

Abstract
Mutant myocilin aggregation is associated with inherited open angle glaucoma, a prevalent optic neuropathy leading to blindness. Comprehension of mutant myocilin aggregation is of fundamental importance to glaucoma pathogenesis and ties glaucoma to amyloid diseases such as Alzheimer’s. Here we probe the aggregation properties of peptides derived from the myocilin olfactomedin domain. Peptides P1 (residues 326–337) and P3 (residues 426–442) were identified previously to form amyloid. Coarse-grained discontinuous molecular dynamics simulations using the PRIME20 force field (DMD/PRIME20) predict that P1 and P3 are aggregation-prone; P1 consistently forms fibrillar aggregates with parallel in-register β-sheets whereas P3 forms β-sheet-containing aggregates without distinct order. Natural abundance 13C solid-state NMR spectra validate that aggregated P1 exhibits amyloid signatures and is less heterogeneous than aggregated P3. DMD/PRIME20 simulations provide a viable method to predict peptide aggregation propensities and aggregate’s structure/order which cannot be accessed by bioinformatics or readily attained experimentally.
TOC Graphic
Introduction
A recent addition to the list of proteins associated with amyloid-based diseases is the myocilin olfactomedin domain (mOLF)1–2. Inherited non-synonymous mutations in mOLF are causative for early-onset open angle glaucoma, an ocular neurodegenerative disease leading to blindness3. Mutant myocilins aggregate instead of being secreted to the trabecular meshwork (TM), a key anatomical structure responsible for maintaining eye pressure that is dysregulated in most cases of glaucoma4. Whereas cells typically deal with mutant proteins with an efficient degradation system mediated by molecular chaperones, this process is significantly compromised in TM cells expressing mutant myocilin. Mutant myocilins accumulate intracellularly5–7 where they exhibit aberrant interactions with molecular chaperones8–9, leading to TM cell death that hastens glaucoma onset. Cumulatively, such behavior suggests that mutant myocilins might be aggregating into amyloid, which are well-known for their high thermochemical stability and resistance to degradation.10–13
Initial evidence for the amyloid behavior of mOLF includes thioflavin-T (ThT) positive aggregates generated in vitro, demonstrating a sigmoidal growth curve composed of a lag phase followed by exponential growth, as well as a positive ThT signal from full-length mutant myocilin that has accumulated within mammalian cells2. The mOLF amyloid aggregation parameters and associated biophysical features have been evaluated further1 (Fig. 1), leading to the identification of two different fibril morphologies, long straight fibrils (Fig. 1A) and more unusual lassoed oligomers (Fig. 1B). These morphologies are also observed in aggregates of glaucoma-associated variants, A427T and D380A, respectively (not shown). Subsequent bioinformatics analysis (Fig. S1) to determine the amyloidogenic regions on full-length mOLF protein revealed three consensus sequences1: G326AVVYSGSLYFQ (P1), G387LWVIYSTDEAKGAIVLSK (P2) and V426ANAFIICGTLYTVSSY (P3). Experimentally, two of these peptides, P3 and P1, formed ThT-positive aggregates (Fig. 1C) whereas P2 remained soluble. At low concentrations, P1 aggregation exhibits the expected lag phase, whereas conditions for observing a lag phase for P3 aggregation have not yet been obtained (data not shown). Upon solving the mOLF crystal structure14, we learned that P1 and P3 are in the interior of the β-propeller, confirming our earlier finding that fibril formation is initiated from a partially folded state1. The fibril morphologies of P1 and P3 (Figs. 1E, F) recapitulate morphologies seen for the full mOLF protein domain (Figs. 1A, B), suggesting that P1 and P3 are the core stretches responsible for the observed morphologies adopted by the full protein1.
Figure 1.

mOLF peptide stretches P1 and P3 recapitulate disparate morphologies of amyloid aggregates derived from full-length mOLF seen by AFM. (A) mOLF fibrils grown at 37 °C with mechanical rocking exhibit straight morphology by AFM while (B) mOLF fibrils grown at 42 °C without rocking exhibit disparate circular morphology. (C) Fibrilization of predicted amyloidogenic peptide stretches, P1, P2 and P3, incubated in 10mM Na2HPO4/KH2PO4 pH 7.2 buffer containing 200mM NaCl at 36 °C, monitored by ThT fluorescence. (D) Amyloidogenic peptide stretches P1 (green) and P3 (purple) identified in (C) highlighted in the context of the native mOLF structure (PDB code 4WXS). (E) P1 fibrils appear straight while (F) P3 fibrils are circular when visualized by AFM. Scale bar is 300 nm for images A, B, E, and F. Images A, B, C, E, F reproduced from ref1. Reprinted from Journal of Molecular Biology, 426, S. E. Hill, R. K. Donegan and R. L. Lieberman, The Glaucoma-Associated Olfactomedin Domain of Myocillin Forms Polymorphic Fibrils that are Constrained by Partial Unfolding and Peptide Sequence, pages 921–935, Copyright (2014), with permission from Elsevier.
To further comprehend amyloidogenesis by mOLF-derived peptides and its molecular relationship to better-studied amyloids like amyloid-β15–17, we performed discontinuous molecular dynamics simulations (DMD) combined with the PRIME20 force field18–20 to predict fibrillar structure, as well as solid-state NMR to experimentally evaluate the extent of structural order of aggregated P1 and P3. In addition to providing new molecular insight into mOLF aggregation, our study demonstrates proof-of-concept for the applicability of DMD/PRIME20 to predict peptide amyloidogenicity and fibril structure.
Methods
DMD/PRIME20 simulations.
PRIME20 is a 4-sphere-per-residue coarse-grained protein model (three backbone spheres Cα, NH, CO and one sidechain sphere) developed in the Hall group that was specifically designed for DMD-based simulation of protein aggregation18–20, and has unique geometric and energetic parameters for each of the 20 different amino acids. Specifically, each sidechain sphere of the 20 amino acids has a distinct hard sphere diameter (effective van der Waals radius), and distinct sidechain-to-backbone distances (R-Cα, R-NH, R-CO). The potential energy function between two amino acid sidechain beads is modeled as a square well potential. The potential energy parameters between the 20 different amino acids (except for glycine) include 210 independent square well widths and 19 independent square well depths. The backbone hydrogen bonding interactions are modeled as a directional square well potential. The detailed description of the derivation of the PRIME20 geometric and energetic parameters along with their values are given in our earlier work.19, 21–22 The reduced temperature is defined as T*=kBT/εHB. It is related to a real temperature by T [K] = 2288.46T*−115.79; this equation was obtained by matching the folding temperature of alanine-rich polypeptides in DMD simulation in our previous work23 to the experimental values24. The reduced time unit is Δtreduced = 0.96 ns; this was obtained by matching the self-diffusion coefficient of Aβ(16–22) obtained from DMD simulations to that calculated from atomistic MD simulation.23
The simulation system consists of eight peptides in a cubic box with peptide concentration C=20mM at T=342K. The simulation temperature is chosen to optimize the likelihood that P1 and P3 aggregate to form stable ordered β-sheet-rich protofilament. If the temperature is too high, the peptides do not aggregate and if it is too low they get trapped in local energy minimum states. The peptides are initially at random locations and in random coil conformations. We performed ten independent runs for the P1, P2 and P3 peptides. Over the course of the simulation, the peptides spontaneously aggregate, constantly rearranging into more-stable structures as they move towards the equilibrium state. The simulation times for the P1, P2 and P3 systems are 223μs, 353μs and 476μs, respectively.
Peptide synthesis and experimentation fibril formation.
P1 and P3 peptides were synthesized by CPC Scientific (Sunnyvale, CA) to >95% purity for P1 and 88.8% for P3. Both peptides were prepared as a 5 mg mL−1 stock solution in DMSO and stored at room temperature. Fibrils were generated by dissolving 500 μM peptide into 10 mM Na2HPO4/KH2PO4 buffer at pH 7.2 containing 200 mM NaCl plus 10 μM ThT and incubating at 36 °C for 24–48 hours, as previously published.1 For P1 the products of three 4-mL reactions were combined for NMR while for P3 a one 4-mL reaction was found to be sufficient for a robust signal. Insoluble aggregates were isolated for solid-state NMR characterization by ultracentrifugation (164,700 xg for 15 minutes at 25°C).
Solid-state NMR measurements.
The 1H-13C CPMAS experiments25–26 were performed on a narrow-bore Bruker 11.75 Tesla (500-MHz 1H NMR frequency) solid-state NMR system with an Avance III console and a 3.2 mm magic angle spinning (MAS) HX NMR probe. The MAS spin rate was kept at 10 kHz for all measurements. For the 2 ms cross polarization, a 50 kHz pulse on 13C channel and a calibrated ramped pulse on 1H channel was used. After the cross polarization, decoupling of 1H was employed with a radio-frequency field power of 100 kHz during the detection period, with two-pulse-phase modulation27 (TPPM). The predicted CPMAS spectrum was generated by a summation of all the Gaussian peaks at the chemical shifts of all 13C in the peptides. The line width (full width at half maximum, FWHM) of each Gaussian peak was identical.
Results & Discussion
P1 peptides aggregated into ordered β-sheet-rich oligomers in all ten independent DMD/PRIME20 simulation runs (Figs. 2 and S2), predicting that P1 has a strong amyloid-forming propensity. P1 peptides formed a U-shaped protofilament (Figs. 2A (red curve, top panel), B, D) in two out of ten runs and an S-shaped protofilament (Fig. 2A (black curve, bottom panel), C, E) in the remaining eight runs. P1 aggregates formed within the first 10μs (Fig. 2A), indicating fast aggregation kinetics during simulations. The kinetics are consistent with other simulation18, 28 studies indicating that, in general, amyloidogenic peptides tend to form fibrils with fast aggregation kinetics if they contain sixteen or fewer amino acids. The U-shaped P1 protofilament adopted a backbone turn at 332Gly (Figs. 2B, D), forming a hydrophobic core containing residues 328Val, 330Tyr, 334Leu and 336Phe. The S-shaped P1 protofilament adopted two turns, one at 332Gly and the other near the C-terminal (Fig. 2C, E). Analysis of the average number of inter-peptide backbone hydrogen bonds formed within the last half of the ten trajectories by each of the residues reveals that the whole P1 sequence, except C-terminal 336Phe and 337Gln, has a high propensity to form β-sheet structure (Fig. 3A). The DMD/PRIME20 result is consistent with bioinformatics predictions (Fig. S1) and experiments (Fig. 1C, E) indicating that the P1 region 326GAVVYSGSLY335 is an amyloid forming peptide within mOLF, and adds key new molecular insight into possible arrangements of the amyloid.
Figure 2.

DMD/PRIME20 simulations of P1. (A) A plot of potential energy of P1 peptide aggregation versus simulation time. Also shown are simulation snapshots of eight random coil P1 peptides aggregating to form U-shaped (top panel corresponding to the red curve) and S-shaped (bottom panel corresponding to the black curve) P1 protofilaments. Each of the eight peptides has a distinct color. (B) and (C) are the final simulation snapshots of the S-shaped (B) and U-shaped (C) P1 protofilaments, respectively. (D) and (E) are the schematic representation of peptide conformation in the U-shaped and S-shaped P1 protofilaments. Hydrophobic and polar residues are shown in white and green, respectively.
Figure 3.

Average number of inter-peptide backbone hydrogen bonds formed per residue of P1 (A) P2 (B) P3 (C). (D) β-sheet propensities calculated for P1, P2 and P3 peptides.
In DMD/PRIME20 simulations, P2 formed disordered oligomers (Fig. S3) containing a small amount of β-hairpin or β-sheet structure in nine out of ten simulation runs. In just one of ten runs, P2 aggregated into a fibrillar structure. In contrast to bioinformatics analysis, but in line with experimental results1, P2 has a low aggregation propensity as it forms only a small number of inter-peptide hydrogen bonds along the whole sequence (Fig. 3B).
P3 peptides aggregated in DMD/PRIME20 simulations to form parallel in-register U-shaped protofilaments in two out of ten runs and polymorphic β-sheet-rich oligomers in the remaining eight runs (Fig. 4, S4). From Fig. 4A(black curve, bottom panel), P3 aggregated into a disordered β-sheet-rich oligomer at t =70.6μs, formed a U-shaped trimer at t =149.5μs and grew into a stable U-shaped octamer protofilament at t= 176.3μs (Figs. 4C, D). Note that the potential energy dropped precipitously as P3 grew from a trimer to an octamer. In contrast when P3 forms polymorphic structures (Fig. 4A (red curve, top panel), B) the potential energy is relatively constant. Note that all the disordered β-sheet-rich P3 oligomers (Fig. S4) are in local minimum states that are metastable with respect to ordered fibrillar state (global minimum). These P3 oligomers may eventually come out of the local minimum states and form a more ordered fibrillar structure, but, based on current computation resources, it’s hard to predict the occurrence probability of such events. P3 is found to have a much slower aggregation kinetics than P1. One reason is that P3 has six more amino acids than P1 and thus has more degrees of freedom in its backbone chain movement which adds complexity to the aggregation pathways and increases nucleation time23, 29–30. Within the U-shaped P3 protofilament, the sidechains of residues 430Phe, 432Ile, 436Leu, 439Val on P3 closely contact each other to form a hydrophobic fibril core (Fig. 4D). In addition, the stretch of P3 that forms a large number of inter-peptide hydrogen bonds is 428NAFIICGTLYTVS440 (Fig. 3C), consistent with bioinformatics analysis (Fig. S1). Simulations support experimental results in that ThT positive aggregates with variable-sized, lassoed oligomer morphologies were observed (Fig. 1C, F), but these aggregates grew quickly in experiment. The ThT kinetic data further suggests that P3 forms less-ordered aggregate structures than P1, based on the intensity of the ThT fluorescence which was lower than P1 even though qualitatively the two peptides appeared to aggregate at sufficient quantities with similar amounts of insoluble material. The simulations support this hypothesis, as the polymorphic β-sheet rich oligomers observed in the P3 simulations likely bind ThT, but possibly with a lower fluorescence emission intensity or affinity when compared to better ordered P1 aggregates. In addition, our simulations suggest that a high level of templated order in a P3 fibril may be achievable if experimental conditions can be driven towards the U-shaped conformation.
Figure 4.

DMD/PRIME20 simulations of P3. (A) A plot of potential energy of P3 peptide aggregation versus simulation time. Also shown are simulation snapshots of eight random coil P3 peptides aggregating to form a disordered oligomer (top panel corresponding to the red curve) and a U-shaped P3 protofilament (bottom panel corresponding to the black curve). Each of the eight peptides has a distinct color. (B) and (C) are final simulation snapshots of the disordered P3 oligomer and U-shaped P3 protofilament, respectively. (D) The schematic representation of peptide conformation in the U-shaped P3 protofilaments. Hydrophobic and polar residues are shown in white and green, respectively.
In order to quantitatively compare the amyloid forming propensities of P1, P2 and P3 peptides based on DMD/PRIME20 simulation results, we define β(Pj) as the propensity of a specific peptide sequence Pj (e.g. P1, P2 or P3) to aggregate and form a β-sheet amyloid:
where is the total number of inter-peptide hydrogen bonds formed by the peptide in the aggregate, and is the total number of backbone hydrogen bonding sites (NH and C=O beads) on the peptide in the aggregate. N is the amino acid length of peptide Pj. ranges from 1 for a perfect β-sheet structure with strong amyloid forming propensity, to 0 for a monomeric state or disordered oligomer with weak amyloid forming propensity. The values of for P1, P2 and P3 with standard deviations over the last one-third of the trajectories of ten runs are = 0.64±0.06, = 0.25±0.07 and = 0.44±0.07, indicating that the order of amyloid forming propensity of the three mOLF peptides is P1>P3>P2 (Fig. 3D), which is consistent with experimental data1 (Fig. 1C, E, F) that P1 and P3 form fibrils but P2 does not.
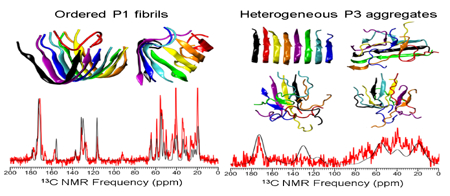
To experimentally evaluate the extent of molecular order for aggregated P1 and P3, we aggregated peptides P1 and P3 under physiological conditions as before1 and then acquired 1H-13C Cross-Polarization Magic Angle Spinning (CPMAS) solid-state NMR spectra. Consistent with DMD/PRIME20 predictions, P1 and P3 showed dramatically different CPMAS spectra (Figs. 5A, C), confirming dissimilar structures. The spectrum of P1 fibrils is consistent with structurally ordered amyloid fibrils12, 31–32, namely, narrow NMR line widths (~1.2 ppm, Fig. 5A). In addition, though many 13C peaks on the P1 CPMAS spectrum overlap with others, some peaks with special chemical shifts can be assigned to a residue type based on the known chemical shift ranges of different 13C sites33–35 (Fig. 5B, Table 1). Indeed, by comparing the chemical shifts of the distinguishable peaks with those from random coil peptides, all the secondary chemical shifts imply a strong trend to β-strand structure33. The observed structural order in the P1 CPMAS spectrum suggests that this sample will be compatible with future solid-state NMR structural methodologies. By contrast, the spectrum of P3 fibrils provides experimental evidence for a heterogeneous aggregate mixture, namely, broad line widths (> 14 ppm, Fig. 5B). In contrast to P1, the P3 sample is unlikely to yield readily interpretable structural constraints, unless experimental aggregation kinetics can be modified to yield a homogeneous sample.
Figure 5.

1H-13C CPMAS 13C spectrum of P1 and P3 aggregates with no 13C enrichment. (A) The spectrum of P1 fibril (red) and the predicted spectrum (generated by summing all Gaussian peaks at the chemical shifts of all 13C in the peptides) with FWHM = 1.2 ppm (black). (B) The region of aliphatic carbons in Panel A. (C) The predicted CPMAS spectrum of P3 fibril (red) and the predicted spectrum with FWHM = 14 ppm (black).
Table 1. The secondary chemical shifts of the distinguishable 13C peaks in the CPMAS spectrum of P1 aggregates.
For residues within β-strands, CO and Cα peak frequencies are expected to be at least 0.5ppm under corresponding random coil values (measured from random coil model peptides)33–34 and Cβ peak frequencies are expected to be at least 0.5 ppm above corresponding random coil values.
| Carbon | Random Coil Shift (ppm) | Measured Chemical Shift (ppm) | Secondary Chemical Shift (ppm) |
|---|---|---|---|
| S Cβ | 62.1 | 64.3 | +2.2 |
| V Cα | 60.5 | 58.9 | −1.6 |
| A Cα | 50.8 | 49.8 | −1.0 |
| Q Cβ | 27.7 | 30.6 | +2.9 |
Conclusion
In summary, this study demonstrates the capability of DMD/PRIME20 to predict peptide amyloidogenicity and fibril structure. DMD/PRIME20 simulations both recapitulated mOLF-derived peptide aggregation propensities seen experimentally1, and predicted the extent of homogeneity in assembly into particular structural arrangements, which we validated by solid-state NMR. Structure predictions obtained from DMD/PRIME20 can now serve as a basis for choices of 13C- and 15N-labeled sites required for P1 structural NMR experiments, and provide direction for optimizing P3 aggregation experiments with the goal of obtaining a homogeneous sample suitable for structure determination. Taken together, DMD/PRIME20 results contribute new insights into cytotoxic myocilin aggregates, specifically, by predicting possible molecular arrangements of the amyloid core structure of the full-length mOLF for further study.
Supplementary Material
Acknowledgements
This work was supported by National Institutes of Health (NIH) Grant RO1EB006006 (to C. K. H), by NIH R01EY021205 (to R.L.L.) and by National Institute on Aging of NIH R01AG045703 (to A. K. P). Partial support was provided by the National Science Foundation Research Triangle Materials Research Science and Engineering Centers Grant DMR-1121107. The authors declare that they have no conflicts of interest.
Footnotes
Supporting Information
Bioinformatics analysis of amyloid forming region of full length myocilin protein, simulation snapshots of P1, P2 and P3 aggregates formed from ten independent simulation runs.
References
- 1.Hill SE; Donegan RK; Lieberman RL The Glaucoma-Associated Olfactomedin Domain of Myocilin Forms Polymorphic Fibrils That Are Constrained by Partial Unfolding and Peptide Sequence. J. Mol. Biol 2014, 426, 921–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Orwig SD; Perry CW; Kim LY; Turnage KC; Zhang R; Vollrath D; Schmidt-Krey I; Lieberman RL Amyloid Fibril Formation by the Glaucoma-Associated Olfactomedin Domain of Myocilin. J. Mol. Biol 2012, 421, 242–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tamm ER Myocilin and Glaucoma: Facts and Ideas. Prog. Retin. Eye Res 2002, 21, 395–428. [DOI] [PubMed] [Google Scholar]
- 4.Acott TS; Kelley MJ; Keller KE; Vranka JA; Abu-Hassan DW; Li XB; Aga M; Bradley JM Intraocular Pressure Homeostasis: Maintaining Balance in a High-Pressure Environment. J. Ocul. Pharmacol. Ther 2014, 30, 94–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yam GHF; Gaplovska-Kysela K; Zuber C; Roth J Aggregated Myocilin Induces Russell Bodies and Causes Apoptosis - Implications for the Pathogenesis of Myocilin-caused Primary Open-angle Glaucoma. Am. J.Pathol 2007, 170, 100–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu YH; Vollrath D Reversal of Mutant Myocilin Non-secretion and Cell Killing: Implications for Glaucoma. Hum. Mol. Genet 2004, 13, 1193–1204. [DOI] [PubMed] [Google Scholar]
- 7.Zhou ZH; Vollrath D A Cellular Assay Distinguishes Normal and Mutant TIGR/Myocilin Protein. Hum. Mol. Genet 1999, 8, 2221–2228. [DOI] [PubMed] [Google Scholar]
- 8.Stothert AR; Suntharalingam A; Huard DJE; Fontaine SN; Crowley VM; Mishra S; Blagg BSJ; Lieberman RL; Dickey CA Exploiting the Interaction Between Grp94 and Aggregated Myocilin to Treat Glaucoma. Hum. Mol. Genet 2014, 23, 6470–6480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Suntharalingam A; Abisambra JF; O’Leary JC; Koren J; Zhang B; Joe MK; Blair LJ; Hill SE; Jinwal UK; Cockman M et al. Glucose-regulated Protein 94 Triage of Mutant Myocilin through Endoplasmic Reticulum-associated Degradation Subverts a More Efficient Autophagic Clearance Mechanism. J. Biol. Chem 2012, 287, 40661–40669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Baldwin AJ; Knowles TPJ; Tartaglia GG; Fitzpatrick AW; Devlin GL; Shammas SL; Waudby CA; Mossuto MF; Meehan S; Gras SL et al. Metastability of Native Proteins and the Phenomenon of Amyloid Formation. J. Am. Chem. Soc 2011, 133, 14160–14163. [DOI] [PubMed] [Google Scholar]
- 11.Hartl FU; Hayer-Hartl M Converging Concepts of Protein Folding in vitro and in vivo. Nat. Struct. Mol. Biol 2009, 16, 574–581. [DOI] [PubMed] [Google Scholar]
- 12.Tycko R Molecular Structure of Amyloid Fibrils: Insights from Solid-state NMR. Q. Rev. Biophys 2006, 39, 1–55. [DOI] [PubMed] [Google Scholar]
- 13.Knowles TPJ; Vendruscolo M; Dobson CM The Amyloid State and its Association with Protein Misfolding Diseases. Nat. Rev. Mol. Cell Biol. 2014, 15, 384–396. [DOI] [PubMed] [Google Scholar]
- 14.Donegan RK; Hill SE; Freeman DM; Nguyen E; Orwig SD; Turnage KC; Lieberman RL Structural Basis for Misfolding in Myocilin-associated Glaucoma. Hum. Mol. Genet 2015, 24, 2111–2124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gremer L; Schölzel D; Schenk C; Reinartz E; Labahn J; Ravelli RBG; Tusche M; Lopez-Iglesias C; Hoyer W; Heise H; Willbold D; Schröder GF Fibril Structure of Amyloid-β(1–42) by Cryo-electron Microscopy. Science 2017, 358, 116–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wälti MA; Ravotti F; Arai H; Glabe CG; Wall JS; Böckmann A; Güntert P; Meier BH; Riek R Atomic-resolution Structure of a Disease-relevant Aβ(1–42) Amyloid Fibril. Proc. Natl. Acad. Sci. U. S. A 2016, 113, E4976–E4984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Colvin MT; Silvers R; Ni QZ; Can TV; Sergeyev I; Rosay M; Donovan KJ; Michael B; Wall J; Linse S; Griffin RG Atomic Resolution Structure of Monomorphic Aβ42 Amyloid Fibrils. J. Am. Chem. Soc 2016, 138, 9663–9674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nguyen HD; Hall CK Molecular Dynamics Simulations of Spontaneous Fibril Formation by Random-coil Peptides. Proc. Natl. Acad. Sci. U. S. A 2004, 101, 16180–16185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cheon M; Chang I; Hall CK Extending the PRIME Model for Protein Aggregation to All 20 Amino Acids. Proteins: Struct., Funct., Bioinf 2010, 78, 2950–2960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Alder BJ; Wainwright TE Stuides in Molecular Dynamics .1. General Method. J. Chem. Phys 1959, 31, 459–466. [Google Scholar]
- 21.Smith AV; Hall CK Alpha-helix Formation: Discontinuous Molecular Dynamics on an Intermediate-resolution Protein Model. Proteins: Struct., Funct., Genet 2001, 44, 344–360. [DOI] [PubMed] [Google Scholar]
- 22.Nguyen HD; Hall CK Spontaneous Fibril Formation by Polyalanines; Discontinuous Molecular Dynamics Simulations. J. Am. Chem. Soc 2006, 128, 1890–1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang Y; Shao Q; Hall CK N-terminal Prion Protein Peptides (PrP(120–144)) Form Parallel In-register β-Sheets via Multiple Nucleation-dependent Pathways. J. Biol. Chem 2016, 291, 22093–22105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Muñoz V; Serrano L Elucidating the Folding Problem of Helical Peptides using Empirical Parameters. III.Temperature and pH Dependence. J. Mol. Biol 1995, 245, 297–308. [DOI] [PubMed] [Google Scholar]
- 25.Schaefer J; Stejskal EO; Buchdahl R High-resolution Carbon-13 Nuclear Magnetic Resonance Study of Some Solid, Glassy Polymers. Macromolecules 1975, 8, 291–296. [Google Scholar]
- 26.Yannoni CS High-resolution NMR in Solids: the CPMAS Experiment. Acc. Chem. Res 1982, 15 (7), 201–208. [Google Scholar]
- 27.Bennett AE; Rienstra CM; Auger M; Lakshmi KV; Griffin RG Heteronuclear Decoupling in Rotating Solids. J. Chem. Phys 1995, 103, 6951–6958. [Google Scholar]
- 28.Cheon M; Chang I; Hall CK Spontaneous Formation of Twisted A beta(16–22) Fibrils in Large-Scale Molecular-Dynamics Simulations. Biophys. J 2011, 101, 2493–2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cheon M; Hall CK; Chang I Structural Conversion of A beta(17–42) Peptides from Disordered Oligomers to U-Shape Protofilaments via Multiple Kinetic Pathways. PLOS Comput. Biol 2015, 11, e1004258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang Y; Latshaw DC; Hall CK Aggregation of Aβ(17–36) in the Presence of Naturally Occurring Phenolic Inhibitors Using Coarse-Grained Simulations. J. Mol. Biol 2017, 429, 3893–3908. [DOI] [PubMed] [Google Scholar]
- 31.Paravastu AK; Leapman RD; Yau WM; Tycko R Molecular Structural Basis for Polymorphism in Alzheimer’s Beta-amyloid Fibrils. Proc. Natl. Acad. Sci. U. S. A 2008, 105, 18349–18354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Petkova AT; Ishii Y; Balbach JJ; Antzutkin ON; Leapman RD; Delaglio F; Tycko R A Structural Model for Alzheimer’s Beta-amyloid Fibrils Based on Experimental constraints from solid state NMR. Proc. Natl. Acad. Sci. U. S. A 2002, 99, 16742–16747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fritzsching KJ; Yang Y; Schmidt-Rohr K; Hong M Practical Use of Chemical Shift Databases for Protein Solid-state NMR: 2D Chemical Shift Maps and Amino-acid Assignment with Secondary-structure Information. J. Biomol. NMR 2013, 56, 155–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wishart DS; Bigam CG; Holm A; Hodges RS; Sykes BD 1H, 13C and 15N Random Coil NMR Chemical Shifts of the Common Amino Acids. I. Investigations of Nearest-Neighbor Effects. J. Biomol. NMR 1995, 5, 67–81. [DOI] [PubMed] [Google Scholar]
- 35.Ulrich EL; Akutsu H; Doreleijers JF; Harano Y; Ioannidis YE; Lin J; Livny M; Mading S; Maziuk D; Miller Z et al. BioMagResBank. Nucleic Acids Res. 2008, 36, D402–D408. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.