

Abstract
Mechanism-based treatments for schizophrenia are needed, and increasing evidence suggests that oxidative stress may be a target. Previous research has shown that N-acetylcysteine (NAC), an antioxidant and glutathione (GSH) precursor almost devoid of side effects, improved negative symptoms, decreased the side effects of antipsychotics, and improved mismatch negativity and local neural synchronization in chronic schizophrenia. In a recent double-blind randomized placebo-controlled trial by Conus et al., early psychosis patients received NAC add-on therapy (2700 mg/day) for 6 months. Compared with placebo-treated controls, NAC patients showed significant improvements in neurocognition (processing speed) and a reduction of positive symptoms among patients with high peripheral oxidative status. NAC also led to a 23% increase in GSH levels in the medial prefrontal cortex (GSHmPFC) as measured by 1H magnetic resonance spectroscopy. A subgroup of the patients in this study were also scanned with multimodal MR imaging (spectroscopy, diffusion, and structural) at baseline (prior to NAC/placebo) and after 6 months of add-on treatment. Based on prior translational research, we hypothesized that NAC would protect white matter integrity in the fornix. A group × time interaction indicated a difference in the 6-month evolution of white matter integrity (as measured by generalized fractional anisotropy, gFA) in favor of the NAC group, which showed an 11% increase. The increase in gFA correlated with an increase in GSHmPFC over the same 6-month period. In this secondary study, we suggest that NAC add-on treatment may be a safe and effective way to protect white matter integrity in early psychosis patients.
Introduction
Mechanism-based treatments for schizophrenia are needed, given that the available treatments have limited efficacy and are often associated with serious side effects. Several lines of evidence show that redox dysregulation and oxidative stress may be a common final pathway in the pathophysiology of psychosis1. Abnormalities in other systems are also involved, including NMDA receptor hypofunction, neuroinflammation, and dopamine dysregulation, all of which interact in a feedforward process2. These mechanisms are thought to belong to a central pathophysiological hub in which an imbalance in any of these systems can lead to microscale (parvalbumin interneurons) and macroscale (white matter tracts) circuit alterations underlying disconnectivity and psychopathology3,4. Although some patients may lack primary redox dysregulation, any point of entry (NMDA receptor hypofunction, neuroinflammation, or dopamine dysregulation) can favor oxidative stress, which may be a common consequence of distinct etiologies (see review by Steullet et al.2).
Oxidative stress results from an imbalance between reactive oxygen/nitrogen species and antioxidants, resulting in macromolecular damage. Indeed, the brain is particularly vulnerable to oxidative stress given its high oxygen consumption and high content of oxidizable polyunsaturated fatty acids. Convergent evidence supports the role of oxidative stress in schizophrenia:5 consequences of oxidative stress, including decreased phospholipids and increased lipid peroxidation, as well as the decline of antioxidant defence systems in both the periphery and the central nervous system, were reported6,7. Critically, glutathione (GSH), the major non-enzymatic antioxidant and redox regulator, was decreased in the cerebrospinal fluid and prefrontal cortex in vivo8 and in postmortem tissue samples9 and was associated with negative symptoms10 in schizophrenia. Genetic evidence includes association with polymorphisms and copy number variations of genes related to GSH synthesis and metabolism1,2,11,12.
In vitro studies revealed that GSH deficit led to impairments in the proliferation and maturation of oligodendrocyte precursors13. Transgenic mice with a GSH deficit (GCLM-KO) present a decrease in mature oligodendrocytes and myelin-associated proteins in the anterior cingulate at peripuberty13. In another translational study from our group, we observed a decrease in fractional anisotropy in the fornix–fimbria bundle in an animal model of redox dysregulation14. Accordingly, we also found reduced generalized fractional anisotropy (gFA) in the fornix of early psychosis patients (EPP)15. Interestingly, in early psychosis, volume loss in the hippocampus correlated positively with fractional anisotropy in the fornix15, indicating that the integrity of these two structures is closely linked in disease. Further, smaller hippocampal volume was associated with higher blood glutathione peroxidase (GPx) activity15, which reflects high central oxidative status (low brain GSH), at least in male EPP16.
Collectively, these findings indicate that GSH and redox regulation have a central role in myelination and white matter maturation. Given that white matter alteration is a core feature of schizophrenia, these observations may lead to new, innovative treatments for use in early psychosis intervention6,13.
Taken together, the evidence appears to show GSH deficits and oxidative stress as promising targets in schizophrenia. Given that GSH is poorly transported across the blood-brain barrier, agents such as N-acetylcysteine (NAC) have attracted great interest as potential therapeutic tools to normalize brain GSH levels and/or redox systems. In particular, NAC, an antioxidant and precursor of GSH, is a promising candidate because it is available over the counter and almost devoid of side effects.
In a proof-of-concept clinical trial, supplementation of NAC in chronic schizophrenia patients (N = 140) led to improvements of negative symptoms17, auditory mismatch negativity18, and local neural synchronization in electroencephalography (EEG)19 as well as decreased side effects of antipsychotics17. Improvement in negative symptoms and total Positive and Negative Syndrome Scale (PANSS) score was replicated in three independent studies20–22 and two studies showed improvement in cognition in chronic schizophrenia22,23. Building upon this prior research, a recent double-blind randomized placebo-controlled add-on trial with NAC was carried out in EPP (n = 63) by Conus et al.24. Patients showed improved cognition (processing speed factor), and a subgroup of patients with a high baseline for peripheral oxidative status also improved in positive symptoms. However, there was no improvement in negative symptoms, potentially owing to the low rate of negative symptoms in this group. Moreover, in a subgroup of patients in the same study who underwent EEG, NAC supplementation led to improved auditory evoked potentials, known to be impaired in schizophrenia25.
Despite the mounting evidence that NAC may be a sustainable strategy to restore GSH deficiency and fight against oxidative stress in schizophrenia, the targets engaged in the brain by NAC have not been elucidated26 and have not been tested in vivo in EPP.
Recently, in vivo measurement of GSH in humans has been demonstrated on a 3-T clinical MRI scanner using short-TE 1H magnetic resonance spectroscopy (1H-MRS)16,27. Therefore, an essential step was achieved in the trial by Conus and colleagues24. In EPP who agreed to participate (n = 24), 1H-MRS was applied and revealed that NAC supplementation for 6 months actually led to an elevation of medial prefrontal GSH levels by 23%24.
No research has investigated whether this increase in brain GSH levels owing to NAC supplementation is accompanied by restoration/protection of white matter integrity in early psychosis6. Given the limited sample size of our study, we decided to focus specifically on the effect of NAC on the white matter integrity of the fornix bundle, which has been shown to be vulnerable to oxidative stress early in the time course of the illness. Moreover, we studied whether the changes in fornix integrity were linked to the changes in medial prefrontal GSH (GSHmPFC) levels and explained the changes in processing speed.
Patients and methods
Clinical trial protocol and study medication
The EPP in the present study (N = 20), representing a subsample of those in the original study trial24, have consented to be assessed with multimodal brain imaging. The flow diagram is shown in supplementary figure 1. A detailed description of the main 6-month, randomized, placebo-controlled, double-blind NAC add-on trial; the patient cohort; the study design; the sample size calculation and assessment procedures; the side effects; the efficacy; and the outcome measures has been published elsewhere24.
In brief, NAC (2700 mg/day) or placebo was administered to each EP patient for 6 months following a double-blinded randomized placebo-controlled design. As previously shown17,28, tolerability was excellent, with NAC patients showing no more side effects on the UKU scale29 compared with placebo24.
Diffusion spectrum imaging (DSI), T1 structural imaging (T1), and 1H-MRS were performed before NAC/placebo intake (baseline measurements) and after 6 months of NAC/placebo intake (follow-up). Symptoms were assessed with the PANSS. The processing speed factor (verbal fluency and the trail making A test, symbol coding) was extracted from the MATRICS Consensus Cognitive Battery (MCCB)30,31, which was administered at baseline and follow-up. Antipsychotic doses at the time of the study were converted to chlorpromazine (CPZ) equivalents in milligrams32 for each patient.
Following their recruitment, patients were given ID numbers, and both patients and investigators were blinded until the time of analysis, when data pooling necessitated unblinding. The hypothesis for the current study was recorded in a statistical plan prior to unblinding. The study was registered at Swiss Medic (2008DR2308) and at ClinicalTrials.gov (NCT01354132).
Participants
The study was conducted from 2009 to 2014. All patients were recruited from TIPP (the Treatment and Early Intervention in Psychosis Program, University Hospital, Lausanne)33, a 3-year program specializing in the treatment of early-phase psychosis. Out of the 63 patients who participated in the original study trial, 20 patients (14 men; aged 25 ± 6.7 years) agreed to undergo DSI/T1, and 17 agreed to DSI/T1/1H-MRS scanning. The inclusion criteria were as follows: (1) male or female, aged 18–38 years; (2) having a psychotic disorder, defined by the “Psychosis threshold” subscale on the Comprehensive Assessment of at Risk Mental States scale (CAARMS);34 (3) having received under 12 months of treatment for psychosis; (4) capability to provide informed consent; (5) sufficient stability to participate in the study. The exclusion criteria were: (1) presence of clinically significant medical illnesses (including peptic ulcers), (2) organic mental disease/organic psychosis, (3) severe cerebral trauma, (4) mental retardation (intelligence quotient < 70), (5) pregnancy or lactation, (6) allergy to NAC, (7) current treatment with antioxidants, (8) poor command of French, (9) and substance-induced psychosis. All participants provided written informed consent, and the procedure was approved by the Ethics Committee of Lausanne University (10th July 2008).
NAC and placebo were kindly provided by Bioadvantex Pharma Inc. (Mississauga, Ontario, Canada) and produced under Good Manufacturing Practice conditions. All participants were randomized (by blocks of four, according to randomization lists known only to the pharmacist) in a 1:1 allocation ratio and assigned to take either effervescent NAC tablets (900 mg) at a dosage of 2700 mg/day (morning: 1800 mg; evening: 900 mg) or matching placebo tablets before meals.
Multimodal imaging acquisition and analysis
gFA measured by MRI
MRI sessions were performed on a 3-Tesla scanner (Magnetom TrioTim, Siemens Medical Solutions, Erlangen, Germany) equipped with a 32-channel head coil. Each scanning session included a magnetization-prepared rapid acquisition gradient echo (MPRAGE) T1-weighted sequence with 1-mm in-plane resolution and 1.2-mm slice thickness, covering 240 × 257 × 160 voxels. The repetition (TR), echo (TE), and inversion (TI) times were 2300, 2.98, and 900 ms, respectively. The DSI sequence included 128 diffusion-weighted images with a maximum b-value of 8000 s mm−2 and one b0 reference image. The acquisition volume was made of 96 × 96 × 34 voxels with 2.2 × 2.2 × 3 mm resolution. TR and TE were 6800 and 144 ms, respectively.
White matter diffusion properties were estimated using gFA computed from DSI as described by Tuch35. Each gFA map was normalized to MNI (Montreal Neurological Institute) standard space using nonlinear registration procedures and smoothed with a Gaussian kernel of SD = 1 mm in FSL 5.0.8 (http://fsl.fmrib.ox.ac.uk/fsl/fslwiki/). Quality control included manual inspection of each gFA image for abnormalities or registration failure. The region of interest for the fornix was extracted from the JHU (Johns Hopkins University) white matter atlas36.
GSHmPFC levels measured by in vivo 1H-MRS
The 1H-MRS method was described in detail in the original study trial24 and previous works27,37. In brief, the levels of GSHmPFC were assessed by localized 1H-MRS measurements performed on a 3-T MR scanner (Magnetom TimTrio, Siemens Healthcare) with a transverse electromagnetic (TEM 3000) head coil (MR Instruments, Inc., Minneapolis, MN, USA). The magnetic field homogeneity was optimized by adjusting first- and second-order shims using FAST(EST) MAP. Single-voxel 1H MR spectra were acquired from a volume of interest (VOI = 20 × 20 × 25 mm3) in the medial prefrontal cortex using a short-TE spin-echo full-intensity acquired localized single voxel spectroscopy technique (SPECIAL) with the following scan parameters: TE/TR = 6/4000 ms, acquisition bandwidth = 2 kHz, number of averages = 148, vector size = 2048. Outer volume suppression and water suppression with variable pulse power and optimized relaxation delays were applied prior to the SPECIAL localization sequence. GSHmPFC concentrations were quantified by analyzing water suppressed in vivo 1H MR spectra using LCModel (Stephen Provencher, Inc., Oakville, ON, Canada) with a basis set consisting of 20 simulated individual metabolite spectra and an experimentally measured macromolecule baseline. Unsuppressed water 1H NMR spectra were used as an internal reference. The spectral range for analysis was set to 0.2–4.2 ppm, and the Cramer-Rao bounds for GSHmPFC were 10 ± 3% (mean ± s.d.).
Statistical analysis
Statistical analyses for demographic and clinical data were performed in Prism for Mac OS X (Version 7.0c, March 1, 2017). Differences between the NAC and placebo group were assessed with t tests or Fisher’s exact test. A paired t test was used to test for interactions between time and treatment status (NAC or placebo) in each white matter voxel within the fornix. gFA was the dependent variable, whereas treatment and time were the independent variables. Age and gender were set as nuisance factors in the general linear model. Correction for multiple comparisons across all white matter voxels in the fornix was performed using a non-parametric cluster-based procedure, namely, “threshold-free cluster enhancement” with “Randomise” in FSL 5.0.8, which avoids inflation of false positives38. The corrected p value for the cluster was calculated from 10,000 permutations, and a p value < 0.05 was considered significant.
Results
Demographics, clinical characteristics, and longitudinal clinical changes
Among the 20 patients, 10 were part of the group that received NAC, whereas 10 received placebo. Clinical and demographic characteristics were not different between the 2 groups (Table 1). Patients treated with NAC had lower baseline GSH levels than those treated with placebo.
Table 1.
Demographic and clinical characteristics of early psychosis patients (NAC vs placebo) at baseline (if not specified otherwise)
| NAC (n = 10) | Placebo (n = 10) | P value | |
|---|---|---|---|
| Age (years) | 25.3 ± 5.7 | 24.8 ± 7.9 | 0.3622 |
| Gender (M/F) | 9/1 | 5/5 | 0.1409 |
| Mean CPZ, mg/day (baseline) | 264.5 ± 69.29 | 307.8 ± 64.45 | 0.6531 |
| Mean CPZ, mg/day (follow-up) | 271.2 ± 65.04 | 418.4 ± 80.27 | 0.1711 |
| PANSS baseline: positive symptoms | 13.8 ± 4.8 | 18.0 ± 6.8 | 0.1249 |
| PANSS baseline: negative symptoms | 14.6 ± 4.6 | 18.6 ± 6.7 | 0.1377 |
| PANSS baseline: total | 32.6 ± 8.5 | 39.4 ± 9.8 | 0.1152 |
| Duration of illness (days) | 981.6 ± 810.2 | 663.9 ± 665.3 | 0.3743 |
| Diagnosis | |||
| Schizophrenia | 6 | 6 | |
| Schizoaffective disorder | 2 | 1 | |
| Bipolar disorder | 1 | ||
| Major depression with psychotic features | 1 | ||
| Brief psychotic episode | 1 | 1 | |
| Psychosis not otherwise specified | 1 | ||
| Antipsychotic medication | |||
| Quetiapine | 5 | 3 | |
| Clozapine | 1 | ||
| Aripiprazole | 1 | 2 | |
| Amisulpride | 2 | ||
| Risperidone | 1 | 2 | |
| Olanzapine | 1 | 1 | |
| No medication | 1 | ||
| GSHmPFC | n = 9 | n = 8 | |
| Baseline (mM) | 0.8432 ± 0.0708 | 1.144 ± 0.06967 | 0.0087 |
| Follow-up (mM) | 1.013 ± 0.08315 | 1.096 ± 0.07635 | 0.4803 |
If not otherwise specified, the mean ± SD is provided. CPZ chlorpromazine equivalents, GSHmPFC glutathione concentration in the medial prefrontal cortex, NAC N-acetylcysteine, PANSS Positive and Negative Syndrome Scale
Six-month longitudinal gFA changes in the fornix: NAC vs placebo
There was no significant difference in mean baseline gFA between NAC (0.1669 ± 0.01044) and placebo (0.1911 ± 0.01893). There was a group × time interaction, which reached significance (corrected p < 0.04) in the body of the fornix (size of the cluster = 10 voxels). gFA values for the subjects of each group (NAC and placebo) at baseline and follow-up are plotted (Fig. 1). Placebo subjects exhibited a decrease in mean gFA values (mean difference = − 0.02063; % change = mean difference gFA/baseline gFA × 100 = −10,795%), whereas NAC subjects showed an increase (mean difference = 0.01932; % change = + 11.576%).
Fig. 1.

Six-month longitudinal changes in gFA: NAC vs placebo. Coronal, sagittal, and axial views (scalar gFA map) show voxel-wise analysis results indicating location (body of the fornix) of the group × time interaction, which reached significance (corrected p value < 0.04) (red cluster). Size of the cluster = 10 voxels. The graph shows extracted gFA values for each subject of each group (placebo, left; NAC, right) at baseline (T0) and follow-up (T1). Most placebo subjects show a decrease in gFA values, whereas NAC subjects show an increase
Correlation between gFA changes and GSHmPFC changes over a 6-month period
Correlation analysis (Fig. 2) revealed a positive relationship between longitudinal change in gFA in the identified cluster in the fornix and longitudinal change in GSHmPFC in the whole group (NAC and placebo; r = 0.67; p = 0.0031). When the treatment (NAC) group and the control (placebo) group were analyzed separately, the NAC group remained significant (r = 0.76; p = 0.0186), whereas the placebo group did not (r = 0.48; p = 0.2245).
Fig. 2.
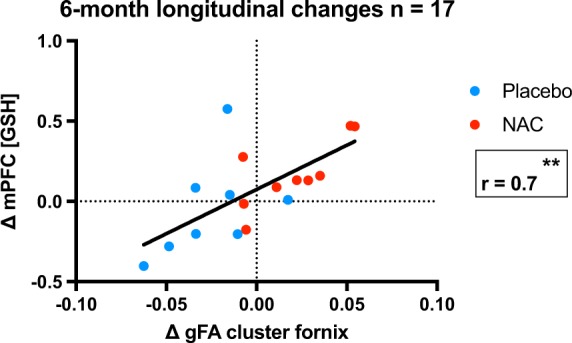
Relationship between change in gFA and change in GSHmPFC. Placebo patients are represented in blue, and NAC patients in red. Changes in gFA correlate with the change in GSHmPFC over the 6-month period (r = 0.67; p = 0.0031)
When gFA was averaged over the whole fornix (supplementary figure 2), the relationship between change in gFA and change in GSHmPFC in the whole sample (NAC and placebo combined) reached trend-level significance (r = 0.4623; p = 0.0617) but was nonsignificant when analyzed separately in the NAC group (r = 0.5254; p = 0.1463) and the placebo group (r = 0.2307; p = 0.5825).
There was no significant correlation between longitudinal change in average gFA extracted from the identified cluster in the fornix and longitudinal change in blood cell GPx (r = 0.195; p = 0.4101) in the whole sample (NAC and placebo combined) or in the groups analyzed separately (NAC group: r = 0.297; p = 0.4069; placebo group: r = 0.3476; p = 0.3224).
Correlation between changes in gFA and processing speed over a 6-month period
Correlations between longitudinal change in processing speed and gFA were nonsignificant in the whole group (r = 0.2771; p = 0.2989), in the NAC group (r = 0.5476; p = 0.1710) and in the placebo group (r = − 0.511; p = 0.1956). (Supplementary figure 3).
Discussion
We observed for the first time that the administration of NAC, a precursor of GSH, to EPP increases white matter integrity in the fornix as measured by gFA. Furthermore, longitudinal change in GSHmPFC (i.e., the difference between baseline and 6 months of NAC/placebo add-on treatment) correlates with the change in gFA along the fornix bundle in NAC patients and in NAC and placebo patients pooled together. These results suggest that a GSH increase through NAC supplementation may improve/protect white matter integrity, at least in the fornix. Thus, our findings highlight that fornix integrity may improve and could represent a valid target for early psychosis intervention. The lack of significance when the whole fornix was analyzed may be linked to the decrease in sensitivity with this approach as well as the small sample size, which is the main limitation of this very demanding study for EPP in terms of scanning (multimodal MRI), design, and duration (6 months).
In the current study, we focused on the fornix bundle because its implication in schizophrenia as well as in early psychosis is well documented15,39–41. The fornix, as a major output of the hippocampus, has important role in cognitive processing, especially memory41. Notably, the hippocampus is the brain structure most robustly implicated in schizophrenia42. In addition, the fornix was listed among the most affected white matter tracts in a recent meta-analysis including 2000 patients with schizophrenia43. Progressive dysfunction in two hippocampal areas, which give rise to the fornix bundle (i.e., CA1 and subiculum), has been also very recently highlighted44.
Data from a GCLM-KO mouse model (with low GSH levels, a consequence of gene inactivation of the modulatory subunit of glutamate–cysteine ligase, the rate-limiting enzyme of GSH synthesis) was an additional strong incentive. A study by Corcoba et al.14 revealed a reduction in FA in the fornix–fimbria in peripubertal GCLM-KO mice, which remained so throughout adulthood. Furthermore, in the same KO mice, the conduction velocity of the fornix bundle was reduced in the slow-conducting fibers. This indicates that the fimbria–fornix is particularly vulnerable to GSH deficit-induced oxidative stress.
The role of redox control in white matter integrity and oligodendrocyte development has been previously documented in several ways (see review by Monin and colleagues6). One proposed mechanism is that redox balance regulates oligodendrocyte maturation and the switch between proliferation and differentiation6,13,45. Interestingly, in a mouse model, GSH deficit conditions led to impairments of proliferation and maturation of oligodendrocytes13.
GSH deficit is not the only mechanism that can generate oxidative stress. Indeed, it can be triggered by the perturbation of a variety of systems known to be implicated in schizophrenia, which include the redox, neuroimmune, glutamatergic, and dopaminergic systems1,2. As mentioned previously, these different systems do not function in isolation but interact reciprocally in a feedforward process, leading to a vicious cycle2. Notably, inflammatory pathways are activated by oxidative stress and vice versa2, and both are implicated in schizophrenia and may lead to impairment of myelination and white matter development.
NAC, as a molecule with multifaceted functions28, may restore or protect white matter integrity by several mechanisms. The most apparent mechanism is that NAC acts as a cysteine donor, which can be used to synthesize and replenish GSH, which, in turn, acts as a free radical scavenger. Anti-inflammatory properties have also been described for NAC, probably conferred at least in part by its antioxidant properties2. In preclinical models, NAC attenuates white matter injuries following a maternal immune challenge46. In clinical studies, NAC was added to other putative neuroprotective compounds in infantile neuronal ceroid lipofuscinosis47 or traumatic brain injury48. However, the absence of a randomized placebo-controlled design with NAC alone prevented the specific effects of NAC from being highlighted in these studies. An alternative hypothesis is that NAC may limit potential side effects of antipsychotic medication, which may impact white matter integrity49.
In the current study, we used a DSI sequence, characterized by strong diffusion weighting and high angular resolution. DSI is thought to be more sensitive and specific than classical diffusion tensor imaging to white matter microstructure, crossing fibers and the slow diffusion compartment50,51. Although it is tempting to conclude from this imaging study that NAC improves myelination, no firm conclusions can be drawn regarding the exact mechanisms. We can only speculate that “myelin maintenance and repair”52 is influenced by NAC. Reduction of FA may not be specific to changes in myelin content; other factors such as axonal size and coherence and changes in the volume of water spaces surrounding axons are also important53. Interestingly, reduction of FA may also result from inflammation54.
The effectiveness of NAC in early psychosis was studied by Conus et al.24 in the whole cohort included in the two-center trial. NAC was demonstrated to have a significant effect on neurocognition (processing speed) but not on negative symptoms. Given the efficacy of NAC in improving processing speed, we studied its relationship with gFA in the fornix; this relationship was positive, although not statistically significant.
In the study by Conus et al.24, subgroup exploration revealed that patients who showed improvements in their positive symptoms had higher baseline blood GPx activity than those whose positive symptoms did not improve. In other words, patients with high peripheral oxidative status benefited the most from NAC. In this context, it is interesting to note that in a previous study, high blood GPx activity was associated with small hippocampal volume15 and with low prefrontal GSH levels16. High GPx activity and/or antioxidant/redox system dysregulation may thus be a marker of response to NAC as well as a marker of small hippocampal size, which is relevant to the current study given the anatomical relationship between the fornix and the hippocampus. We thus studied the relationship between the change in GPx and the change in gFA in the fornix but did not find a significant effect. Given the small sample size, it is difficult to draw any final conclusions on this matter.
The limited sample size of the current study deserves further consideration. First, despite the absence of statistically significant difference, the two groups were not well-matched regarding sex (i.e., one female only in the NAC group) and disease severity. Second, an excess significance bias has been reported in voxel-based studies55 and especially in small samples56. This is partly owing to the wide use of parametric tests for cluster-based statistics56. Parametric tests rely on the assumption of a normal distribution of the data, which is often not the case when sample size is limited57. Here, we used non-parametric testing that does not rely on data normality and that has been shown to limit the rate of false positives in neuroimaging studies38. Nevertheless, these findings need to be interpreted with caution and replication in a larger randomized controlled trial is needed.
One further limitation that must be mentioned is that GSHmPFC was measured in the medial prefrontal region, whereas gFA was measured in the fornix; there may be differences in GSH concentrations between brain regions. Nevertheless, we measured a longitudinal change in GSH levels, which is more likely to be proportional across brain regions. In addition, we cannot rule out the possibility that a spontaneous increase in brain GSH contributed to the observed effect in NAC-treated patients, as their basal levels were lower than those of the placebo group.
Although it is recognized that white matter anomalies are a hallmark of schizophrenia present before initiation of treatment, it has been suggested that antipsychotic medication may contribute to brain atrophy, including changes in white matter49. Haloperidol or olanzapine administered to macaque monkeys resulted in a tendency toward a 12.9% decrease in oligodendrocyte count58. In the current study, CPZ equivalents were stable in the NAC group, whereas there was a nonsignificant increase in the placebo group. Previous findings regarding the putative effects of antipsychotics on FA in patients are heterogeneous and even contradictory, with studies reporting a positive59, negative60, or null effect61 of antipsychotics on white matter integrity. Nevertheless, we cannot exclude that the observed effect was driven by the nonsignificant group difference in CPZ equivalents.
NAC add-on treatment is safe24,28 and if this preliminary study is confirmed, NAC may prove to be efficient in promoting white matter integrity in EPP, even with a mere 6 months of treatment. Further diffusion MRI studies investigating white matter changes to monitor NAC treatment response are warranted.
Electronic supplementary material
Acknowledgements
We express our gratitude to all patients for their continued participation. This work was supported by grants from the Swiss National Science Foundation (320030_122419 to P.C. and K.Q.D., 310030_156874 to P.H.) and by the National Center of Competence in Research (NCCR) “SYNAPSY – The Synaptic Bases of Mental Diseases”, financed by the Swiss National Science Foundation (no. 51AU40_125759). P.S.B. is financially supported by the Leenaards Foundation. P.K. is financially supported by the Adrian and Simone Frutiger Foundation. Magnetic resonance spectroscopy was performed at the Centre d’Imagerie BioMédicale (CIBM) of the UNIL, UNIGE, HUG, CHUV, EPFL. We are grateful for the support we received from the Damm-Etienne Foundation, the Alamaya Foundation and Banque Lombard Odier & Cie SA. We thank Bioadvantex Pharma, Inc., for providing NAC and placebo.
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Paul Klauser and Lijing Xin.
These seniors authors contributed equally: Kim Q. Do and Philipp S. Baumann.
Electronic supplementary material
Supplementary Information accompanies this paper at (10.1038/s41398-018-0266-8).
References
- 1.Steullet P, et al. Oxidative stress-driven parvalbumin interneuron impairment as a common mechanism in models of schizophrenia. Mol. Psychiatry. 2017;22:936–943. doi: 10.1038/mp.2017.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Steullet P, et al. Redox dysregulation, neuroinflammation, and NMDA receptor hypofunction: a “central hub” in schizophrenia pathophysiology. Schizophr. Res. 2016;176:41–51. doi: 10.1016/j.schres.2014.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hardingham GE, Do KQ. Linking early-life NMDAR hypofunction and oxidative stress in schizophrenia pathogenesis. Nat. Rev. Neurosci. 2016;17:125–134. doi: 10.1038/nrn.2015.19. [DOI] [PubMed] [Google Scholar]
- 4.Do KQ, Cabungcal JH, Frank A, Steullet P, Cuenod M. Redox dysregulation, neurodevelopment, and schizophrenia. Curr. Opin. Neurobiol. 2009;19:220–230. doi: 10.1016/j.conb.2009.05.001. [DOI] [PubMed] [Google Scholar]
- 5.Koga M, Serritella AV, Sawa A, Sedlak TW. Implications for reactive oxygen species in schizophrenia pathogenesis. Schizophr. Res. 2016;176:52–71. doi: 10.1016/j.schres.2015.06.022. [DOI] [PubMed] [Google Scholar]
- 6.Monin Aline, Fournier Margot, Baumann Philipp S., Cuénod Michel, Do Kim Q. Handbook of Behavioral Neuroscience. 2016. Role of Redox Dysregulation in White Matter Anomalies Associated with Schizophrenia; pp. 481–500. [Google Scholar]
- 7.Flatow J, Buckley P, Miller BJ. Meta-analysis of oxidative stress in schizophrenia. Biol. Psychiatry. 2013;74:400–409. doi: 10.1016/j.biopsych.2013.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Do KQ, et al. Schizophrenia: glutathione deficit in cerebrospinal fluid and prefrontal cortex in vivo. Eur. J. Neurosci. 2000;12:3721–3728. doi: 10.1046/j.1460-9568.2000.00229.x. [DOI] [PubMed] [Google Scholar]
- 9.Yao JK, Leonard S, Reddy R. Altered glutathione redox state in schizophrenia. Dis. Markers. 2006;22:83–93. doi: 10.1155/2006/248387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Matsuzawa D, et al. Negative correlation between brain glutathione level and negative symptoms in schizophrenia: a 3T 1H-MRS study. PLoS. ONE. 2008;3:e1944. doi: 10.1371/journal.pone.0001944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gysin R, et al. Impaired glutathione synthesis in schizophrenia: convergent genetic and functional evidence. Proc. Natl. Acad. Sci. USA. 2007;104:16621–16626. doi: 10.1073/pnas.0706778104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rodríguez-Santiago B, et al. Association of common copy number variants at the glutathione S-transferase genes and rare novel genomic changes with schizophrenia. Mol. Psychiatry. 2010;15:1023–1033. doi: 10.1038/mp.2009.53. [DOI] [PubMed] [Google Scholar]
- 13.Monin A, et al. Glutathione deficit impairs myelin maturation: relevance for white matter integrity in schizophrenia patients. Mol. Psychiatry. 2015;20:827–838. doi: 10.1038/mp.2014.88. [DOI] [PubMed] [Google Scholar]
- 14.Corcoba A, et al. Glutathione deficit affects the integrity and function of the fimbria/fornix and anterior commissure in mice: relevance for schizophrenia. Int. J. Neuropsychopharmacol. 2015;19:pyv110. doi: 10.1093/ijnp/pyv110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Baumann PS, et al. Impaired fornix-hippocampus integrity is linked to peripheral glutathione peroxidase in early psychosis. Transl. Psychiatry. 2016;6:e859. doi: 10.1038/tp.2016.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xin L, et al. Genetic polymorphism associated prefrontal glutathione and its coupling with brain glutamate and peripheral redox status in early psychosis. Schizophr. Bull. 2016;42:1185–1196. doi: 10.1093/schbul/sbw038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Berk M, et al. N-acetyl cysteine as a glutathione precursor for schizophrenia--a double-blind, randomized, placebo-controlled trial. Biol. Psychiatry. 2008;64:361–368. doi: 10.1016/j.biopsych.2008.03.004. [DOI] [PubMed] [Google Scholar]
- 18.Lavoie S, et al. Glutathione precursor, N-acetyl-cysteine, improves mismatch negativity in schizophrenia patients. Neuropsychopharmacology. 2008;33:2187–2199. doi: 10.1038/sj.npp.1301624. [DOI] [PubMed] [Google Scholar]
- 19.Carmeli C, Knyazeva MG, Cuénod M, Do KQ. Glutathione precursor N-acetyl-cysteine modulates EEG synchronization in schizophrenia patients: a double-blind, randomized, placebo-controlled trial. PLoS. ONE. 2012;7:e29341. doi: 10.1371/journal.pone.0029341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Farokhnia M, et al. N-acetylcysteine as an adjunct to risperidone for treatment of negative symptoms in patients with chronic schizophrenia: a randomized, double-blind, placebo-controlled study. Clin. Neuropharmacol. 2013;36:185–192. doi: 10.1097/WNF.0000000000000001. [DOI] [PubMed] [Google Scholar]
- 21.Breier Alan, Liffick Emily, Hummer Tom A., Vohs Jenifer L., Yang Ziyi, Mehdiyoun Nicole F., Visco Andrew C., Metzler Emmalee, Zhang Ying, Francis Michael M. Effects of 12-month, double-blind N-acetyl cysteine on symptoms, cognition and brain morphology in early phase schizophrenia spectrum disorders. Schizophrenia Research. 2018;199:395–402. doi: 10.1016/j.schres.2018.03.012. [DOI] [PubMed] [Google Scholar]
- 22.Sepehrmanesh Z, Heidary M, Akasheh N, Akbari H, Heidary M. Therapeutic effect of adjunctive N-acetyl cysteine (NAC) on symptoms of chronic schizophrenia: a double-blind, randomized clinical trial. Prog. Neuropsychopharmacol. Biol. Psychiatry. 2018;82:289–296. doi: 10.1016/j.pnpbp.2017.11.001. [DOI] [PubMed] [Google Scholar]
- 23.Rapado-Castro M, et al. Cognitive effects of adjunctive N-acetyl cysteine in psychosis. Psychol. Med. 2017;47:866–876. doi: 10.1017/S0033291716002932. [DOI] [PubMed] [Google Scholar]
- 24.Conus P, et al. N-acetylcysteine in a double-blind randomized placebo-controlled trial: toward biomarker-guided treatment in early psychosis. Schizophr. Bull. 2018;44:317–327. doi: 10.1093/schbul/sbx093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Retsa Chrysa, Knebel Jean-François, Geiser Eveline, Ferrari Carina, Jenni Raoul, Fournier Margot, Alameda Luis, Baumann Philipp S., Clarke Stephanie, Conus Philippe, Do Kim Q., Murray Micah M. Treatment in early psychosis with N-acetyl-cysteine for 6 months improves low-level auditory processing: Pilot study. Schizophrenia Research. 2018;191:80–86. doi: 10.1016/j.schres.2017.07.008. [DOI] [PubMed] [Google Scholar]
- 26.Shungu DC. N-acetylcysteine for the treatment of glutathione deficiency and oxidative stress in schizophrenia. Biol. Psychiatry. 2012;71:937–938. doi: 10.1016/j.biopsych.2012.03.025. [DOI] [PubMed] [Google Scholar]
- 27.Mekle R, et al. MR spectroscopy of the human brain with enhanced signal intensity at ultrashort echo times on a clinical platform at 3T and 7T. Magn. Reson. Med. 2009;61:1279–1285. doi: 10.1002/mrm.21961. [DOI] [PubMed] [Google Scholar]
- 28.Deepmala SlatteryJ, et al. Clinical trials of N-acetylcysteine in psychiatry and neurology: a systematic review. Neurosci. Biobehav. Rev. 2015;55:294–321. doi: 10.1016/j.neubiorev.2015.04.015. [DOI] [PubMed] [Google Scholar]
- 29.Lingjaerde O, Ahlfors UG, Bech P, Dencker SJ, Elgen K. The UKU side effect rating scale. A new comprehensive rating scale for psychotropic drugs and a cross-sectional study of side effects in neuroleptic-treated patients. Acta Psychiatr. Scand. Suppl. 1987;334:1–100. doi: 10.1111/j.1600-0447.1987.tb10566.x. [DOI] [PubMed] [Google Scholar]
- 30.Nuechterlein KH, et al. The MATRICS Consensus Cognitive Battery, part 1: test selection, reliability, and validity. Am. J. Psychiatry. 2008;165:203–213. doi: 10.1176/appi.ajp.2007.07010042. [DOI] [PubMed] [Google Scholar]
- 31.Kern RS, et al. The MATRICS Consensus Cognitive Battery, part 2: co-norming and standardization. Am. J. Psychiatry. 2008;165:214–220. doi: 10.1176/appi.ajp.2007.07010043. [DOI] [PubMed] [Google Scholar]
- 32.Andreasen NC, Pressler M, Nopoulos P, Miller D, Ho BC. Antipsychotic dose equivalents and dose-years: a standardized method for comparing exposure to different drugs. Biol. Psychiatry. 2010;67:255–262. doi: 10.1016/j.biopsych.2009.08.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Baumann PS, et al. Treatment and early intervention in psychosis program (TIPP-Lausanne): implementation of an early intervention programme for psychosis in Switzerland. Early Interv. Psychiatry. 2013;7:322–328. doi: 10.1111/eip.12037. [DOI] [PubMed] [Google Scholar]
- 34.Yung AR, et al. Mapping the onset of psychosis: the Comprehensive Assessment of At-Risk Mental States. Aust. N. Z. J. Psychiatry. 2005;39:964–971. doi: 10.1080/j.1440-1614.2005.01714.x. [DOI] [PubMed] [Google Scholar]
- 35.Tuch DS. Q-ball imaging. Magn. Reson. Med. 2004;52:1358–1372. doi: 10.1002/mrm.20279. [DOI] [PubMed] [Google Scholar]
- 36.Wakana S, et al. Reproducibility of quantitative tractography methods applied to cerebral white matter. Neuroimage. 2007;36:630–644. doi: 10.1016/j.neuroimage.2007.02.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tkác I, Oz G, Adriany G, Uğurbil K, Gruetter R. In vivo 1H NMR spectroscopy of the human brain at high magnetic fields: metabolite quantification at 4T vs. 7T. Magn. Reson. Med. 2009;62:868–879. doi: 10.1002/mrm.22086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Eklund A, Nichols TE, Knutsson H. Cluster failure: Why fMRI inferences for spatial extent have inflated false-positive rates. Proc. Natl. Acad. Sci. USA. 2016;113:7900–7905. doi: 10.1073/pnas.1602413113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Luck D, Malla AK, Joober R, Lepage M. Disrupted integrity of the fornix in first-episode schizophrenia. Schizophr. Res. 2010;119:61–64. doi: 10.1016/j.schres.2010.03.027. [DOI] [PubMed] [Google Scholar]
- 40.Fitzsimmons J, et al. Diffusion tractography of the fornix in schizophrenia. Schizophr. Res. 2009;107:39–46. doi: 10.1016/j.schres.2008.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nestor PG, et al. Episodic memory and neuroimaging of hippocampus and fornix in chronic schizophrenia. Psychiatry Res. 2007;155:21–28. doi: 10.1016/j.pscychresns.2006.12.020. [DOI] [PubMed] [Google Scholar]
- 42.Heckers S, Konradi C. Hippocampal pathology in schizophrenia. Curr. Top. Behav. Neurosci. 2010;4:529–553. doi: 10.1007/7854_2010_43. [DOI] [PubMed] [Google Scholar]
- 43.Kelly S., et al. Widespread white matter microstructural differences in schizophrenia across 4322 individuals: results from the ENIGMA Schizophrenia DTI Working Group. Mol. Psychiatry23,1261–1269 (2017). [DOI] [PMC free article] [PubMed]
- 44.Lieberman J A, Girgis R R, Brucato G, Moore H, Provenzano F, Kegeles L, Javitt D, Kantrowitz J, Wall M M, Corcoran C M, Schobel S A, Small S A. Hippocampal dysfunction in the pathophysiology of schizophrenia: a selective review and hypothesis for early detection and intervention. Molecular Psychiatry. 2018;23(8):1764–1772. doi: 10.1038/mp.2017.249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Noble M, Smith J, Power J, Mayer-Pröschel M. Redox state as a central modulator of precursor cell function. Ann. N. Y. Acad. Sci. 2003;991:251–271. doi: 10.1111/j.1749-6632.2003.tb07481.x. [DOI] [PubMed] [Google Scholar]
- 46.Paintlia MK, Paintlia AS, Contreras MA, Singh I, Singh AK. Lipopolysaccharide-induced peroxisomal dysfunction exacerbates cerebral white matter injury: attenuation by N-acetyl cysteine. Exp. Neurol. 2008;210:560–576. doi: 10.1016/j.expneurol.2007.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Levin SW, et al. Oral cysteamine bitartrate and N-acetylcysteine for patients with infantile neuronal ceroid lipofuscinosis: a pilot study. Lancet Neurol. 2014;13:777–787. doi: 10.1016/S1474-4422(14)70142-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Amen DG, Wu JC, Taylor D, Willeumier K. Reversing brain damage in former NFL players: implications for traumatic brain injury and substance abuse rehabilitation. J. Psychoact. Drugs. 2011;43:1–5. doi: 10.1080/02791072.2011.566489. [DOI] [PubMed] [Google Scholar]
- 49.Zipursky Robert B., Reilly Thomas J., Murray Robin M. The Myth of Schizophrenia as a Progressive Brain Disease. Schizophrenia Bulletin. 2012;39(6):1363–1372. doi: 10.1093/schbul/sbs135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Baumann PS, et al. High b-value diffusion-weighted imaging: a sensitive method to reveal white matter differences in schizophrenia. Psychiatry Res. 2012;201:144–151. doi: 10.1016/j.pscychresns.2011.08.003. [DOI] [PubMed] [Google Scholar]
- 51.Wedeen VJ, et al. The geometric structure of the brain fiber pathways. Science. 2012;335:1628–1634. doi: 10.1126/science.1215280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Davis KL, et al. White matter changes in schizophrenia: evidence for myelin-related dysfunction. Arch. Gen. Psychiatry. 2003;60:443–456. doi: 10.1001/archpsyc.60.5.443. [DOI] [PubMed] [Google Scholar]
- 53.Kubicki M, Shenton ME. Diffusion tensor imaging findings and their implications in schizophrenia. Curr. Opin. Psychiatry. 2014;27:179–184. doi: 10.1097/YCO.0000000000000053. [DOI] [PubMed] [Google Scholar]
- 54.Assaf Y, Pasternak O. Diffusion tensor imaging (DTI)-based white matter mapping in brain research: a review. J. Mol. Neurosci. 2008;34:51–61. doi: 10.1007/s12031-007-0029-0. [DOI] [PubMed] [Google Scholar]
- 55.Ioannidis JP. Excess significance bias in the literature on brain volume abnormalities. Arch. Gen. Psychiatry. 2011;68:773–780. doi: 10.1001/archgenpsychiatry.2011.28. [DOI] [PubMed] [Google Scholar]
- 56.Fusar-Poli P, et al. Evidence of reporting biases in voxel-based morphometry (VBM) studies of psychiatric and neurological disorders. Hum. Brain Mapp. 2014;35:3052–3065. doi: 10.1002/hbm.22384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Salmond CH, et al. Distributional assumptions in voxel-based morphometry. Neuroimage. 2002;17:1027–1030. doi: 10.1006/nimg.2002.1153. [DOI] [PubMed] [Google Scholar]
- 58.Konopaske GT, et al. Effect of chronic antipsychotic exposure on astrocyte and oligodendrocyte numbers in macaque monkeys. Biol. Psychiatry. 2008;63:759–765. doi: 10.1016/j.biopsych.2007.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Reis Marques T, et al. White matter integrity as a predictor of response to treatment in first episode psychosis. Brain. 2014;137:172–182. doi: 10.1093/brain/awt310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Szeszko PR, et al. White matter changes associated with antipsychotic treatment in first-episode psychosis. Neuropsychopharmacology. 2014;39:1324–1331. doi: 10.1038/npp.2013.288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zeng B, et al. Abnormal white matter microstructure in drug-naive first episode schizophrenia patients before and after eight weeks of antipsychotic treatment. Schizophr. Res. 2016;172:1–8. doi: 10.1016/j.schres.2016.01.051. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.