestry in Present-Day Humans. Current Biology 26, 1241–1247 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kim Bernard Y., Lohmueller Kirk E., Selection and Reduced Population Size Cannot Explain Higher Amounts of Neandertal Ancestry in East Asian than in European Human Populations. American Journal of Human Genetics 96, 454–461 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Vernot B, Akey JM, Complex History of Admixture between Modern Humans and Neandertals. American Journal of Human Genetics 96, 448–453 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Fu Q et al. , The genetic history of Ice Age Europe. Nature 534, 200–205 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.The Chimpanzee Sequencing and Analysis Consortium, Initial sequence of the chimpanzee genome and comparison with the human genome. Nature 437, 69–87 (2005). [DOI] [PubMed] [Google Scholar]
- 83.Gibbs RA et al. , Evolutionary and biomedical insights from the rhesus macaque genome. Science 316, 222–234 (2007). [DOI] [PubMed] [Google Scholar]
- 84.Locke DP et al. , Comparative and demographic analysis of orang-utan genomes. Nature 469, 529–533 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Scally A et al. , Insights into hominid evolution from the gorilla genome sequence. Nature 483, 169–175 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Prüfer K et al. , The bonobo genome compared with the chimpanzee and human genomes. Nature 486, 527–531 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kong A et al. , Fine-scale recombination rate differences between sexes, populations and individuals. Nature 467, 1099–1103 (2010). [DOI] [PubMed] [Google Scholar]
- 88.Staab PR, Zhu S, Metzler D, Lunter G, scrm: efficiently simulating long sequences using the approximated coalescent with recombination. Bioinformatics 31, 1680–1682 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Terhorst J, Kamm JA, Song YS, Robust and scalable inference of population history from hundreds of unphased whole genomes. Nat Genet 49, 303–309 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Sankararaman S, Patterson N, Li H, Paabo S, Reich D, The date of interbreeding between Neandertals and modern humans. PLoS Genet 8, e1002947 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Fu Q et al. , An early modern human from Romania with a recent Neanderthal ancestor. Nature 524, 216–219 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Sudmant PH et al. , Evolution and diversity of copy number variation in the great ape lineage. Genome Res 23, 1373–1382 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Sudmant PH et al. , Global diversity, population stratification, and selection of human copy-number variation. Science 349, aab3761 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Sudmant PH et al. , An integrated map of structural variation in 2,504 human genomes. Nature 526, 75–81 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Dennis MY et al. , Evolution of Human-Specific Neural SRGAP2 Genes by Incomplete Segmental Duplication. Cell 149, (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Florio M et al. , Human-specific gene ARHGAP11B promotes basal progenitor amplification and neocortex expansion. Science 347, 1465–1470 (2015). [DOI] [PubMed] [Google Scholar]
- 97.Nuttle X et al. , Emergence of a Homo sapiens-specific gene family and chromosome 16p11.2 CNV susceptibility. Nature 536, 205–209 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Hach F et al. , mrsFAST: a cache-oblivious algorithm for short-read mapping. Nat Methods 7, 576–577 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Fiskerstrand T et al. , Mutations in ABHD12 cause the neurodegenerative disease PHARC: An inborn error of endocannabinoid metabolism. Am J Hum Genet 87, 410–417 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Knowlton WM, McKemy DD, TRPM8: from cold to cancer, peppermint to pain. Curr Pharm Biotechnol 12, 68–77 (2011). [DOI] [PubMed] [Google Scholar]
- 101.Ugarte M et al. , Overview of mutations in the PCCA and PCCB genes causing propionic acidemia. Hum Mutat 14, 275–282 (1999). [DOI] [PubMed] [Google Scholar]
- 102.Danecek P et al. , The variant call format and VCFtools. Bioinformatics 27, 2156–2158 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Welter D et al. , The NHGRI GWAS Catalog, a curated resource of SNP-trait associations. Nucleic Acids Res 42, D1001–1006 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kuhn RM, Haussler D, Kent WJ, The UCSC genome browser and associated tools. Brief Bioinform 14, 144–161 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Huerta-Sanchez E et al. , Altitude adaptation in Tibetans caused by introgression of Denisovan-like DNA. Nature 512, 194–197 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.C. Global Lipids Genetics et al. , Discovery and refinement of loci associated with lipid levels. Nat Genet 45, 1274–1283 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Mozaffarian D et al. , Genetic loci associated with circulating phospholipid trans fatty acids: a meta-analysis of genome-wide association studies from the CHARGE Consortium. Am J Clin Nutr 101, 398–406 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Lutz SM et al. , A genome-wide association study identifies risk loci for spirometric measures among smokers of European and African ancestry. BMC Genet 16, 138 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Chen PL et al. , Genetic determinants of antithyroid drug-induced agranulocytosis by human leukocyte antigen genotyping and genome-wide association study. Nat Commun 6, 7633 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Lauc G et al. , Loci associated with N-glycosylation of human immunoglobulin G show pleiotropy with autoimmune diseases and haematological cancers. PLoS Genet 9, e1003225 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Sullivan PF et al. , Genomewide association for schizophrenia in the CATIE study: results of stage 1. Mol Psychiatry 13, 570–584 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Aberg K et al. , Genomewide association study of movement-related adverse antipsychotic effects. Biol Psychiatry 67, 279–282 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Okada Y et al. , Genetics of rheumatoid arthritis contributes to biology and drug discovery. Nature 506, 376–381 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Wade TD et al. , Genetic variants associated with disordered eating. Int J Eat Disord 46, 594–608 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Fox CS et al. , Genome-wide association for abdominal subcutaneous and visceral adipose reveals a novel locus for visceral fat in women. PLoS Genet 8, e1002695 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Anderson D et al. , Genome-wide association study of vitamin D levels in children: replication in the Western Australian Pregnancy Cohort (Raine) study. Genes Immun 15, 578–583 (2014). [DOI] [PubMed] [Google Scholar]
- 117.Park SL et al. , Mercapturic Acids Derived from the Toxicants Acrolein and Crotonaldehyde in the Urine of Cigarette Smokers from Five Ethnic Groups with Differing Risks for Lung Cancer. PLOS ONE 10, e0124841 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Williams SR et al. , Shared genetic susceptibility of vascular-related biomarkers with ischemic and recurrent stroke. Neurology 86, 351–359 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.