

Abstract
Glucocorticoids are steroid hormones that regulate multiple aspects of glucose homeostasis. Glucocorticoids promote gluconeogenesis in liver, whereas in skeletal muscle and white adipose tissue they decrease glucose uptake and utilization by antagonizing insulin response. Therefore, excess glucocorticoid exposure causes hyperglycemia and insulin resistance. Glucocorticoids also regulate glycogen metabolism. In liver, glucocorticoids increase glycogen storage, whereas in skeletal muscle they play a permissive role for catecholamine-induced glycogenolysis and/or inhibit insulin-stimulated glycogen synthesis. Moreover, glucocorticoids modulate the function of pancreatic α and β cells to regulate the secretion of glucagon and insulin, two hormones that play a pivotal role in the regulation of blood glucose levels. Overall, the major glucocorticoid effect on glucose homeostasis is to preserve plasma glucose for brain during stress, as transiently raising blood glucose is important to promote maximal brain function. In this chapter we will discuss the current understanding of the mechanisms underlying different aspects of glucocorticoid-regulated mammalian glucose homeostasis.
Keywords: Glucocorticoids, Glucocorticoid receptor, Gluconeogenesis, Insulin, Glucose utilization, Glycogen, Pancreas, Glucose metabolism
Introduction
Glucocorticoids (GC) are stress hormones that play a key role in the regulation of mammalian glucose homeostasis. The name “glucocorticoids” originates from their profound effects on plasma glucose levels. GC regulate multiple aspects of glucose homeostasis (Fig. 5.1). First, GC promote hepatic gluconeogenesis [1, 2] and reduce glucose uptake and utilization in skeletal muscle and white adipose tissue (WAT) [3, 4]. These effects are critical for metabolic adaptation during stress, such as fasting/starvation, when plasma glucose needs to be preserved because it is the brains’ pri-mary energy source, and transiently raising blood glucose is important to promoting maximal brain functions [5]. Insulin, a hormone secreted from pancreatic β cells, exerts opposite effects on these physiological processes by inhibiting hepatic gluco-neogenesis and promoting glucose utilization in skeletal muscle and WAT. Thus, to exert their responses, GC need to antagonize insulin actions. These effects are critical during stress, which in short term does not affect or even enhances glucose tolerance. However, chronic GC exposure results in hyperglycemia and insulin resistance [3, 4, 6]. Second, GC exert tissue-specific effects on glycogen metabolism. In liver, GC increase glycogen storage, whereas in skeletal muscle GC play a permissive role for catecholamine-induced glycogenolysis or inhibit insulin-stimulated glyco-gen synthesis [7–9]. Third, GC modulate insulin and glucagon secretion from pan-creas. GC treatment increases plasma glucagon levels [10, 11], whereas the effects of GC on insulin secretion are complex [12–16]. GC induce pancreas islet hyperpla-sia in vivo that leads to hyperinsulinemia [12, 17–19] and have been shown to exert cytotoxic effects on β cells [20, 21]. Overall, in this chapter we will discuss the current understanding of the mechanisms underlying these distinct aspects of GC-regulated glucose homeostasis.
Fig. 5.1.
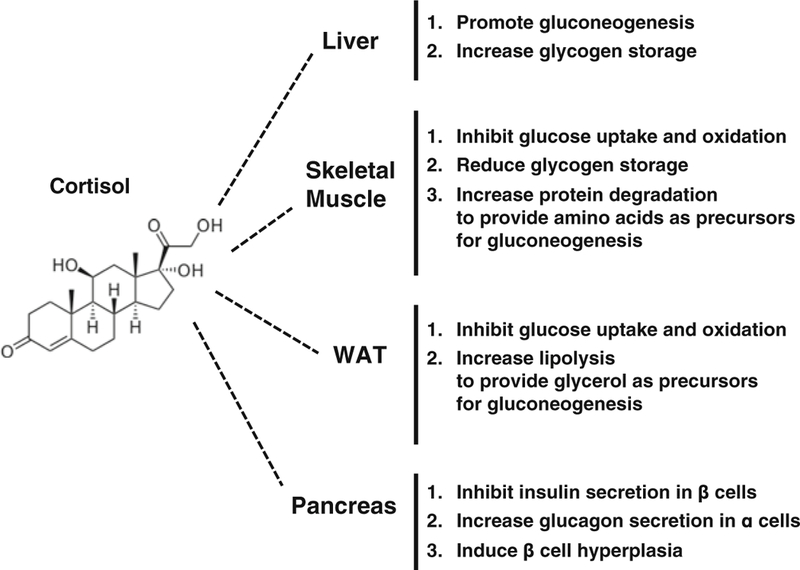
Glucocorticoid effects on glucose homeostasis. The effects of cortisol on glucose homeo-stasis in peripheral tissues
Gluconeogenesis
The gluconeogenic pathway generates glucose from non-carbohydrate substrates (Fig. 5.2) [22, 23]. Gluconeogenesis mainly occurs in liver, though kidney and intestine are also contributors. The major gluconeogenic precursors are lactate, glycerol and gluconeogenic amino acids, such as alanine. Both lactate and alanine can be converted to pyruvate, which is then carboxylated to oxaloacetate (OAA) by pyruvate carboxylase (PC) [24, 25], a step that occurs in mitochondria. OAA needs to be converted to malate to be shuttled to the cytoplasm where it is converted back to OAA. Cytosolic phosphoenolpyruvate carboxykinase (PCK1) [26, 27] then cata-lyzes OAA to phosphoenolpyruvate (PEP), which then enters the gluconeogenic pathway. There is also a mitochondrial form of PCK1 (m-PCK1) that can directly convert OAA to PEP in mitochondria, which is then transported to cytoplasm and participates in gluconeogenesis. Recent studies suggest that m-PCK1 could also play a role in gluconeogenesis [28–31].
Fig. 5.2.
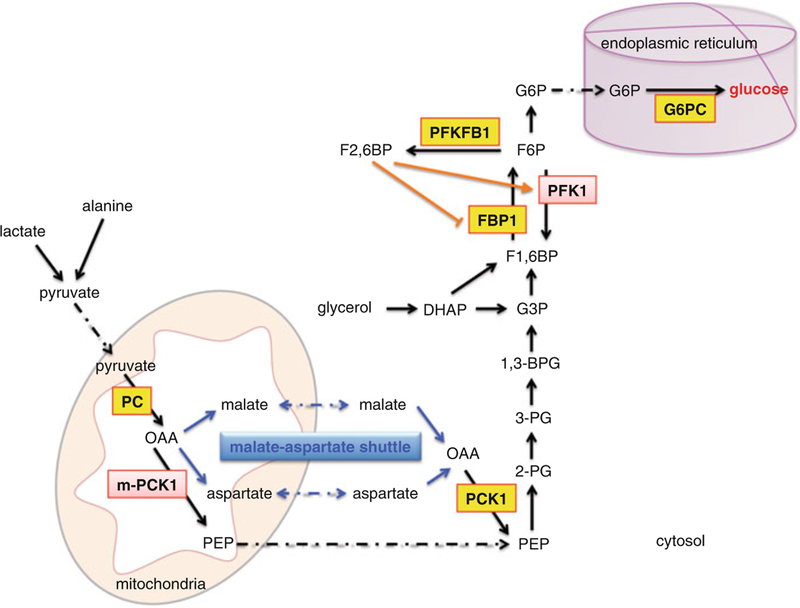
Gluconeogenic pathway in hepatocytes. Lactate and alanine are converted to pyruvate, which enters the mitochondria and is then converted to OAA by enzyme PC. Through malate-aspartate shuttle, OAA exits the mitochondria to form PEP. OAA can also be converted to PEP directly within the mitochondria. PEP then feeds into the gluconeogenic pathway. In addition, glycerol is metabolized to DHAP, which is then converted directly or indirectly through G3P to F1,6BP. The final product, glucose, is produced in the ER by enzyme G6PC. The key enzymes are boxed, with GR primary targets shown in yellow. Abbreviation: OAA oxaloacetate, PEP phospho-enolpyruvate, DHAP dihydroxyacetone phosphate, G3P glyceraldehyde-3-phosphate, F1,6BP fructose-1,6-bisphosphate, 2-PG 2-phosphoglycerate, 3-PG 3-phosphoglycerate, 1,3-BPG 1,3-bisphosphoglycerate, G3P glyceraldehyde-3-phosphate, F1,6BP fructose-1,6-bisphosphate, F2,6BP fructose-2,6-bisphosphate, F6P fructose-6-phosphate, and G6P glucose-6-phosphate. Enzyme abbreviation: PC pyruvate carboxylase, m-PCK1 mitochondrial phosphoenolpyruvate carboxykinase, PCK1 cytosolic phosphoenolpyruvate carboxykinase, FBP1 fructose-1,6-bisphosphatase 1, PFK1 phosphofructokinase 1, PFKFB1 phosphofructokinase 2/fructose bispho-sphatase 2, G6PC glucose-6-phosphatase catalytic subunit
Conversion of PEP to fructose-1,6-phosphate (F1,6BP) requires five enzymatic steps that are essentially the reverse of glycolysis (Fig. 5.2). The “bifunctional” enzyme, phosphofructokinase 2 (PFK2)/fructose bisphosphatase 2 (FBPase2) (a.k.a. PFKFB1) [32, 33], plays a critical role in the switch between gluconeogen-esis and glycolysis. PFKFB1 regulates the production of fructose 2,6 bisphosphate (F2,6BP), which is an allosteric activator of phosphofructokinase 1 (PFK1), an enzyme in the glycolytic pathway. When circulating glucose levels are low, such as during fasting and starvation, glucagon inactivates PFK2, which allows FBPase2 activity to be favored. This results in decreased production of F2,6BP, and reduced glycolysis and enhanced gluconeogenesis. Fructose 1,6-bisphosphatase (FBP1) converts F1,6BP to fructose-6-phosphate (F6P) [34], which is then converted to glucose-6-phosphate (G6P). G6P enters the endoplasmic reticulum (ER), where the enzyme glucose-6-phosphatase (G6PC) [35–37] converts G6P to glucose (Fig. 5.2). Notably, distinct gluconeogenic amino acids can be converted to specific intermedi-ates in the tricarboxylic acid (TCA) cycle. These TCA cycle intermediates are con-verted to OAA to enter the gluconeogenic cycle. Glycerol enters gluconeogenesis through conversion to dihydroxyacetone phosphate (DHAP), which can then be metabolized to glycerol-3-phosphate (G3P) or directly to F1,6BP (Fig. 5.2) thus entering the gluconeogenic cycle above PCK1.
The positive effect of GC on hepatic gluconeogenesis is well established and has been under extensive study for several decades. Injecting GC into humans [38, 39] and rodents increases hepatic gluconeogenesis. In addition, GC are important for other hormones to activate gluconeogenesis. Although glucagon is regarded as a major hormone that activates gluconeogenesis during fasting, fasting-induced gluconeogenesis is reduced in adrenalectomized mice, an effect that is restored by treating mice with GC [40–42]. In fact, glucagon-, epinephrine-, or cyclic AMP (cAMP)-induced gluconeogenesis are all attenuated in adrenalectomized mice. Giving GC to adrenalectomized mice restores the ability of these hormones to induce gluconeogenesis. Thus, GC play a “permissive” role promoting the optimal ability of these hormones in gluconeogenesis. GC also provide gluconeogenic precursors by promoting protein degradation in skeletal muscle to generate gluconeogenic amino acids [1, 4]. They also enhance lipolysis in WAT. This releases glycerol and fatty acids. Glycerol is used as a gluconeogenic precursor, whereas fatty acids provide energy to drive the gluconeogenic pathway [8, 43, 44].
GC promote gluconeogenesis mainly through activation of the transcription of genes encoding enzymes in the gluconeogenic pathway. The transcription of PC, PCK1, FBP1, PFKFB1, G6PC and G6P transporter (SLC37A4) are all stimulated by GC (Fig. 5.2). GC convey their signals mainly through an intracellular receptor, the glucocorticoid receptor (GR). Before binding to GC, GR resides in cytoplasm and is associated with Hsp90 chaperone complex. Upon binding to GC, GR dissoci-ates from Hsp90 chaperone complex and enters the nucleus, where the GR is recruited to specific genomic sequences, called glucocorticoid response elements (GREs). The GR:GRE association could be a direct GR:DNA interaction or an indi-rect association through other DNA-binding transcription factors. In any case, once GR associates with a specific GRE, it recruits a number of transcriptional coregula-tors. While they do not bind to DNA directly, these coregulators are able to assist the GR in modulation of the transcriptional rate of nearby genes through distinct mech-anisms: altering chromatin structure, inducing histone modifications; recruiting RNA polymerase II containing basal transcription machinery; and modulating tran-scriptional elongation. Notably, most genomic GREs are “composite” GREs (also called glucocorticoid response units, GRUs) that consist of multiple cis-acting elements, which include binding sites for GR and other transcription factors (called accessory elements and accessory factors, respectively), to mediate a complete GC response. Because different DNA binding transcription factors other than GR are involved in the regulation of distinct GR primary target genes, the multi-protein transcriptional regulatory complex assembled on each GRE is likely distinct. Composite GREs could allow GC to differentially regulate distinct target genes in different cell types depending on the presence of cell type specific accessory factors and transcriptional coregulators. Another advantage of employing composite GREs is to allow specific cross-talk between GC and other signaling pathways at the accessory elements and their respective accessory factors. Thus, cross-talk does not need to occur directly through GR.
The GREs of Pck1, Pfkfb1, and G6pc genes have been identified. In particular, the mechanism of GR-regulated Pck1 gene transcription has been extensively stud-ied. By contrast, the mechanisms governing GC-activated Pc and Fbp1 gene are unclear. Below we will discuss the mechanisms of GR-stimulated Pck1, Pfkfb1, and G6pc gene transcription.
PCK1
Rat Pck1 gene contains two GREs, GRE1 and GRE2 (Fig. 5.3) [45]. GRE1 is located between −388 and −374 (relative to transcription start site, TSS), whereas GRE2 is located between and −367 and −354 in the Pck1 gene promoter. When GRE1 or GRE2 is placed in front of TATA box in a synthetic reporter gene, neither mediates a GC response [46]. The combination of GRE1 and GRE2 also fails to confer GC-induced transcription [46]. In fact, both GRE1 and GRE2 bind GR very weakly in vitro [46]. However, in cooperation with other accessory elements on the Pck1 promoter, they confer a robust GC response. These accessory elements include gAF1 [45], gAF2 [45], gAF3 [47] and the cAMP response element (CRE) [48]. Both gAF1 (between −451 and −434 of rat PCK1 promoter) and gAF2 (−416 and −407) are located 5′ from GRE1 and GRE2, whereas gAF3 and CRE are located in 3′ of these GREs (Fig. 5.3). Hepatic nuclear factor 4 (HNF4, NR2A1) and chicken ovalbumin upstream transcription factor (COUP-TF, NR2F2) bind to gAF1 and serve as accessory factors for a complete GC response [49]. The gAF1 element also serves as a retinoic acid response element (RARE). An all-trans retinoic acid recep-tor (RAR) and 9-cis retinoic acid receptor (RXR) heterodimer binds to gAF1 and confers retinoic acid (RA)-activated Pck1 gene transcription [50]. RA has been shown to synergize with GC to stimulate Pck1 gene transcription [51].
Fig. 5.3.
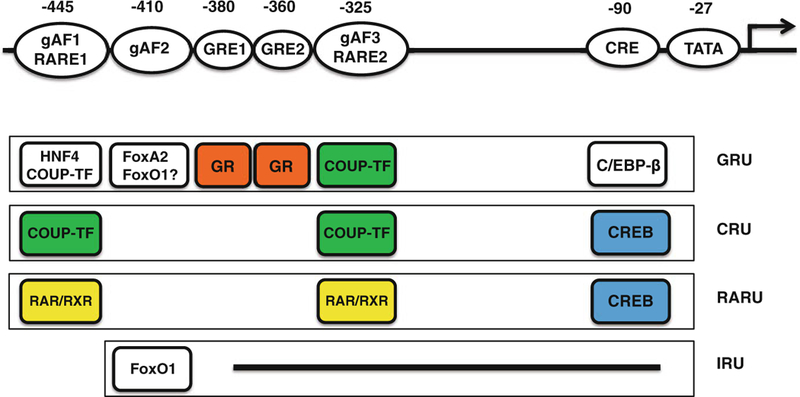
Hormone response units in the PEPCK gene. Binding sites for various regulatory and transcription factors are shown in the top row, with the number indicating the center nucleotide position of each element with respect to the transcription start site. Four hormone-specific response units are drawn: proximal glucocorticoid response unit (GRU), cyclic AMP response unit (CRU), retinoic acid response unite (RARU), and insulin response unit (IRU). In the absence of the other hormones, the components of each response unit are depicted. These units interact functionally, cooperating or competing, to comprise the PEPCK promoter. Except for gAF2, DNA elements involved in IRU are not yet identified
The gAF2 element binds to members of the forkhead box transcription factor fam-ily that include FoxA1 (also called hepatic nuclear factor 3 α, HNF3α), FoxA2 (hepatic nuclear factor 3 β, HNF3β), FoxO1 (FKHR) and FoxO3A (FKHRL1). FoxA2 have been shown to act as accessory factors for GR-regulated Pck1 gene tran-scription in vitro [52]. Liver specific deletion of FoxO1 but not FoxO3A significantly reduces fasting-induced Pck1 gene expression [53]. Because GC play an important role in fasting-induced Pck1 gene transcription, these results suggest that FoxO1 may serve as an accessory factor for GR in vivo. The gAF2 element also serves as an insu-lin response sequence (IRS) that confers at least part of repressive effect of insulin on Pck1 gene transcription [54, 55]. The ability of insulin to suppress Pck1 gene expres-sion is compromised in liver specific FoxO1 knockout mice [53] or mice overex-pressed dominant negative FoxO1 [56]. Notably, the ability of insulin to reduce Pck1 gene expression is not affected in FoxO3A deletion mice [53]. These results support the key role of FoxO1 in the regulation of Pck1 gene expression in vivo. The potential role of FoxA2 inhibiting insulin effects has been reported [57], although more studies are needed to confirm the importance of FoxA2 in insulin-suppressed Pck1 gene expression.
Streptozotocin is a relatively specific pancreas β cell cytotoxin. Treatment of mice with streptozotocin induces a state that mimics type 1 diabetes. Circulating GC levels are increased in such animals and Pck1 gene expression is augmented. In mice bearing a reporter gene containing a construct containing −2 kb rat Pck1 gene promoter with a mutation at gAF2, streptozotocin-induced reporter gene expression is markedly reduced [58]. These results confirm the importance of the gAF2 ele-ment in GC-activated Pck1 gene in vivo.
The gAF3 element (−337 and −321) binds to COUP-TF [47] and, like gAF1, also serves as a RARE that binds to RAR/RXR heterodimer [59]. The cAMP response element (CRE, between −90 and −82) is also required for a complete GC-stimulated Pck1 gene transcription [48]. CCAAT enhancer binding protein β (C/EBPβ) binds to the CRE to mediate the accessory activity for the GC response [60].
Chromatin immunoprecipitation (ChIP) was used to monitor the recruitment of GR and various accessory factors to their respective binding sites in rat H4IIE hepa-toma cells. GC treatment increases the recruitment of GR, FoxO1, FoxO3A and RNA polymerase II (Pol II) to the Pck1 promoter [61]. FoxA2, C/EBPβ, HNF4 and COUP-TF occupy the Pck1 promoter before GC treatment and their occupancy is not altered after GC treatment [61]. The recruitment of transcriptional coregulators, SRC-1, p300 and CREB binding protein (CBP), to the Pck1 GRU is markedly increased by GC treatment [61]. This suggests that GC treatment initiates the assembly of multi-protein transcriptional regulatory complex on the Pck1 promoter. Insulin treatment for just 3 min markedly decreases the GC-induced recruitment of GR, FoxO1, FoxO3A, FoxA2, SRC-1, p300, CBP, and Pol II, and only the occupancy of C/EBPβ, HNF-4 and COUP-TF remains unchanged [61]. Thus, insulin treatment rapidly dis-rupts the assembly of GC-induced transcriptional complex on the Pck1GRU.Analyzing epigenetic marks showed that the most significant change is the methylation at his-tone H3 arginine residue 17, which is significantly increased upon GC treatment and is abolished by insulin [61]. CARM1/PRMT4, is the histone methyltransferase that methylates histone H3 tail arginine 17 residue (H3R17) [62, 63]. CARM1 has been shown to serve as a transcriptional coactivator for GR. However, the occupancy of CARM1 on the Pck1 promoter is not significantly changed upon GC or insulin treat-ment. One way to explain these results is that CARM1 is present on the Pck1 GRU before GC treatment and the activity of CARM1 is modulated by insulin treatment. The activity of CARM1 is regulated by post-translational modifications [64]. Alternatively, it is possible that an unknown histone methyltransferase is involved in the elevation of H3R17 methylation on the Pck1 GRU.
An analysis of the rat Pck1 promoter in human hepatoma HepG2 cells identified two additional accessory elements (dAF1 and dAF2) that are involved in GC-stimulated Pck1 gene transcription. The dAF1 element is located at −993 and has sequence similarity to the gAF1 element, whereas the dAF2 element, located at −1365, resembles more proximal gAF2 [65]. HNF4 and peroxisome proliferator activated receptor α (PPARα) and RXR heterodimer (PPARα/RXR) bind to dAF1, whereas FoxA1, FoxA2 and FoxO1 bind to dAF2 [65]. ChIP experiments showed that GC treatment increases FoxO1 and PPARα recruitment to the gAF2 and the dAF2, and HNF4 to the dAF1 in mouse liver [65]. Overexpressing PPARα or HNF4 synergizes with GR to activate a reporter gene harboring 2 kb of rat Pck1 promoter [65]. A role for PPARα in GC-induced Pck1 expression in vivo is consistent with the observation that GC-induced Pck1 gene expression is markedly reduced in Pparα null mice [66]. Notably, streptozotocin-induced Pck1 gene expression and blood glucose levels are reduced in transgenic mice that harbor a targeted ablation of the dAF1 in the Pck1 gene promoter. These results support the importance of the dAF1 in GC-activated Pck1 gene expression in vivo [65].
Overall, GC activate Pck1 gene transcription through a complex GRU. Intriguingly, all accessory elements in the Pck1 GRU are involved in the responses of other hor-mones that provide potential cross-talk between GC and other hormones, including glucagon, retinoic acid and insulin (Fig. 5.3). The complexity of GRU also allows various signaling pathways to fine tune the transcription levels of Pck1 gene. Interestingly, GC repress the transcription of Pck1 gene in adipocytes [67, 68] where the major metabolic role of Pck1 is glyceroneogenesis [69, 70]. It is not entirely clear why Pck1 GRU is not functional in adipocytes, although it is proposed that GR inhibits Pck1 gene transcription through antagonizing C/EBP family of transcription factors in adipocytes [67, 68]. Nonetheless, the requirement of accessory fac-tors to act with GR on “weak” GREs provides the flexibility for GC to regulate Pck1 gene transcription in a tissue specific manner. While it is unknown how accessory factors participate in GR-regulated Pck1 gene transcription, there are two potential mechanisms. First, accessory factors may aid in recruitment of transcriptional coregulators to the GRU to stimulate the transcription. Previous studies have shown that the transactivation domain of HNF4 and FoxA2 are required for their accessory activities [71]. When gAF1, gAF2 or gAF3 is replaced by the binding site of a yeast transcription factor Gal4, a fusion protein that consists of GAL4 DNA binding domain and a transcriptional coregulator, SRC1, is able to provide accessory activ-ity [72]. Moreover, another transcriptional coregulator, peroxisome proliferator activated receptor γ coactivator-1α (PGC1α), has been shown to interact with HNF4 to participate in GC-activated Pck1 gene transcription. PGC1α also interacts with and coactivates FoxO1 [73] that binds to the gAF2 element. But the role of PGC1α-FoxO1 interaction in GC-stimulated Pck1 gene transcription has not been exam-ined. Second, accessory factors can potentiate GR:GRE association. Using quantitative, real time equilibrium and stopped-flow fluorescence anisotropy mea-surements of nuclear protein-DNA interactions it was shown that GR binds to the Pck1 GREs poorly. However, the presence of the gAF1 and the gAF2 elements markedly enhanced the association between GR and the Pck1 GREs [74]. It is pos-sible that the assembly of a multi-protein complex that includes GR, accessory fac-tors and transcriptional coregulators enhances the association between GR and the two Pck1 GREs.
G6PC
G6PC gene transcription is induced by GC, whereas insulin suppresses both basal and GC-activated G6PC gene transcription. Mouse G6pc also contains a complex GRU in the proximal promoter region. This GRU consists of three GREs, a CRE, a HNF4 binding site, a hepatic nuclear factor 1 (HNF1) binding site, and multiple FoxO/FoxA binding sites (FREs) [75]. GC significantly activate the expression of a reporter gene that contains the G6pc GRU. Mutation at any of these accessory ele-ments in the G6pc GRU reporter gene reduces the GC response [75]. Mutations at FREs that bind FoxO1 and FoxO3A also reduce both the inhibitory effect of insulin and basal expression of G6pc gene [76]. In liver specific FoxO1 deletion mice the expression of G6pc in 18 h fasted mice is markedly lower than that of 18 h fasted wild type mice [53]. Moreover, the ability of insulin to suppress the expression of G6pc is abolished in liver specific FoxO1 deletion mice [53] or mice overexpressed dominant negative FoxO1 [56]. In contrast, liver specific FoxO3A deletion does not affect basal expression of G6pc and the ability of insulin to inhibit G6pc remains intact [53]. These results are reminiscent of the regulation of Pck1 gene and sug-gests that FoxO1 plays a key role in the regulation of gluconeogenic genes in vivo. Also, similar to the regulation of Pck1 gene, transcription coregulator PGC1α posi-tively regulates basal G6pc gene transcription and enhances GC-stimulated G6pc gene transcription through interaction with HNF4 [77, 78]. FoxO1 and PGC1α appear to synergistically activate G6pc gene transcription [73, 79].
GC also activate the transcription of the G6P transporter (SLC37A4) gene, which encodes a protein that is responsible for shuttling G6P from the cytoplasm to the ER lumen. The mouse Slc37a4 gene promoter contains a GRE [80, 81] and a FoxO1 binding site is identified nearby the GRE [81]. GC increase the activity of a luciferase reporter gene under the control of the Slc37a4 gene promoter in 293 cells, whereas the mutation at the FoxO1 binding site reduces the ability of GC to potentiate this reporter gene activity [81]. Overexpression of FoxO1 in 293 cells potentiates the ability of GC to activate the reporter gene activity [81]. These results suggest that GC stimulate the Slc37a4 gene through a GRU that contains at least a GRE and a FoxO1 binding site.
PFKFB1
Hepatic rat Pfkfb1 gene transcription is stimulated by GC and a GRU has been identified in the intronic region of this gene. In addition to the GRE, this GRU con-sists of binding sites for FoxA2, hepatic nuclear factor 6 (HNF6, a.k.a. Onecut1), C/EBP and Nuclear factor 1 (NF1) [82]. Insulin antagonizes the stimulatory effect of GC on Pfkfb1 gene [82, 83]. While insulin acts through PI3K and Akt to inhibit Pck1 and G6pc gene expression [84, 85], this pathway is apparently not involved in the suppressive effect of insulin on GC-induced Pfkfb1 gene. Instead, insulin acti-vates the Jun N-terminal Kinase (JNK) pathway to inhibit GC-induced Pfkfb1 gene expression [86].
Factors Regulating GC-Stimulated Hepatic Gluconeogenesis
Many other factors regulate gluconeogenesis by modulating GC signaling. Liver X receptor β (LXRβ) is involved in GC-activated Pck1 gene expression. In Lxrβ null mice (Lxrβ−/−) GC-induced Pck1 gene expression and glucose production are reduced, and GC-induced recruitment of GR to the Pck1 GRE is impaired [87]. In contrast, in Lxrα null mice (Lxra−/−) GC-regulated Pck1 gene expression is not affected. Moreover, GC-stimulated expression of tyrosine aminotransferase (Tat) gene, which encodes the enzyme that converts tyrosine to 4-hydroxyphenolpyruvate, is not affected in Lxrb−/− mice. Thus, the role of LXRβ in GC action is relatively specific to the Pck1 gene. The mechanisms governing LXRβ action in GC-activated Pck1 gene transcription are unclear, especially in view of another study, which showed that treating hepatoma cells with LXR ligands suppresses GC-stimulated Pck1 and G6pc gene expression [88]. Microarray analyses showed that LXR ligands affect only a subset of GC-regulated genes. Both gel shift and ChIP experiments sug-gest that an LXRα/RXRα heterodimer competes with GR for binding at rat G6pc GRE [88]. In agreement with its effects on GC-induced gluconeogenic gene expres-sion is the observation that treating rats with an LXR ligand attenuates GC-augmented plasma glucose levels [88]. In summary, unliganded LXRβ is required for maximal GC-induced Pck1 gene transcription and is necessary for GC-induced recruitment of GR to the Pck1 GRE. By contrast, LXR ligands suppress GC-activated gluconeo-genic gene transcription by inhibiting the recruitment of GR to the GREs of these genes.
The expression of transcription factor ying yang 1 (YY1) is increased upon fast-ing and in insulin resistant state [89]. Overexpression of YY1 in mouse liver increases gluconeogenesis [89]. In contrast, the deletion of YY1 in mouse liver results in hypoglycemia. YY1 potentiates gluconeogenesis through the increase of hepatic GR expression, which in turn augments the expression of gluconeogenic genes [89].
The expression of farnesoid x receptor (FXR, NR1H4), a bile acid receptor, is also increased during fasting [90]. Fxr null mice become hypoglycemic during fasting and have a reduced glucose production after a pyruvate challenge and a decreased expres-sion of Pck1 and G6pc [90]. The treatment of fasted mice with an FXR ligand, 6α-ethylchenodeoxycholic acid (6E-CDCA), increases hepatic glucose production and Pck1 and G6pc gene expression. These effects are not observed in Fxr null mice, nor are they seen in fed mice [90]. 6E-CDCA elevates the expression of GR. By con-trast, the expression of GR is decreased in Fxr null mice. Reducing GR expression in liver abolishes 6E-CDCA-induced glucose production and the expression of Pck1 and G6pc [90]. Thus, FXR activation elevates GR expression, which in turn results in enhanced gluconeogenesis.
Ubiquitin-specific protease 2 (USP2) expression is induced by fasting by both GC and glucagon, and by PGC1α overexpression [91]. Overexpression of Usp2 in mouse liver increases glucose production and exacerbates high fat diet-induced glu-cose intolerance, whereas the reduction of Usp2 expression in mouse liver improves systemic glycemic control [91]. Usp2 induces the expression of 11β-hydroxysteroid dehydrogenase type 1 (11β-HSD1), the enzyme that converts the inactive GC, 11-DHC (rodents) or cortisone (humans), to active corticosterone (rodents) or cor-tisol (humans) in liver. Thus, Usp2 increases hepatic gluconeogenesis by increasing the active GC levels in hepatocytes. Usp2 is a ubiquitin specific protease. How Usp2 increases the expression of 11β-HSD1 is unclear.
Transforming growth factor β (TGFβ) decreases hepatic gluconeogenesis. TGFβ increases the expression of SMAD6, which directly associates with the N-terminus of GR [92]. When SMAD6 associates with GRE-bound GR, it recruits histone deacetylase 3 (HDAC3) which antagonizes the acetylation of histone H3 and H4 on genomic regions near the GREs [92]. This results in an inhibition of the transactivation activity of GR. Overexpression of SMAD6 in liver reduces GC-induced Pck1 transcription and blood glucose levels, which mimics TGFβ effects [92].
GR associates with the Hsp90-containing chaperone complex in the cytoplasm, an interaction that induces a GR conformation favorable for binding GC. The Hsp90-containing chaperone complex also plays a role in the translocation of GR into the nucleus. Acetylation of the lysine 294 residue of Hsp90 reduces its interac-tion with GR [93]. Therefore, keeping lysine 294 in a deacetylated state is critical for the GR-Hsp90 interaction. Histone deacetylase 6 (HDAC6) deacetylates lysine 294 of Hsp90. Ablation of Hdac6 results in a decreased GR response due to defec-tive GR translocation into the nucleus [93]. In Hdac6 knockout mice, GC-induced hepatic Pck1 and G6pc gene expression and glucose production are reduced [93]. GC-induced glucose production in primary hepatocytes isolated from Hdac6 knock-out mice is markedly lower than in primary hepatocytes isolated from wild type mice [93]. Notably, Hdac6 ablation generally reduces GR signaling. Not surpris-ingly, other GR-regulated processes, including GC-induced insulin resistance and lipolysis in adipocytes, are also affected when Hdac6 is deleted.
Glucose Utilization
GC inhibit glucose utilization by reducing both glucose uptake and glucose oxida-tion in skeletal muscle and WAT, two major tissues involved in insulin-responsive glucose utilization [94, 95]. These GC effects counteract those of insulin, which promote glucose uptake, glycolysis and glucose oxidation. These result in a tran-sient increase of circulating glucose, which is considered beneficial during stress [5]. In both mouse and human myotubes GC reduce insulin-stimulated glucose uptake [96–98] by attenuating insulin-induced GLUT4 translocation to the cell membrane. By contrast, reduced GC signaling improves insulin sensitivity and glu-cose utilization in mouse and human skeletal muscle. Circulating GC levels are increased in genetically obese ob/ob, db/db and lipotrophic A-ZIP/F-1 [99] mice as compared to wild type. These mice are insulin resistant, but adrenalectomy improves insulin-stimulated muscle glucose disposal [100, 101]. In high fat diet-induced obese mice, adrenalectomy or treatment with the GR antagonist, RU-486, improves insulin sensitivity and increases glucose utilization in skeletal muscle. Moreover, 11β-HSD1-specific inhibitors, which reduce corticosterone levels in various tissues, improve insulin sensitivity and skeletal muscle glucose utilization in animal models of diabetes or insulin resistance [96].
The ability of GC to inhibit glucose uptake and glucose oxidation in skeletal mus-cle is due to, at least in part, a direct effect of GCs on myotubes. Treating cultured myotubes with GC inhibits insulin-stimulated glucose utilization [96, 102]. One mechanism by which GC reduce glucose utilization is the inhibition of insulin sig-naling [96, 103, 104]. Insulin binds to and activates the cell-surface insulin receptor (IR) tyrosine kinase, which in turn phosphorylates the members of insulin receptor substrate (IRS) protein family [105] (Fig. 5.4). Tyrosine-phosphorylated IRS proteins associate with the IR and initiate downstream signaling events [105] (Fig. 5.4). Mice treated with GCs have reduced levels of tyrosine-phosphorylated IR and total IRS-1 proteins in skeletal muscle [96], and the activities of phosphoinositide-3-kinase (PI3K) and Akt, two signaling molecules downstream of IRS-1, are decreased [96, 106, 107] (Fig. 5.4).
Fig. 5.4.
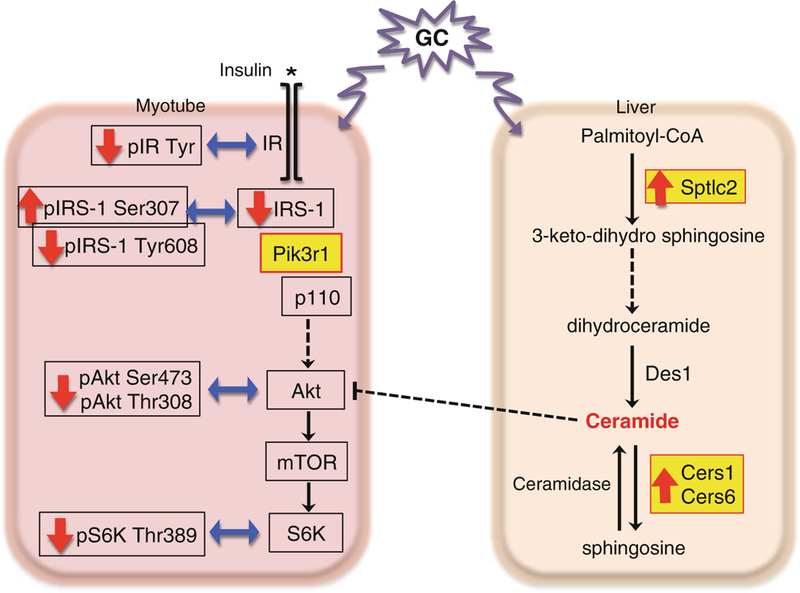
Models of glucocorticoid-regulated insulin action. Mechanisms of glucocorticoid-induced insulin resistance are depicted. In myotubes, glucocorticoids (GC) decrease the tyrosine phosphorylation of insulin receptor (IR) and the expression of IRS1. They increase the serine 307 phosphorylation while decrease the tyrosine 608 phosphorylation of IRS1. GC also increase the expression of Pik3r1, which results in decreased activity of Akt and p70 S6 kinase (S6K). In the liver, GC increase the gene expression of enzymes involved in ceramide synthesis, including Sptlc2, Cers1 and Cers6, which results in increased levels of ceramides. These ceramides then interfere with insulin signaling. The GC-regulated genes are shown in yellow
The ability of GC to inhibit glucose utilization in myotubes requires protein synthesis. A list of potential GR primary target genes that can suppress insulin action has been identified in mouse C2C12 myotubes [108]. Among these genes, the role of Pik3r1 (a.k.a. p85α) in the GC response was examined in vitro. Pik3r1 encodes the regulatory subunit of PI3K, which binds to activate IRS1 (Fig. 5.4) through its SRC homology 2 (SH2) domain to bring the catalytic subunit of PI3K, Pik3ca (a.k.a. p110), to the plasma membrane [109, 110]. Pik3ca then catalyzes the conversion of phosphatidylinositol (4,5)-bisphosphate (PIP2) to phosphatidylinositol (3,4,5)-bispho-sphate (PIP3) [109, 110]. PIP3 anchors the protein kinase Akt protein kinase family to the plasma membrane and thus initiates downstream signaling events [110]. Pik3r1 is a key component in the insulin signaling pathway. But monomeric Pik3r1 is thought to compete with the Pik3r1/Pik3ca heterodimer to interact with IRS-1 to suppress insulin action [111, 112]. In addition, Pik3r1 is required for the maximal activity of phosphatase and tensin homolog (PTEN) [113], which antagonizes PI3K activity. In C2C12 mouse myotubes, GC treatment reduces the activity of several components of the insulin signaling pathway. These GC effects are markedly decreased in C2C12 myotubes that have reduced Pik3r1 expression [108]. Thus, Pik3r1 is a potential GR primary target gene in the mediation of the suppressive effect of GC on glucose utili-zation in skeletal muscle, though this notion needs to be confirmed in vivo. Interestingly, global heterozygous deletion of Pik3r1 gene in mice has improved whole body insulin sensitivity [114, 115]. Elevated expression of PIK3R1 is found in patients with insulin resistance [116]. Notably, Pik3r1 is likely not the only GR target gene that mediating suppressive effects of GC in insulin response, and additional GR primary target genes could also participate in this process.
GC also modulate insulin sensitivity and glucose utilization through the genera-tion of specific lipid mediators. GC treatment increases ceramide levels in mouse liver and portal circulation [117]. These ceramides cause hepatic insulin resistance [118]. They are also delivered to skeletal muscle so one might expect systemic effects. In fact, hyperinsulinemic-euglycemic clamp studies in mice show that GC decrease the glucose infusion rate required to maintain euglycemia, prevent insulin-inhibited hepatic glucose output, and inhibit 2-deoxyglucose uptake into skeletal muscle, all evidence of reduced insulin sensitivity, whereas mice pretreat with myriocin, an inhibitor of serine palmitoyaltransferase (Sptlc1 and Sptlc2), an enzyme in the ceramide synthetic pathway, have reduced GC-induced insulin resistance in skeletal muscle and hepatic glucose output (Fig. 5.4) [117]. Notably, GC augment the expression of enzymes in the ceramide synthetic and metabolic pathway in liver, such as Sptlc2, ceramide synthase 1 (Cers1, a.k.a. Lass1), and ceramide synthase 6 (Cers6, a.k.a. Lass6) (Fig. 5.4) [117]. However, it is not yet clear whether the genes encoding these enzymes are primary targets of GR signaling.
In addition to increased ceramide levels in liver, GC-promoted lipolysis in WAT could impair whole body glucose homeostasis. Acipimox, an inhibitor of lipolysis in adipocytes, improves whole body glucose homeostasis in human subjects treated with GC [119]. Although it is not clear how GC-induced lipolysis affects glucose homeostasis, it is likely that the fatty acids generated from lipolysis are mobilized to skeletal muscle and liver and converted to lipid mediators, such as diacylglycerol (DAG) and ceramides, that cause insulin resistance.
Notably, Brennan-Speranza et. al. also reported that GC inhibit the expression of osteocalcin, a secreted protein from bone that reduce adiposity and hepatic steato-sis, to decrease insulin sensitivity.
GC also inhibit glucose oxidation by stimulating the expression of several mem-bers of the pyruvate dehydrogenase kinase family (PDK). PDK regulates glucose oxidation by inhibiting the pyruvate dehydrogenase complex that converts pyruvate to acetyl-CoA [120]. Among the PDK family, PDK4 is a GR primary target gene; GREs have been identified in human PDK4 and rat Pdk4 genes [121, 122]. FoxO1 binding sites have been identified near the human PDK4 gene GRE and they are required for the maximal induction of PDK4 gene transcription by GC. These FREs also mediate the inhibitory response of insulin on PDK4 gene transcription [122]. For the rat Pdk4 gene, an FRE located approximately 6 kb away from the GREs is thought to participate in both the insulin and GC responses [121]. Thus, the mecha-nisms governing the transcriptional regulation of the PDK4 gene by GC appear to be similar to GC-activated Pck1 and G6pc gene transcription discussed above.
In addition to skeletal muscle, GC reduce glucose uptake and glucose oxidation in many other tissues. GC inhibit insulin-stimulated glucose uptake in both mouse 3T3-L1 and primary adipocytes. The mechanisms governing these GC effects are mostly unknown. Overexpression of dual specificity protein phosphatase 1 (Dusp1, a.k.a. MAP kinase phosphatase 1, Mkp1), a primary GR target gene, inhibits insulin-stimulated glucose uptake in 3T3-L1 adipocytes [123]. However, the exact role of Dusp1 in GC-induced insulin resistance in adipocytes has not been established. Most reports indicate that GC inhibit glucose uptake by antagonizing the insulin response, though the direct inhibition of glucose transporter 4 (Glut4) trafficking process by GC in 3T3-L1 adipocytes has also been reported [124]. Pik3r1 expression is also increased by GC in adipocytes [125]. Thus, Pik3r1 may also participate in the GC-inhibited insulin response in adipocytes. Studies of human adipocytes found that GC inhibit insulin-stimulated glucose uptake and signaling in omental but not subcu-taneous adipocytes [126]. In fact, studies in human primary subcutaneous adipocytes show that GC pre-treatment potentiates insulin-stimulated glucose uptake [102, 127]. This suggests that GC affect insulin signaling in a depot-specific manner in humans.
The ability of GC to inhibit glucose oxidation has been linked to GC-induced apoptosis in leukemia cells. GC inhibit the expression of glucose transporter 1 (GLUT1) that results in a decreased glucose uptake into leukemia cells [128]. GC also suppress glucose uptake and oxidation in certain regions of brain, such as the hypothalamus and hippocampus [129–131]. The exact mechanisms of these GC effects on these cell types are mostly unclear.
Glycogen Metabolism
GC regulate glycogen metabolism in a tissue-specific manner. In liver the adminis-tration of GC to fasted mice increases liver glycogen content [40, 132]. GC induce the activity of glycogen synthase [7, 133, 134]. Glycogen synthase activity is regulated by post-translational modification. Protein kinase A (PKA) and glycogen synthase kinase 3 (GSK3) phosphorylates and inactivates glycogen synthase [135–137]. In contrast, protein phosphatase 1 (PP1) dephosphorylates glycogen synthase, which potentiates its activity [138, 139]. The active form of glycogen phosphory-lase, glycogen phosphorylase a, breaks down glycogen to glucose units. Glycogen phosphorylase a also inhibits the dephosphorylation of glycogen synthase by PP1. PKA can also inhibit PP1 activity. Data suggest that the activation of glycogen syn-thase phosphatase, PP1, by GC [140, 141], though the exact the mechanism of this activation is unclear.
Epinephrine plays a key role in skeletal muscle glycogenolysis. This effect is blunted in adrenalectomized rats [142]. The stimulatory effects of epinephrine on muscle glycogen phosphorylase and phosphorylase kinase are all attenuated in the skeletal muscle of adrenalectomized rats. PP1 dephosphorylates glycogen phos-phorylase and phosphorylase kinase and suppresses their activities. PP1 activity is increased in skeletal muscle of adrenalectomized rats and the ability of epinephrine to inhibit PP1 is reduced in the skeletal muscle of adrenalectomized rats. Cortisol treatment in adrenalectomized rats restores normal epinephrine effects through the activation of phosphorylase kinase and glycogen phosphorylase and the inhibition of PP1. The induction of PDK4 gene transcription by GR may contribute to GC-regulated glycogen metabolism in myotubes. One report shows that a reduction of the expression of PDK4 in human primary myotubes diminishes GC-repressed glycogen synthesis [143]. Notably, GC also inhibits insulin-stimulated glycogen synthesis and the activity of glycogen synthase [9]. These GC effects are mainly due to their ability to reduce insulin signaling in skeletal muscle. Interestingly, one report shows that glycogen storage is actually increased in soleus muscle by dexa-methasone treatment despite a decrease of glycogen synthesis [9]. The mechanisms underlying these phenotypes are not clear.
In cardiac muscle, adrenalectomy also blocks epinephrine-induced glycogenoly-sis [144]. GC treatment, however, has been shown to facilitate glycogen storage in cardiac muscle [145]. GC increase AMP-activated protein kinase (AMPK) activity that leads to the elevation of glucose uptake, the induction of glycogen synthase, and the reduction of glycogen phosphorylase. These contribute to an augmentation of glycogen content in cardiac muscle. Brain stores certain amounts of glycogen, though the levels are lower than in skeletal muscle or liver. Astrocytes store most of the brain glycogen, whose levels are increased by norepinephrine. In primary astrocyte culture norepinephrine induces glycogen synthesis, an effect which is suppressed by GC [146].
GC Effects on Pancreas
As an endocrine organ, the pancreas secretes several hormones that include insulin (from β cells in the islets of Langerhans), glucagon (from α cells), and somatostatin (from δ cells). Insulin secreted during the fed state promotes glucose uptake and utilization, and inhibits gluconeogenesis. In contrast, glucagon secreted during fasting stimulates gluconeogenesis and glycogenolysis. Somatostatin suppresses both insulin and glucagon secretion.
The effect of GC on insulin secretion is an area of vigorous research; however, the direct mechanistic relationship between GC and β cell function in vivo remains elu-sive. This is largely due to the difficulty of separating the indirect effects of GC-induced peripheral insulin resistance from the direct actions of GC on pancreatic cells. Hypercortisolism induces insulin resistance in tissues such as liver, adipose and skeletal muscle, as discussed above. In murine models and human studies, this GC-induced insulin resistance can lead to compensatory β cell hyperplasia and hyperinsulinemia, and consequent normoglycemia. However, long-term GC treat-ment that exceeds the β cell compensatory capacity leads to insufficient insulin secre-tion, hyperglycemia, and eventually Type 2 diabetes [15, 17, 147–150]. Overall, the effects of GC on β cell function are dependent on the length and dosage of the treat-ment, the experimental animal model under investigation, as well as the susceptibil-ity of the strain exposed [151]. It is important to note that short-term effects of GC on cells are likely reversible [152]. To begin to dissect the direct in vivo effects of GC on β cells, a mouse model of insulin promoter-driven GR overexpression was gener-ated [14]. These mice show glucose intolerance due to blunted insulin secretion.
Insulin secretion is mainly triggered by a postprandial increase in the concentra-tion of plasma glucose, which enters pancreatic β cells through GLUT2 transport-ers. Glucokinase phosphorylates glucose to produce glucose-6-phosphate, which enters the glycolytic cycle. Through mitochondrial β-oxidation, the ratio of ATP to ADP increases, which leads to the closure of ATP-sensitive potassium channels and plasma membrane depolarization. The increase in cellular electrical conductivity drives the opening of voltage-sensitive calcium ion channels. The consequent influx of calcium ions eventually leads to exocytosis of insulin-containing granules.
Compared to islets isolated from GC-treated animals, isolated islets directly treated with GC present opposite results. First, synthetic GC dexamethasone pro-motes posttranslational degradation of GLUT2 and decreases glucose-stimulated insulin secretion (GSIS) in isolated rat pancreatic islets [153]. Second, dexametha-sone inhibits glucokinase mRNA expression in RIN cells, a rat pancreatic islet tumor cell line [154]. GC have also been reported to suppress the gene expression of insulin [155] and pancreatic and duodenal homeobox 1 (Pdx1) in β cells [156]. Pdx1 is a transcription factor essential for pancreatic development and β cell matu-ration. Third, in ob/ob mice, GC enhance the activity of glucose-6-phosphatase, which dephosphorylates glucose-6-phosphate to produce glucose, resulting in a futile cycle [157]. Fourth, in the INS-1 rat pancreatic β cell line, the transcription of voltage-gated potassium channel Kv1.5 is upregulated by dexamethasone treatment [158]. This results in an increase in the repolarizing outward current and reduces the influx of calcium ions, which compromises GSIS. On the contrary, islets purified from GC-treated rats display enhanced insulin secretion, likely due to compensa-tory hyperinsulinemia [159–161], and have improved glucose sensitivity and oxidative metabolism [162]. Furthermore, transmission electron microscopy shows an increased amount of docked secretory granules in GC-treated β cells [148].
GC also regulates the expression of genes that modulate insulin secretion. The blunting of GSIS by dexamethasone is significantly restored in islets isolated from serum and glucocorticoid-induced kinase (Sgk1) knockout mice compared to wild type [158]. Sgk1 is a well-established GR primary target gene. Moreover, GC treat-ment of MIN6 mouse insulinoma cells and mouse islets augments the expression of corticotropin-releasing factor receptor type 2α (CRFR2α) and inhibits the expression of CRFR type 1 (CRFR1) [163]. CRFR1 potentiates both glucose-induced insulin secretion in vitro and in vivo and the proliferation of neonatal rat β cells [164].
GC can stimulate β cell proliferation in vivo; however, GC exposure could also have cytotoxic effects on mouse isolated pancreatic islets of Langerhans and MIN6 cells [165]. Interestingly, increased intracellular cAMP levels have previously been shown to attenuate dexamethasone-induced β cell death using exendin-4 [20] and forskolin [166]. Co-treatment with RU486, an antagonist of GC, abolished these GC effects. Mitogen-activated protein kinases (MAPK) such as p38 MAPK and JNK regulate apoptosis. In MIN6 cells, inhibition of p38 MAPK reduces glucocorticoid-induced apoptosis [167]. This may be due to decreased phosphoryla-tion of mouse GR at serine residue 220 by p38 MAPK. Phosphorylation of GR at serine 220 is positively associated with GR transcriptional activity [168, 169]. Moreover, in isolated islets, inhibition of p38 MAPK decreases glucocorticoid-induced formation of cleaved caspase 3, which plays a key role in the execution phase of apoptosis [167]. Thus, p38 MAPK is required for glucocorticoid-induced cytotoxicity. In contrast, JNK signaling dampens glucocorticoid-induced cytotoxic-ity, as inhibition of JNK potentiates the cytotoxic effect of GC in MIN6 cells [167]. A plausible explanation is that JNK is responsible for phosphorylating GR at serine 234, which is negatively associated with the transcriptional activity of GR [170].
Reactive oxygen species (ROS) may also mediate glucocorticoid-induced cyto-toxicity [171]. Thioredoxin-interacting protein (Txnip) could exert its pro-apoptotic effect by blocking the activity of thioredoxin, which is involved in a major pro-survival thiol-reducing pathway in cells. In db/db mice, overexpression of thiore-doxin suppresses the progression of hyperglycemia, likely through the prevention of the reduction of Pdx1 and V-maf musculoaponeurotic fibrosarcoma oncogene homologue A (MafA) transcription factors [172]. GC induces Txnip expression in human and mouse islets, and in MIN6 cells [165]. Overexpressing Txnip mimicked pro-apoptotic effect of GC, while knocking down Txnip partially rescued this phe-notype in MIN6 cells. Furthermore, thioredoxin overexpression protected MIN6 cells from GC-induced cytotoxicity [165]. Interestingly, GC-induced Txnip is dependent on the presence of p38 MAPK. Studies in leukemia and other cell types have shown that Txnip is a primary GR target gene [173].
In addition to insulin secretion, GC has been shown to modulate glucagon secre-tion. GC treatment causes both hyperinsulinemia and hyperglucagonemia. Rodents treated with GC have unaltered insulin/glucagon ratio from the fasted state to the fed state. They tend to have increased α cell mass, and show impaired high glucose-suppressed glucagon secretion. This hyperglucagonemia leads to hyperglycemia through the activation of hepatic glycogenolysis and gluconeogenesis [174]. Notably, co-treatment of GC and a glucagon receptor antagonist on rats resulted in normoglycemia [11]. The mechanisms underlying the effects of GC on glucagon secretion, however, are unclear.
Conclusion and Future Directions
The physiological responses of GC that modulate glucose homeostasis have been well documented. Significant progress has been made to understand these GC actions in the last three decades. However, the precise manner by which GC regu-lates glucose homeostasis is still unclear. The key to understanding the mechanisms of GC-regulated glucose homeostasis is the identification of GR primary target genes that mediate GC actions, and understanding how these primary target genes are transcriptionally regulated by GR in various processes, including gluconeogen-esis, glucose uptake and utilization, glycogen metabolism, and pancreatic endocrine secretions. For transcriptional regulation, GC-activated Pck1 gene transcription has received the most attention. Nevertheless, further studies are needed to understand how different accessory factors coordinate with GR and transcriptional coregulators to activate Pck1 gene transcription. Analyzing the transcriptional regulatory mecha-nisms of these GC-regulated gluconeogenic genes should allow the identification of common molecular features in GR-stimulated gluconeogenesis. In vivo, GC actions are affected by other hormones and environmental cues. Understanding the cross-talk and integration of these signals should be the focus of future work.
The emergence of advanced techniques and technologies during the last decade have facilitated the characterization of molecular features of GC-regulated glucose homeostasis. Using high throughput DNA sequencing technology, genomic GR binding regions, transcriptional coregulator occupancy sites, and global chromatin structure changes were identified. The discovery of GC-regulated non-coding RNA and enhancer RNA, using genomic run-on sequencing (GRO-seq) and RNA sequencing (RNA-seq), unveiled another layer of glucose homeostasis regulation [175–178]. Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) genome editing technology can now be used to efficiently ablate a GR primary tar-get gene, and modify a particular residue of GRE to study its role in the natural chromosomal context [179–183]. These new approaches in combination with refined genetic, physiological and biochemical tools will play a critical role in improving our understanding of GC-regulated biology in metabolic tissues.
Ultimately, the goal of the studying the basic mechanisms of GC-regulated glu-cose homeostasis is to develop a pharmacotherapy targeting GC signaling for treat-ing metabolic diseases. Reducing GC signaling in vivo improves insulin sensitivity and decreases plasma glucose levels, which would largely benefit type 2 diabetes patients. However, reducing GC actions globally is not ideal. For example, the body’s inflammatory status would be elevated with reduced GC signaling. In fact, as inflammation promotes insulin resistance, the anti-inflammatory activity of GR could also improve insulin sensitivity. Overall, identifying approaches to dissociate metabolic and anti-inflammatory actions of GR could provide novel ways to treat metabolic disease.
Acknowledgment
We thank Drs. Daryl Granner and Richard O’Brien for their insightful comments and suggestions for this chapter. The Wang laboratory is supported by NIH DK83591.
Contributor Information
Taiyi Kuo, Department of Nutritional Sciences and Toxicology, University of California Berkeley, 212 Morgan Hall, Berkeley, CA 94720, USA
Allison McQueen, Department of Nutritional Sciences and Toxicology, University of California Berkeley, 212 Morgan Hall, Berkeley, CA 94720, USA
Tzu-Chieh Chen, Department of Nutritional Sciences and Toxicology, University of California Berkeley, 212 Morgan Hall, Berkeley, CA 94720, USA
Jen-Chywan Wang, Department of Nutritional Sciences and Toxicology, University of California Berkeley, 119 Morgan Hall, Berkeley, CA 94720-3104, USA
References
- 1.Exton JH. Regulation of gluconeogenesis by glucocorticoids. Monogr Endocrinol 1979;12: 535–46. [DOI] [PubMed] [Google Scholar]
- 2.Kraus-Friedmann N Hormonal regulation of hepatic gluconeogenesis. Physiol Rev 1984;64:170–259. [DOI] [PubMed] [Google Scholar]
- 3.Di Dalmazi G, Pagotto U, Pasquali R, Vicennati V. Glucocorticoids and type 2 diabetes: from physiology to pathology. J Nutr Metab 2012;2012:525093 10.1155/2012/525093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kuo T, Harris CA, Wang JC. Metabolic functions of glucocorticoid receptor in skeletal muscle. Mol Cell Endocrinol 2013;380:79–88. 10.1016/j.mce.2013.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Charmandari E, Tsigos C, Chrousos G. Endocrinology of the stress response. Annu Rev Physiol 2005;67:259–84. 10.1146/annurev.physiol.67.040403.120816. [DOI] [PubMed] [Google Scholar]
- 6.Andrews RC, Walker BR. Glucocorticoids and insulin resistance: old hormones, new targets. Clin Sci (Lond) 1999;96:513–23. [DOI] [PubMed] [Google Scholar]
- 7.Stalmans W, Laloux M. Glucocorticoids and hepatic glycogen metabolism. Monogr Endocrinol 1979;12:517–33. [DOI] [PubMed] [Google Scholar]
- 8.Exton JH, Friedmann N, Wong EH, Brineaux JP, Corbin JD, Park CR. Interaction of gluco-corticoids with glucagon and epinephrine in the control of gluconeogenesis and glycogenolysis in liver and of lipolysis in adipose tissue. J Biol Chem 1972;247:3579–88. [PubMed] [Google Scholar]
- 9.Ruzzin J, Wagman AS, Jensen J. Glucocorticoid-induced insulin resistance in skeletal muscles: defects in insulin signalling and the effects of a selective glycogen synthase kinase-3 inhibitor. Diabetologia 2005;48:2119–30. 10.1007/s00125-005-1886-0. [DOI] [PubMed] [Google Scholar]
- 10.Wise JK, Hendler R, Felig P. Influence of glucocorticoids on glucagon secretion and plasma amino acid concentrations in man. J Clin Invest 1973;52:2774–82. 10.1172/JCI107473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rafacho A, Goncalves-Neto LM, Santos-Silva JC, Alonso-Magdalena P, Merino B, Taboga SR, Carneiro EM, Boschero AC, Nadal A, Quesada I. Pancreatic alphacell dysfunction con-tributes to the disruption of glucose homeostasis and compensatory insulin hypersecretion in glucocorticoid-treated rats. PLoS One 2014;9:e93531 10.1371/journal.pone.0093531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Beaudry JL, Riddell MC. Effects of glucocorticoids and exercise on pancreatic betacell function and diabetes development. Diabetes Metab Res Rev 2012;28:560–73. 10.1002/dmrr.2310. [DOI] [PubMed] [Google Scholar]
- 13.Longano CA, Fletcher HP. Insulin release after acute hydrocortisone treatment in mice. Metabolism 1983;32:603–8. [DOI] [PubMed] [Google Scholar]
- 14.Delaunay F, Khan A, Cintra A, Davani B, Ling ZC, Andersson A, Ostenson CG, Gustafsson J, Efendic S, Okret S. Pancreatic beta cells are important targets for the diabetogenic effects of glucocorticoids. J Clin Invest 1997;100:2094–8. 10.1172/JCI119743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ogawa A, Johnson JH, Ohneda M, McAllister CT, Inman L, Alam T, Unger RH. Roles of insulin resistance and beta-cell dysfunction in dexamethasone induced diabetes. J Clin Invest 1992;90:497–504. 10.1172/JCI115886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dinneen S, Alzaid A, Miles J, Rizza R. Metabolic effects of the nocturnal rise in cortisol on carbohydrate metabolism in normal humans. J Clin Invest 1993;92:2283–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rafacho A, Cestari TM, Taboga SR, Boschero AC, Bosqueiro JR. High doses of dexametha-sone induce increased beta-cell proliferation in pancreatic rat islets. Am J Physiol Endocrinol Metab 2009;296:E681–9. 10.1152/ajpendo.90931.2008. [DOI] [PubMed] [Google Scholar]
- 18.Morisset J, Jolicoeur L. Effect of hydrocortisone on pancreatic growth in rats. Am J Physiol 1980;239:G95–8. [DOI] [PubMed] [Google Scholar]
- 19.Like AA, Chick WL. Pancreatic beta cell replication induced by glucocorticoids in subhuman primates. Am J Pathol 1974;75:329–48. [PMC free article] [PubMed] [Google Scholar]
- 20.Ranta F, Avram D, Berchtold S, Dufer M, Drews G, Lang F, Ullrich S. Dexamethasone induces cell death in insulin-secreting cells, an effect reversed by exendin-4. Diabetes 2006;55:1380–90. [DOI] [PubMed] [Google Scholar]
- 21.Weinhaus AJ, Bhagroo NV, Brelje TC, Sorenson RL. Dexamethasone counteracts the effect of prolactin on islet function: implications for islet regulation in late pregnancy. Endocrinology 2000;141:1384–93. 10.1210/endo.141.4.7409. [DOI] [PubMed] [Google Scholar]
- 22.Pilkis SJ, el-Maghrabi MR, Claus TH. Hormonal regulation of hepatic gluconeogenesis and glycolysis. Annu Rev Biochem 1988;57:755–83. 10.1146/annurev.bi.57.070188.003543. [DOI] [PubMed] [Google Scholar]
- 23.Pilkis SJ, Granner DK. Molecular physiology of the regulation of hepatic gluconeogenesis and glycolysis. Annu Rev Physiol 1992;54:885–909. 10.1146/annurev.ph.54.030192.004321. [DOI] [PubMed] [Google Scholar]
- 24.Jitrapakdee S, Wallace JC. Structure, function and regulation of pyruvate carboxylase. Biochem J 1999;340(Pt 1):1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Menefee AL, Zeczycki TN. Nearly 50 years in the making: defining the catalytic mechanism of the multifunctional enzyme, pyruvate carboxylase. FEBS J 2014;281:1333–54. 10.1111/febs.12713. [DOI] [PubMed] [Google Scholar]
- 26.Hanson RW, Garber AJ. Phosphoenolpyruvate carboxykinase. I. Its role in gluconeogenesis. Am J Clin Nutr 1972;25:1010–21. [DOI] [PubMed] [Google Scholar]
- 27.Hanson RW, Patel YM. Phosphoenolpyruvate carboxykinase (GTP): the gene and the enzyme. Adv Enzymol Relat Areas Mol Biol 1994;69:203–81. [DOI] [PubMed] [Google Scholar]
- 28.Mendez-Lucas A, Duarte JA, Sunny NE, Satapati S, He T, Fu X, Bermudez J, Burgess SC, Perales JC. PEPCK-M expression in mouse liver potentiates, not replaces, PEPCK-C medi-ated gluconeogenesis. J Hepatol 2013;59:105–13. 10.1016/j.jhep.2013.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stark R, Guebre-Egziabher F, Zhao X, Feriod C, Dong J, Alves TC, Ioja S, Pongratz RL, Bhanot S, Roden M, Cline GW, Shulman GI, Kibbey RG. A role for mitochondrial phospho-enolpyruvate carboxykinase (PEPCK-M) in the regulation of hepatic gluconeogenesis. J Biol Chem 2014;289:7257–63. 10.1074/jbc.C113.544759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Arinze IJ, Garber AJ, Hanson RW. The regulation of gluconeogenesis in mammalian liver. The role of mitochondrial phosphoenolpyruvate carboxykinase. J Biol Chem 1973;248:2266–74. [PubMed] [Google Scholar]
- 31.Cheng SC, Cheng RH. A mitochondrial phosphoenolpyruvate carboxykinase from rat brain. Arch Biochem Biophys 1972;151:501–11. [DOI] [PubMed] [Google Scholar]
- 32.Rider MH, Bertrand L, Vertommen D, Michels PA, Rousseau GG, Hue L. 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase: head-to-head with a bifunctional enzyme that controls glycolysis. Biochem J 2004;381:561–79. 10.1042/BJ20040752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rousseau GG, Hue L. Mammalian 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase: a bifunctional enzyme that controls glycolysis. Prog Nucleic Acid Res Mol Biol 1993;45: 99–127. [DOI] [PubMed] [Google Scholar]
- 34.Marcus F, Rittenhouse J, Gontero B, Harrsch PB. Function, structure and evolution of fructose-1,6-bisphosphatase. Arch Biol Med Exp 1987;20:371–8. [PubMed] [Google Scholar]
- 35.Burchell A, Waddell ID. The molecular basis of the hepatic microsomal glucose-6-phosphatase system. Biochim Biophys Acta 1991;1092:129–37. [DOI] [PubMed] [Google Scholar]
- 36.Hutton JC, O’Brien RM. Glucose-6-phosphatase catalytic subunit gene family. J Biol Chem 2009;284:29241–5. 10.1074/jbc.R109.025544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.van Schaftingen E, Gerin I. The glucose-6-phosphatase system. Biochem J 2002;362:513–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schneiter P, Tappy L. Kinetics of dexamethasone-induced alterations of glucose metabolism in healthy humans. Am J Physiol 1998;275:E806–13. [DOI] [PubMed] [Google Scholar]
- 39.Tounian P, Schneiter P, Henry S, Delarue J, Tappy L. Effects of dexamethasone on hepatic glucose production and fructose metabolism in healthy humans. Am J Physiol 1997;273:E315–20. [DOI] [PubMed] [Google Scholar]
- 40.Exton JH, Miller TB, Harper SC, Park CR. Carbohydrate metabolism in perfused livers of adrenalectomized and steroid-replaced rats. Am J Physiol 1976;230:163–70. [DOI] [PubMed] [Google Scholar]
- 41.Exton JH, Park CR. Control of gluconeogenesis in the perfused liver of normal and adrenal-ectomized rats. J Biol Chem 1965;240:955–7. [PubMed] [Google Scholar]
- 42.Sistare FD, Haynes RC Jr. Acute stimulation by glucocorticoids of gluconeogenesis from lactate/pyruvate in isolated hepatocytes from normal and adrenalectomized rats. J Biol Chem 1985;260:12754–60. [PubMed] [Google Scholar]
- 43.Exton JH, Mallette LE, Jefferson LS, Wong EH, Friedmann N, Miller TB Jr, Park CR. The hormonal control of hepatic gluconeogenesis. Recent Prog Horm Res 1970;26:411–61. [DOI] [PubMed] [Google Scholar]
- 44.Menon RK, Sperling MA. Carbohydrate metabolism. Semin Perinatol 1988;12:157–62. [PubMed] [Google Scholar]
- 45.Imai E, Stromstedt PE, Quinn PG, Carlstedt-Duke J, Gustafsson JA, Granner DK. Characterization of a complex glucocorticoid response unit in the phosphoenolpyruvate carboxykinase gene. Mol Cell Biol 1990;10:4712–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Scott DK, Stromstedt PE, Wang JC, Granner DK. Further characterization of the glucocorti-coid response unit in the phosphoenolpyruvate carboxykinase gene. The role of the glucocor-ticoid receptor-binding sites. Mol Endocrinol 1998;12:482–91. [DOI] [PubMed] [Google Scholar]
- 47.Scott DK, Mitchell JA, Granner DK. The orphan receptor COUP-TF binds to a third gluco-corticoid accessory factor element within the phosphoenolpyruvate carboxykinase gene pro-moter. J Biol Chem 1996;271:31909–14. [DOI] [PubMed] [Google Scholar]
- 48.Imai E, Miner JN, Mitchell JA, Yamamoto KR, Granner DK. Glucocorticoid receptor-cAMP response element-binding protein interaction and the response of the phosphoenolpyruvate carboxykinase gene to glucocorticoids. J Biol Chem 1993;268:5353–6. [PubMed] [Google Scholar]
- 49.Hall RK, Sladek FM, Granner DK. The orphan receptors COUP-TF and HNF-4 serve as accessory factors required for induction of phosphoenolpyruvate carboxykinase gene tran-scription by glucocorticoids. Proc Natl Acad Sci U S A 1995;92:412–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hall RK, Scott DK, Noisin EL, Lucas PC, Granner DK. Activation of the phosphoenolpyru-vate carboxykinase gene retinoic acid response element is dependent on a retinoic acid recep-tor/coregulator complex. Mol Cell Biol 1992;12:5527–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang XL, Herzog B, Waltner-Law M, Hall RK, Shiota M, Granner DK. The synergistic effect of dexamethasone and all-trans-retinoic acid on hepatic phosphoenolpyruvate car-boxykinase gene expression involves the coactivator p300. J Biol Chem 2004;279:34191– 200. 10.1074/jbc.M403455200. [DOI] [PubMed] [Google Scholar]
- 52.Wang JC, Stromstedt PE, O’Brien RM, Granner DK. Hepatic nuclear factor 3 is an accessory factor required for the stimulation of phosphoenolpyruvate carboxykinase gene transcription by glucocorticoids. Mol Endocrinol 1996;10:794–800. [DOI] [PubMed] [Google Scholar]
- 53.Zhang K, Li L, Qi Y, Zhu X, Gan B, DePinho RA, Averitt T, Guo S. Hepatic suppression of Foxo1 and Foxo3 causes hypoglycemia and hyperlipidemia in mice. Endocrinology 2012;153:631–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.O’Brien RM, Lucas PC, Forest CD, Magnuson MA, Granner DK. Identification of a sequence in the PEPCK gene that mediates a negative effect of insulin on transcription. Science 1990;249:533–7. [DOI] [PubMed] [Google Scholar]
- 55.Forest CD, O’Brien RM, Lucas PC, Magnuson MA, Granner DK. Regulation of phospho-enolpyruvate carboxykinase gene expression by insulin. Use of the stable transfection approach to locate an insulin responsive sequence. Mol Endocrinol 1990;4:1302–10. 10.1210/mend-4-9-1302. [DOI] [PubMed] [Google Scholar]
- 56.Nakae J, Kitamura T, Silver DL, Accili D. The forkhead transcription factor Foxo1 (Fkhr) confers insulin sensitivity onto glucose-6-phosphatase expression. J Clin Invest 2001;108: 1359–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wolfrum C, Asilmaz E, Luca E, Friedman JM, Stoffel M. Foxa2 regulates lipid metabolism and ketogenesis in the liver during fasting and in diabetes. Nature 2004;432:1027–32. 10.1038/nature03047. [DOI] [PubMed] [Google Scholar]
- 58.Lechner PS, Croniger CM, Hakimi P, Millward C, Fekter C, Yun JS, Hanson RW. The use of transgenic mice to analyze the role of accessory factor two in the regulation of phosphoenolpyruvate carboxykinase (GTP) gene transcription during diabetes. J Biol Chem 2001;276:22675–9. 10.1074/jbc.M102422200. [DOI] [PubMed] [Google Scholar]
- 59.Scott DK, Mitchell JA, Granner DK. Identification and characterization of the second retinoic acid response element in the phosphoenolpyruvate carboxykinase gene promoter. J Biol Chem 1996;271:6260–4. [DOI] [PubMed] [Google Scholar]
- 60.Yamada K, Duong DT, Scott DK, Wang JC, Granner DK. CCAAT/enhancer-binding protein beta is an accessory factor for the glucocorticoid response from the cAMP response element in the rat phosphoenolpyruvate carboxykinase gene promoter. J Biol Chem 1999;274:5880–7. [DOI] [PubMed] [Google Scholar]
- 61.Hall RK, Wang XL, George L, Koch SR, Granner DK. Insulin represses phosphoenolpyru-vate carboxykinase gene transcription by causing the rapid disruption of an active transcrip-tion complex: a potential epigenetic effect. Mol Endocrinol 2007;21:550–63. 10.1210/me.2006-0307. [DOI] [PubMed] [Google Scholar]
- 62.Schurter BT, Koh SS, Chen D, Bunick GJ, Harp JM, Hanson BL, Henschen-Edman A, Mackay DR, Stallcup MR, Aswad DW. Methylation of histone H3 by coactivator-associated arginine methyltransferase 1. Biochemistry 2001;40:5747–56. [DOI] [PubMed] [Google Scholar]
- 63.Lee DY, Teyssier C, Strahl BD, Stallcup MR. Role of protein methylation in regulation of transcription. Endocr Rev 2005;26:147–70. [DOI] [PubMed] [Google Scholar]
- 64.Feng Q, He B, Jung SY, Song Y, Qin J, Tsai SY, Tsai MJ, O’Malley BW. Biochemical control of CARM1 enzymatic activity by phosphorylation. J Biol Chem 2009;284:36167–74. 10.1074/jbc.M109.065524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cassuto H, Kochan K, Chakravarty K, Cohen H, Blum B, Olswang Y, Hakimi P, Xu C, Massillon D, Hanson RW, Reshef L. Glucocorticoids regulate transcription of the gene for phosphoenolpyruvate carboxykinase in the liver via an extended glucocorticoid regulatory unit. J Biol Chem 2005;280:33873–84. 10.1074/jbc.M504119200. [DOI] [PubMed] [Google Scholar]
- 66.Bernal-Mizrachi C, Weng S, Feng C, Finck BN, Knutsen RH, Leone TC, Coleman T, Mecham RP, Kelly DP, Semenkovich CF. Dexamethasone induction of hypertension and diabetes is PPAR-alpha dependent in LDL receptor-null mice. Nat Med 2003;9:1069–75. [DOI] [PubMed] [Google Scholar]
- 67.Beale EG, Forest C, Hammer RE. Regulation of cytosolic phosphoenolpyruvate carboxyki-nase gene expression in adipocytes. Biochimie 2003;85:1207–11. [DOI] [PubMed] [Google Scholar]
- 68.Olswang Y, Blum B, Cassuto H, Cohen H, Biberman Y, Hanson RW, Reshef L. Glucocorticoids repress transcription of phosphoenolpyruvate carboxykinase (GTP) gene in adipocytes by inhibiting its C/EBP-mediated activation. J Biol Chem 2003;278:12929–36. 10.1074/jbc.M300263200. [DOI] [PubMed] [Google Scholar]
- 69.Hanson RW, Reshef L. Glyceroneogenesis revisited. Biochimie 2003;85:1199–205. [DOI] [PubMed] [Google Scholar]
- 70.Cadoudal T, Leroyer S, Reis AF, Tordjman J, Durant S, Fouque F, Collinet M, Quette J, Chauvet G, Beale E, Velho G, Antoine B, Benelli C, Forest C. Proposed involvement of adi-pocyte glyceroneogenesis and phosphoenolpyruvate carboxykinase in the metabolic syn-drome. Biochimie 2005;87:27–32. 10.1016/j.biochi.2004.12.005. [DOI] [PubMed] [Google Scholar]
- 71.Wang JC, Stromstedt PE, Sugiyama T, Granner DK. The phosphoenolpyruvate carboxyki-nase gene glucocorticoid response unit: identification of the functional domains of accessory factors HNF3 beta (hepatic nuclear factor-3 beta) and HNF4 and the necessity of proper alignment of their cognate binding sites. Mol Endocrinol 1999;13:604–18. [DOI] [PubMed] [Google Scholar]
- 72.Stafford JM, Waltner-Law M, Granner DK. Role of accessory factors and steroid receptor coactivator 1 in the regulation of phosphoenolpyruvate carboxykinase gene transcription by glucocorticoids. J Biol Chem 2001;276:3811–9. 10.1074/jbc.M009389200. [DOI] [PubMed] [Google Scholar]
- 73.Puigserver P, Rhee J, Donovan J, Walkey CJ, Yoon JC, Oriente F, Kitamura Y, Altomonte J, Dong H, Accili D, Spiegelman BM. Insulin-regulated hepatic gluconeogenesis through FOXO1-PGC-1alpha interaction. Nature 2003;423:550–5. 10.1038/nature01667. [DOI] [PubMed] [Google Scholar]
- 74.Stafford JM, Wilkinson JC, Beechem JM, Granner DK. Accessory factors facilitate the binding of glucocorticoid receptor to the phosphoenolpyruvate carboxykinase gene promoter. J Biol Chem 2001;276:39885–91. [DOI] [PubMed] [Google Scholar]
- 75.Vander Kooi BT, Onuma H, Oeser JK, Svitek CA, Allen SR, Vander Kooi CW, Chazin WJ, O’Brien RM. The glucose-6-phosphatase catalytic subunit gene promoter contains both posi-tive and negative glucocorticoid response elements. Mol Endocrinol 2005;19:3001–22. [DOI] [PubMed] [Google Scholar]
- 76.Onuma H, Vander Kooi BT, Boustead JN, Oeser JK, O’Brien RM. Correlation between FOXO1a (FKHR) and FOXO3a (FKHRL1) binding and the inhibition of basal glucose-6-phosphatase catalytic subunit gene transcription by insulin. Mol Endocrinol 2006;20:2831– 47. 10.1210/me.2006-0085. [DOI] [PubMed] [Google Scholar]
- 77.Yoon JC, Puigserver P, Chen G, Donovan J, Wu Z, Rhee J, Adelmant G, Stafford J, Kahn CR, Granner DK, Newgard CB, Spiegelman BM. Control of hepatic gluconeogenesis through the transcriptional coactivator PGC-1. Nature 2001;413:131–8. [DOI] [PubMed] [Google Scholar]
- 78.Boustead JN, Stadelmaier BT, Eeds AM, Wiebe PO, Svitek CA, Oeser JK, O’Brien RM. Hepatocyte nuclear factor-4 alpha mediates the stimulatory effect of peroxisome proliferator-activated receptor gamma co-activator-1 alpha (PGC-1 alpha) on glucose-6-phosphatase catalytic subunit gene transcription in H4IIE cells. Biochem J 2003;369:17–22. 10.1042/BJ20021382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Matsumoto M, Pocai A, Rossetti L, Depinho RA, Accili D. Impaired regulation of hepatic glucose production in mice lacking the forkhead transcription factor Foxo1 in liver. Cell Metab 2007;6:208–16. [DOI] [PubMed] [Google Scholar]
- 80.Hiraiwa H, Chou JY. Glucocorticoids activate transcription of the gene for the glucose-6-phosphate transporter, deficient in glycogen storage disease type 1b. DNA Cell Biol 2001;20:447–53. 10.1089/104454901316976073. [DOI] [PubMed] [Google Scholar]
- 81.Kallwellis-Opara A, Zaho X, Zimmermann U, Unterman TG, Walther R, Schmoll D. Characterization of cis-elements mediating the stimulation of glucose-6-phosphate trans-porter promoter activity by glucocorticoids. Gene 2003;320:59–66. [DOI] [PubMed] [Google Scholar]
- 82.Pierreux CE, Urso B, De Meyts P, Rousseau GG, Lemaigre FP. Inhibition by insulin of glucocorticoid-induced gene transcription: involvement of the ligand-binding domain of the glucocorticoid receptor and independence from the phosphatidylinositol 3-kinase and mitogen-activated protein kinase pathways. Mol Endocrinol 1998;12:1343–54. 10.1210/mend.12.9.0172. [DOI] [PubMed] [Google Scholar]
- 83.Lemaigre FP, Lause P, Rousseau GG. Insulin inhibits glucocorticoid-induced stimulation of liver 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase gene transcription. FEBS Lett 1994;340:221–5. [DOI] [PubMed] [Google Scholar]
- 84.Sutherland C, O’Brien RM, Granner DK. Phosphatidylinositol 3-kinase, but not p70/p85 ribo-somal S6 protein kinase, is required for the regulation of phosphoenolpyruvate carboxykinase (PEPCK) gene expression by insulin. Dissociation of signaling pathways for insulin and phor-bol ester regulation of PEPCK gene expression. J Biol Chem 1995;270:15501–6. [DOI] [PubMed] [Google Scholar]
- 85.Liao J, Barthel A, Nakatani K, Roth RA. Activation of protein kinase B/Akt is sufficient to repress the glucocorticoid and cAMP induction of phosphoenolpyruvate carboxykinase gene. J Biol Chem 1998;273:27320–4. [DOI] [PubMed] [Google Scholar]
- 86.De Los Pinos E, Fernandez De Mattos S, Joaquin M, Tauler A. Insulin inhibits glucocorticoid-stimulated L-type 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase gene expression by activation of the c-Jun N-terminal kinase pathway. Biochem J 2001;353:267–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Patel R, Patel M, Tsai R, Lin V, Bookout AL, Zhang Y, Magomedova L, Li T, Chan JF, Budd C, Mangelsdorf DJ, Cummins CL. LXRbeta is required for glucocorticoid-induced hypergly-cemia and hepatosteatosis in mice. J Clin Invest 2010;121:431–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Nader N, Ng SS, Wang Y, Abel BS, Chrousos GP, Kino T. Liver X receptors regulate the transcriptional activity of the glucocorticoid receptor: implications for the carbohydrate metabolism. PLoS One 2012;7:e26751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Lu Y, Xiong X, Wang X, Zhang Z, Li J, Shi G, Yang J, Zhang H, Ning G, Li X. Yin Yang 1 promotes hepatic gluconeogenesis through upregulation of glucocorticoid receptor. Diabetes 2013;62:1064–73. 10.2337/db12-0744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Renga B, Mencarelli A, D’Amore C, Cipriani S, Baldelli F, Zampella A, Distrutti E, Fiorucci S. Glucocorticoid receptor mediates the gluconeogenic activity of the farnesoid X receptor in the fasting condition. FASEB J 2012;26:3021–31. 10.1096/fj.11-195701. [DOI] [PubMed] [Google Scholar]
- 91.Molusky MM, Li S, Ma D, Yu L, Lin JD. Ubiquitin-specific protease 2 regulates hepatic gluconeogenesis and diurnal glucose metabolism through 11beta-hydroxysteroid dehydroge-nase 1. Diabetes 2012;61:1025–35. 10.2337/db11-0970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ichijo T, Voutetakis A, Cotrim AP, Bhattachryya N, Fujii M, Chrousos GP, Kino T. The Smad6-histone deacetylase 3 complex silences the transcriptional activity of the glucocorti-coid receptor: potential clinical implications. J Biol Chem 2005;280:42067–77. 10.1074/jbc.M509338200. [DOI] [PubMed] [Google Scholar]
- 93.Winkler R, Benz V, Clemenz M, Bloch M, Foryst-Ludwig A, Wardat S, Witte N, Trappiel M, Namsolleck P, Mai K, Spranger J, Matthias G, Roloff T, Truee O, Kappert K, Schupp M, Matthias P, Kintscher U. Histone deacetylase 6 (HDAC6) is an essential modifier of glucocorticoid-induced hepatic gluconeogenesis. Diabetes 2012;61:513–23. 10.2337/db11-0313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ferrannini E, Simonson DC, Katz LD, Reichard G Jr, Bevilacqua S, Barrett EJ, Olsson M, DeFronzo RA. The disposal of an oral glucose load in patients with non-insulin-dependent diabetes. Metabolism 1988;37:79–85. [DOI] [PubMed] [Google Scholar]
- 95.DeFronzo RA, Tripathy D. Skeletal muscle insulin resistance is the primary defect in type 2 diabetes. Diabetes Care 2009;32 Suppl 2:S157–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Morgan SA, Sherlock M, Gathercole LL, Lavery GG, Lenaghan C, Bujalska IJ, Laber D, Yu A, Convey G, Mayers R, Hegyi K, Sethi JK, Stewart PM, Smith DM, Tomlinson JW. 11beta-hydroxysteroid dehydrogenase type 1 regulates glucocorticoid-induced insulin resistance in skeletal muscle. Diabetes 2009;58:2506–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Dimitriadis G, Leighton B, Parry-Billings M, Sasson S, Young M, Krause U, Bevan S, Piva T, Wegener G, Newsholme EA. Effects of glucocorticoid excess on the sensitivity of glucose transport and metabolism to insulin in rat skeletal muscle. Biochem J 1997;321(Pt 3):707–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Weinstein SP, Wilson CM, Pritsker A, Cushman SW. Dexamethasone inhibits insulin-stimulated recruitment of GLUT4 to the cell surface in rat skeletal muscle. Metabolism 1998;47:3–6. [DOI] [PubMed] [Google Scholar]
- 99.Moitra J, Mason MM, Olive M, Krylov D, Gavrilova O, Marcus-Samuels B, Feigenbaum L, Lee E, Aoyama T, Eckhaus M, Reitman ML, Vinson C. Life without white fat: a transgenic mouse. Genes Dev 1998;12:3168–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Ohshima K, Shargill NS, Chan TM, Bray GA. Effects of dexamethasone on glucose transport by skeletal muscles of obese (ob/ob) mice. Int J Obes 1989;13:155–63. [PubMed] [Google Scholar]
- 101.Haluzik M, Dietz KR, Kim JK, Marcus-Samuels B, Shulman GI, Gavrilova O, Reitman ML. Adrenalectomy improves diabetes in A-ZIP/F-1 lipoatrophic mice by increasing both liver and muscle insulin sensitivity. Diabetes 2002;51:2113–8. [DOI] [PubMed] [Google Scholar]
- 102.Gathercole LL, Bujalska IJ, Stewart PM, Tomlinson JW. Glucocorticoid modulation of insulin signaling in human subcutaneous adipose tissue. J Clin Endocrinol Metab 2007;92:4332–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Pivonello R, De Leo M, Vitale P, Cozzolino A, Simeoli C, De Martino MC, Lombardi G, Colao A. Pathophysiology of diabetes mellitus in Cushing’s syndrome. Neuroendocrinology 2010;92 Suppl 1:77–81. [DOI] [PubMed] [Google Scholar]
- 104.Schakman O, Gilson H, Thissen JP. Mechanisms of glucocorticoid-induced myopathy. J Endocrinol 2008;197:1–10. [DOI] [PubMed] [Google Scholar]
- 105.Lee YH, White MF. Insulin receptor substrate proteins and diabetes. Arch Pharm Res 2004;27:361–70. [DOI] [PubMed] [Google Scholar]
- 106.Saad MJ, Folli F, Kahn JA, Kahn CR. Modulation of insulin receptor, insulin receptor sub-strate-1, and phosphatidylinositol 3-kinase in liver and muscle of dexamethasone-treated rats. J Clin Invest 1993;92:2065–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Giorgino F, Almahfouz A, Goodyear LJ, Smith RJ. Glucocorticoid regulation of insulin receptor and substrate IRS-1 tyrosine phosphorylation in rat skeletal muscle in vivo. J Clin Invest 1993;91:2020–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Kuo T, Lew MJ, Mayba O, Harris CA, Speed TP, Wang JC. Genome-wide analysis of glucocorticoid receptor-binding sites in myotubes identifies gene networks modulating insu-lin signaling. Proc Natl Acad Sci U S A 2012;109:11160–5. 10.1073/pnas.1111334109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Backer JM. The regulation of class IA PI 3-kinases by inter-subunit interactions. Curr Top Microbiol Immunol 2010;346:87–114. 10.1007/82_2010_52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Cantley LC. The phosphoinositide 3-kinase pathway. Science 2002;296:1655–7. 10.1126/science.296.5573.1655. [DOI] [PubMed] [Google Scholar]
- 111.Barbour LA, Shao J, Qiao L, Leitner W, Anderson M, Friedman JE, Draznin B. Human placental growth hormone increases expression of the p85 regulatory unit of phosphatidylinositol 3-kinase and triggers severe insulin resistance in skeletal muscle. Endocrinology 2004;145:1144–50. [DOI] [PubMed] [Google Scholar]
- 112.Draznin B Molecular mechanisms of insulin resistance: serine phosphorylation of insulinreceptor substrate-1 and increased expression of p85alpha: the two sides of a coin. Diabetes 2006;55:2392–7. [DOI] [PubMed] [Google Scholar]
- 113.Chagpar RB, Links PH, Pastor MC, Furber LA, Hawrysh AD, Chamberlain MD, Anderson DH. Direct positive regulation of PTEN by the p85 subunit of phosphatidylinositol 3-kinase. Proc Natl Acad Sci U S A 2010;107:5471–6. 10.1073/pnas.0908899107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Terauchi Y, Tsuji Y, Satoh S, Minoura H, Murakami K, Okuno A, Inukai K, Asano T, Kaburagi Y, Ueki K, Nakajima H, Hanafusa T, Matsuzawa Y, Sekihara H, Yin Y, Barrett JC, Oda H, Ishikawa T, Akanuma Y, Komuro I, Suzuki M, Yamamura K, Kodama T, Suzuki H, Yamamura K, Kodama T, Suzuki H, Koyasu S, Aizawa S, Tobe K, Fukui Y, Yazaki Y, Kadowaki T. Increased insulin sensitivity and hypoglycaemia in mice lacking the p85 alpha subunit of phosphoinositide 3-kinase. Nat Genet 1999;21:230–5. 10.1038/6023. [DOI] [PubMed] [Google Scholar]
- 115.Mauvais-Jarvis F, Ueki K, Fruman DA, Hirshman MF, Sakamoto K, Goodyear LJ, Iannacone M, Accili D, Cantley LC, Kahn CR. Reduced expression of the murine p85alpha subunit of phosphoinositide 3-kinase improves insulin signaling and ameliorates diabetes. J Clin Invest 2002;109:141–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Bandyopadhyay GK, Yu JG, Ofrecio J, Olefsky JM. Increased p85/55/50 expression and decreased phosphotidylinositol 3-kinase activity in insulin-resistant human skeletal muscle. Diabetes 2005;54:2351–9. [DOI] [PubMed] [Google Scholar]
- 117.Holland WL, Brozinick JT, Wang LP, Hawkins ED, Sargent KM, Liu Y, Narra K, Hoehn KL, Knotts TA, Siesky A, Nelson DH, Karathanasis SK, Fontenot GK, Birnbaum MJ, Summers SA. Inhibition of ceramide synthesis ameliorates glucocorticoid-, saturated-fat-, and obesity-induced insulin resistance. Cell Metab 2007;5:167–79. [DOI] [PubMed] [Google Scholar]
- 118.Chavez JA, Summers SA. A ceramide-centric view of insulin resistance. Cell Metab 2012;15:585–94. 10.1016/j.cmet.2012.04.002. [DOI] [PubMed] [Google Scholar]
- 119.Tappy L, Randin D, Vollenweider P, Vollenweider L, Paquot N, Scherrer U, Schneiter P, Nicod P, Jequier E. Mechanisms of dexamethasone-induced insulin resistance in healthy humans. J Clin Endocrinol Metab 1994;79:1063–9. [DOI] [PubMed] [Google Scholar]
- 120.Sugden MC, Holness MJ. Recent advances in mechanisms regulating glucose oxidation at the level of the pyruvate dehydrogenase complex by PDKs. Am J Physiol Endocrinol Metab 2003;284:E855–62. 10.1152/ajpendo.00526.2002. [DOI] [PubMed] [Google Scholar]
- 121.Connaughton S, Chowdhury F, Attia RR, Song S, Zhang Y, Elam MB, Cook GA, Park EA. Regulation of pyruvate dehydrogenase kinase isoform 4 (PDK4) gene expression by glucocor-ticoids and insulin. Mol Cell Endocrinol 2010;315:159–67. 10.1016/j.mce.2009.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Kwon HS, Huang B, Unterman TG, Harris RA. Protein kinase B-alpha inhibits human pyru-vate dehydrogenase kinase-4 gene induction by dexamethasone through inactivation of FOXO transcription factors. Diabetes 2004;53:899–910. [DOI] [PubMed] [Google Scholar]
- 123.Bazuine M, Carlotti F, Tafrechi RS, Hoeben RC, Maassen JA. Mitogen-activated protein kinase (MAPK) phosphatase-1 and −4 attenuate p38 MAPK during dexamethasone-induced insulin resistance in 3T3-L1 adipocytes. Mol Endocrinol 2004;18:1697–707. [DOI] [PubMed] [Google Scholar]
- 124.Sakoda H, Ogihara T, Anai M, Funaki M, Inukai K, Katagiri H, Fukushima Y, Onishi Y, Ono H, Fujishiro M, Kikuchi M, Oka Y, Asano T. Dexamethasone-induced insulin resistance in 3T3-L1 adipocytes is due to inhibition of glucose transport rather than insulin signal trans-duction. Diabetes 2000;49:1700–8. [DOI] [PubMed] [Google Scholar]
- 125.Yu CY, Mayba O, Lee JV, Tran J, Harris C, Speed TP, Wang JC. Genome-wide analysis of glucocorticoid receptor binding regions in adipocytes reveal gene network involved in tri-glyceride homeostasis. PLoS One 2010;5:e15188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Hazlehurst JM, Gathercole LL, Nasiri M, Armstrong MJ, Borrows S, Yu J, Wagenmakers AJ, Stewart PM, Tomlinson JW. Glucocorticoids fail to cause insulin resistance in human subcu-taneous adipose tissue in vivo. J Clin Endocrinol Metab 2013;98:1631–40. 10.1210/jc.2012-3523. [DOI] [PubMed] [Google Scholar]
- 127.Gathercole LL, Morgan SA, Bujalska IJ, Stewart PM, Tomlinson JW. Short- and long-term glucocorticoid treatment enhances insulin signalling in human subcutaneous adipose tissue. Nutr Diabetes 2011;1:e3 10.1038/nutd.2010.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Buentke E, Nordstrom A, Lin H, Bjorklund AC, Laane E, Harada M, Lu L, Tegnebratt T, Stone-Elander S, Heyman M, Soderhall S, Porwit A, Ostenson CG, Shoshan M, Tamm KP, Grander D. Glucocorticoid-induced cell death is mediated through reduced glucose metabo-lism in lymphoid leukemia cells. Blood Cancer J 2011;1:e31 10.1038/bcj.2011.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.de Leon MJ, McRae T, Rusinek H, Convit A, De Santi S, Tarshish C, Golomb J, Volkow N, Daisley K, Orentreich N, McEwen B. Cortisol reduces hippocampal glucose metabolism in normal elderly, but not in Alzheimer’s disease. J Clin Endocrinol Metab 1997;82:3251–9. 10.1210/jcem.82.10.4305. [DOI] [PubMed] [Google Scholar]
- 130.Horner HC, Packan DR, Sapolsky RM. Glucocorticoids inhibit glucose transport in cultured hippocampal neurons and glia. Neuroendocrinology 1990;52:57–64. [DOI] [PubMed] [Google Scholar]
- 131.Cherian AK, Briski KP. Effects of adrenalectomy on neuronal substrate fuel transporter and energy transducer gene expression in hypothalamic and hindbrain metabolic monitoring sites. Neuroendocrinology 2010;91:56–63. 10.1159/000264919. [DOI] [PubMed] [Google Scholar]
- 132.Mersmann HJ, Segal HL. Glucocorticoid control of the liver glycogen synthetase-activating system. J Biol Chem 1969;244:1701–4. [PubMed] [Google Scholar]
- 133.Ray PD, Foster DO, Lardy HA. Mode of action of glucocorticoids. I. Stimulation of gluconeo-genesis independent of synthesis de novo of enzymes. J Biol Chem 1964;239:3396–400. [PubMed] [Google Scholar]
- 134.De Wulf H, Hers HG. The stimulation of glycogen synthesis and of glycogen synthetase in the liver by glucocorticoids. Eur J Biochem 1967;2:57–60. [DOI] [PubMed] [Google Scholar]
- 135.Huang TS, Krebs EG. Amino acid sequence of a phosphorylation site in skeletal muscle glycogen synthetase. Biochem Biophys Res Commun 1977;75:643–50. [DOI] [PubMed] [Google Scholar]
- 136.Rylatt DB, Aitken A, Bilham T, Condon GD, Embi N, Cohen P. Glycogen synthase from rab-bit skeletal muscle. Amino acid sequence at the sites phosphorylated by glycogen synthase kinase-3, and extension of the N-terminal sequence containing the site phosphorylated by phosphorylase kinase. Eur J Biochem 1980;107:529–37. [PubMed] [Google Scholar]
- 137.Pugazhenthi S, Khandelwal RL. Regulation of glycogen synthase activation in isolated hepa-tocytes. Mol Cell Biochem 1995;149–150:95–101. [DOI] [PubMed] [Google Scholar]
- 138.Brady MJ, Nairn AC, Saltiel AR. The regulation of glycogen synthase by protein phosphatase 1 in 3T3-L1 adipocytes. Evidence for a potential role for DARPP-32 in insulin action. J Biol Chem 1997;272:29698–703. [DOI] [PubMed] [Google Scholar]
- 139.Printen JA, Brady MJ, Saltiel AR. PTG, a protein phosphatase 1-binding protein with a role in glycogen metabolism. Science 1997;275:1475–8. [DOI] [PubMed] [Google Scholar]
- 140.Laloux M, Stalmans W, Hers HG. On the mechanism by which glucocorticoids cause the activation of glycogen synthase in mouse and rat livers. Eur J Biochem 1983;136:175–81. [DOI] [PubMed] [Google Scholar]
- 141.Vanstapel F, Dopere F, Stalmans W. The role of glycogen synthase phosphatase in the glucocorticoid-induced deposition of glycogen in foetal rat liver. Biochem J 1980;192: 607–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Green GA, Chenoweth M, Dunn A. Adrenal glucocorticoid permissive regulation of muscle glycogenolysis: action on protein phosphatase(s) and its inhibitor(s). Proc Natl Acad Sci U S A 1980;77:5711–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Salehzadeh F, Al-Khalili L, Kulkarni SS, Wang M, Lonnqvist F, Krook A. Glucocorticoid-mediated effects on metabolism are reversed by targeting 11 beta hydroxysteroid dehydroge-nase type 1 in human skeletal muscle. Diabetes Metab Res Rev 2009;25:250–8. 10.1002/dmrr.944. [DOI] [PubMed] [Google Scholar]
- 144.Miller TB, Exton JH, Park CR. A block in epinephrine-induced glycogenolysis in hearts from adrenalectomized rats. J Biol Chem 1971;246:3672–8. [PubMed] [Google Scholar]
- 145.Puthanveetil P, Rodrigues B. Glucocorticoid excess induces accumulation of cardiac glyco-gen and triglyceride: suggested role for AMPK. Curr Pharm Des 2013;19:4818–30. [DOI] [PubMed] [Google Scholar]
- 146.Baltrons MA, Agullo L, Garcia A. Dexamethasone up-regulates a constitutive nitric oxide synthase in cerebellar astrocytes but not in granule cells in culture. J Neurochem 1995;64: 447–50. [DOI] [PubMed] [Google Scholar]
- 147.Binnert C, Ruchat S, Nicod N, Tappy L. Dexamethasone-induced insulin resistance shows no gender difference in healthy humans. Diabetes Metab 2004;30:321–6. [DOI] [PubMed] [Google Scholar]
- 148.Rafacho A, Marroqui L, Taboga SR, Abrantes JL, Silveira LR, Boschero AC, Carneiro EM, Bosqueiro JR, Nadal A, Quesada I. Glucocorticoids in vivo induce both insulin hypersecre-tion and enhanced glucose sensitivity of stimulus-secretion coupling in isolated rat islets. Endocrinology 2010;151:85–95. 10.1210/en.2009-0704. [DOI] [PubMed] [Google Scholar]
- 149.Besse C, Nicod N, Tappy L. Changes in insulin secretion and glucose metabolism induced by dexamethasone in lean and obese females. Obes Res 2005;13:306–11. 10.1038/oby.2005.41. [DOI] [PubMed] [Google Scholar]
- 150.Fransson L, Franzen S, Rosengren V, Wolbert P, Sjoholm A, Ortsater H. beta-Cell adaptation in a mouse model of glucocorticoid-induced metabolic syndrome. J Endocrinol 2013;219: 231–41. 10.1530/JOE-13-0189. [DOI] [PubMed] [Google Scholar]
- 151.Jeong IK, Oh SH, Kim BJ, Chung JH, Min YK, Lee MS, Lee MK, Kim KW. The effects of dexamethasone on insulin release and biosynthesis are dependent on the dose and duration of treatment. Diabetes Res Clin Pract 2001;51:163–71. [DOI] [PubMed] [Google Scholar]
- 152.van Raalte DH, Nofrate V, Bunck MC, van Iersel T, Elassaiss Schaap J, Nassander UK, Heine RJ, Mari A, Dokter WH, Diamant M. Acute and 2-week exposure to prednisolone impair different aspects of beta-cell function in healthy men. Eur J Endocrinol 2010;162:729–35. 10.1530/EJE-09-1034. [DOI] [PubMed] [Google Scholar]
- 153.Gremlich S, Roduit R, Thorens B. Dexamethasone induces posttranslational degradation of GLUT2 and inhibition of insulin secretion in isolated pancreatic beta cells. Comparison with the effects of fatty acids. J Biol Chem 1997;272:3216–22. [DOI] [PubMed] [Google Scholar]
- 154.Borboni P, Porzio O, Magnaterra R, Fusco A, Sesti G, Lauro R, Marlier LN. Quantitative analysis of pancreatic glucokinase gene expression in cultured beta cells by competitive poly-merase chain reaction. Mol Cell Endocrinol 1996;117:175–81. [DOI] [PubMed] [Google Scholar]
- 155.Goodman PA, Medina-Martinez O, Fernandez-Mejia C. Identification of the human insulin negative regulatory element as a negative glucocorticoid response element. Mol Cell Endocrinol 1996;120:139–46. [DOI] [PubMed] [Google Scholar]
- 156.Sharma S, Jhala US, Johnson T, Ferreri K, Leonard J, Montminy M. Hormonal regulation of an islet-specific enhancer in the pancreatic homeobox gene STF-1. Mol Cell Biol 1997;17: 2598–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Khan A, Ostenson CG, Berggren PO, Efendic S. Glucocorticoid increases glucose cycling and inhibits insulin release in pancreatic islets of ob/ob mice. Am J Physiol 1992;263:E663–6. [DOI] [PubMed] [Google Scholar]
- 158.Ullrich S, Berchtold S, Ranta F, Seebohm G, Henke G, Lupescu A, Mack AF, Chao CM, Su J, Nitschke R, Alexander D, Friedrich B, Wulff P, Kuhl D, Lang F. Serum- and glucocorticoid-inducible kinase 1 (SGK1) mediates glucocorticoid-induced inhibition of insulin secretion. Diabetes 2005;54:1090–9. [DOI] [PubMed] [Google Scholar]
- 159.Negrato CA, Jovanovic L, Rafacho A, Tambascia MA, Geloneze B, Dias A, Rudge MV. Association between different levels of dysglycemia and metabolic syndrome in preg-nancy. Diabetol Metab Syndr 2009;1:3 10.1186/1758-5996-1-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Rafacho A, Quallio S, Ribeiro DL, Taboga SR, Paula FM, Boschero AC, Bosqueiro JR. The adap-tive compensations in endocrine pancreas from glucocorticoid-treated rats are reversible after the interruption of treatment. Acta Physiol 2010;200:223–35. 10.1111/j.1748-1716.2010.02146.x. [DOI] [PubMed] [Google Scholar]
- 161.Rafacho A, Abrantes JL, Ribeiro DL, Paula FM, Pinto ME, Boschero AC, Bosqueiro JR. Morphofunctional alterations in endocrine pancreas of short- and long-term dexamethasone-treated rats. Horm Metab Res 2011;43:275–81. 10.1055/s-0030-1269896. [DOI] [PubMed] [Google Scholar]
- 162.Rafacho A, Giozzet VA, Boschero AC, Bosqueiro JR. Functional alterations in endocrine pancreas of rats with different degrees of dexamethasone-induced insulin resistance. Pancreas 2008;36:284–93. 10.1097/MPA.0b013e31815ba826. [DOI] [PubMed] [Google Scholar]
- 163.Huising MO, Pilbrow AP, Matsumoto M, van der Meulen T, Park H, Vaughan JM, Lee S, Vale WW. Glucocorticoids differentially regulate the expression of CRFR1 and CRFR2alpha in MIN6 insulinoma cells and rodent islets. Endocrinology 2011;152:138–50. 10.1210/en.2010-0791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Huising MO, van der Meulen T, Vaughan JM, Matsumoto M, Donaldson CJ, Park H, Billestrup N, Vale WW. CRFR1 is expressed on pancreatic beta cells, promotes beta cell proliferation, and potentiates insulin secretion in a glucose-dependent manner. Proc Natl Acad Sci U S A 2010;107:912–7. 10.1073/pnas.0913610107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Reich E, Tamary A, Sionov RV, Melloul D. Involvement of thioredoxin-interacting protein (TXNIP) in glucocorticoid-mediated beta cell death. Diabetologia 2012;55:1048–57. 10.1007/s00125-011-2422-z. [DOI] [PubMed] [Google Scholar]
- 166.Avram D, Ranta F, Hennige AM, Berchtold S, Hopp S, Haring HU, Lang F, Ullrich S. IGF-1 protects against dexamethasone-induced cell death in insulin secreting INS-1 cells indepen-dent of AKT/PKB phosphorylation. Cell Physiol Biochem 2008;21:455–62. 10.1159/000129638. [DOI] [PubMed] [Google Scholar]
- 167.Fransson L, Rosengren V, Saha TK, Grankvist N, Islam T, Honkanen RE, Sjoholm A, Ortsater H. Mitogen-activated protein kinases and protein phosphatase 5 mediate glucocorticoid-induced cytotoxicity in pancreatic islets and beta-cells. Mol Cell Endocrinol 2014;383:126– 36. 10.1016/j.mce.2013.12.010. [DOI] [PubMed] [Google Scholar]
- 168.Bodwell JE, Webster JC, Jewell CM, Cidlowski JA, Hu JM, Munck A. Glucocorticoid receptor phosphorylation: overview, function and cell cycle-dependence. J Steroid Biochem Mol Biol 1998;65:91–9. [DOI] [PubMed] [Google Scholar]
- 169.Ismaili N, Garabedian MJ. Modulation of glucocorticoid receptor function via phosphoryla-tion. Ann N Y Acad Sci 2004;1024:86–101. 10.1196/annals.1321.007. [DOI] [PubMed] [Google Scholar]
- 170.Rogatsky I, Logan SK, Garabedian MJ. Antagonism of glucocorticoid receptor transcrip-tional activation by the c-Jun N-terminal kinase. Proc Natl Acad Sci U S A 1998;95: 2050–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Roma LP, Oliveira CA, Carneiro EM, Albuquerque GG, Boschero AC, Souza KL. N-acetylcysteine protects pancreatic islet against glucocorticoid toxicity. Redox Rep 2011;16:173–80. 10.1179/1351000211Y.0000000006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Yamamoto M, Yamato E, Toyoda S, Tashiro F, Ikegami H, Yodoi J, Miyazaki J. Transgenic expression of antioxidant protein thioredoxin in pancreatic beta cells prevents progression of type 2 diabetes mellitus. Antioxid Redox Signal 2008;10:43–9. 10.1089/ars.2007.1586. [DOI] [PubMed] [Google Scholar]
- 173.Wang Z, Rong YP, Malone MH, Davis MC, Zhong F, Distelhorst CW. Thioredoxin-interacting protein (txnip) is a glucocorticoid-regulated primary response gene involved in mediating glucocorticoid-induced apoptosis. Oncogene 2006;25:1903–13. 10.1038/sj.onc.1209218. [DOI] [PubMed] [Google Scholar]
- 174.Quesada I, Tuduri E, Ripoll C, Nadal A. Physiology of the pancreatic alpha-cell and glucagon secretion: role in glucose homeostasis and diabetes. J Endocrinol 2008;199:5–19. 10.1677/JOE-08-0290. [DOI] [PubMed] [Google Scholar]
- 175.Everett LJ, Lazar MA. Cell-specific integration of nuclear receptor function at the genome. Wiley Interdiscip Rev Syst Biol Med 2013;5:615–29. 10.1002/wsbm.1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Hah N, Danko CG, Core L, Waterfall JJ, Siepel A, Lis JT, Kraus WL. A rapid, extensive, and transient transcriptional response to estrogen signaling in breast cancer cells. Cell 2011;145:622–34. 10.1016/j.cell.2011.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Hah N, Kraus WL. Hormone-regulated transcriptomes: lessons learned from estrogen signaling pathways in breast cancer cells. Mol Cell Endocrinol 2014;382:652–64. 10.1016/j.mce.2013.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Madsen JG, Schmidt SF, Larsen BD, Loft A, Nielsen R, Mandrup S. iRNA-seq: computa-tional method for genome-wide assessment of acute transcriptional regulation from total RNA-seq data. Nucleic Acids Res 2015;43(6):e40 10.1093/nar/gku1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Jinek M, East A, Cheng A, Lin S, Ma E, Doudna J. RNA-programmed genome editing in human cells. eLife 2013;2:e00471 10.7554/eLife.00471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, Church GM. RNA-guided human genome engineering via Cas9. Science 2013;339:823–6. 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, Zhang F. Multiplex genome engineering using CRISPR/Cas systems. Science 2013;339: 819–23. 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Cho SW, Kim S, Kim JM, Kim JS. Targeted genome engineering in human cells with the Cas9 RNA-guided endonuclease. Nat Biotechnol 2013;31:230–2. 10.1038/nbt.2507. [DOI] [PubMed] [Google Scholar]
- 183.Yang H, Wang H, Jaenisch R. Generating genetically modified mice using CRISPR/Cas-mediated genome engineering. Nat Protoc 2014;9:1956–68. 10.1038/nprot.2014.134. [DOI] [PubMed] [Google Scholar]