

Abstract
Objective
We aimed to assess whether differences in energy metabolism in fibroblast cell lines derived from patients with Huntington disease were associated with age at onset independent of the cytosine-adenine-guanine (CAG) repeat number in the mutant allele.
Methods
For this study, we selected 9 pairs of patients with Huntington disease matched for mutant CAG repeat size and sex, but with a difference of at least 10 years in age at onset, using the Leiden Huntington disease database. From skin biopsies, we isolated fibroblasts in which we (1) quantified the ATP concentration before and after a hydrogen-peroxide challenge and (2) measured mitochondrial respiration and glycolysis in real time, using the Seahorse XF Extracellular Flux Analyzer XF24.
Results
The ATP concentration in fibroblasts was significantly lower in patients with Huntington disease with an earlier age at onset, independent of calendar age and disease duration. Maximal respiration, spare capacity, and respiration dependent on complex II activity, and indices of mitochondrial respiration were significantly lower in patients with Huntington disease with an earlier age at onset, again independent of calendar age and disease duration.
Conclusions
A less efficient bioenergetics profile was found in fibroblast cells from patients with Huntington disease with an earlier age at onset independent of mutant CAG repeat size. Thus, differences in bioenergetics could explain part of the residual variation in age at onset in Huntington disease.
Huntington disease, a devastating neurodegenerative disorder, is caused by an elongated cytosine-adenine-guanine (CAG) repeat sequence in exon 1 of the huntingtin gene (HTT).1–3 The length of the CAG repeat sequence accounts for 50%–70% of the variation in age at onset, leaving a substantial amount of unexplained variation, which could be attributed to genetic and environmental modifiers.4,5 In addition to progressive motor disturbances, neuropsychiatric symptoms, and cognitive decline, patients with Huntington disease and premanifest mutation carriers suffer from unintended weight loss.6,7 Recently, we demonstrated that this weight loss was associated with a faster rate of disease progression independent of CAG repeat number.8 Various evidence indeed indicates that disturbances in energy metabolism and mitochondrial defects play a role in Huntington disease pathology.9–11 Furthermore, mitochondrial metabolism was found to be impaired in peripheral tissues of patients with Huntington disease, including lymphoblastoid cell lines and skin fibroblasts.12–15 In support of the association between mitochondrial defects and Huntington disease symptomology, Huntington disease characteristics emerged in humans after accidental exposure to 3-nitropropionic acid (3-NP), a mitochondrial toxin that selectively inhibits the activity of mitochondrial complex II.16–19 However, to what extent differences in energy metabolism are associated with the onset of Huntington disease symptoms independent of the CAG repeat number is unknown. Therefore, the aim of our study was to investigate whether differences in energy metabolism were present in fibroblast cell lines from patients with Huntington disease with identical CAG repeat sizes but a large difference in age at onset.
Methods
Participants
From the Leiden Huntington disease database that contained data on 356 patients with Huntington disease, we selected 9 pairs of patients with Huntington disease older than 18 years, matched for sex and CAG repeat length, but with a large difference in age at onset, as defined by an expert neurologist (R.A.C.R.) based on motor, psychiatric, and/or cognitive symptoms. As a cutoff, we used a difference in age at onset of at least 10 years between each pair of patients. Aside from the age at onset derived from the date of clinical Huntington disease, we also noted the age at onset as estimated by the rater based on patient information and the age at onset of different Huntington disease symptoms (table 1). Exclusion criteria were presence of inflammatory diseases, an active infectious disease, and the use of anti-inflammatory or immunosuppressive drugs (e.g., non-steroidal anti-inflammatory drugs, and corticosteroids) or antioxidants (e.g., vitamin C) in case temporary cessation of medication use (for about 7 times the drug's half-life) was not possible before sampling.
Table 1.
Age at onset of Huntington disease symptoms

Standard protocol approvals, registrations, and patient consents
The study protocol was approved by the local ethics committee, and written informed consent was obtained from all participants.
Sampling and cell culture
During a regular visit to our outpatient clinic, we obtained phenotypic data and a small skin sample (i.e., 3 mm diameter) from the upper thigh of each participant via a punch biopsy. Fibroblasts from the skin samples were cultured in minimal essential medium (Gibco #10370-07) supplemented with 15% heat-inactivated fetal bovine serum (Gibco #10270106), 1% penicillin-streptomycin (10,000 U/mL, Gibco #15140122) and 1% GlutaMAX supplement (Gibco #35050061), and stored in a humidified incubator at 37°C with 5% CO2. For the experiments described below, the fibroblasts were grown up to a maximum of 15 passages and harvested by trypsinization with trypsin-EDTA (0.05%, Gibco #25300054) at 37°C.
ATP concentration under oxidative stress
To assess whether bioenergetics differences in fibroblasts' response to oxidative stress between the matched pairs were present, we quantified the ATP concentration in the fibroblasts after subjecting them to 0.5 mM H2O2 for 0, 5, 10, and 15 minutes. In solid 96-well plates, we plated 12 replicates per cell line (i.e., 3 replicates per exposure period) with 30,000 cells in 100 µL regular culturing medium per well. Per plate, we included 14 blank wells (i.e., containing no cells) to which different ATP standard concentrations would later be added. Afterward, we incubated the plate for a minimum of 2 hours in a humidified incubator at 37°C with 5% CO2 to allow the cells to attach.
We applied oxidative stress to the fibroblasts by replacing the regular culturing medium with a medium containing 0.5 mM H2O2 and 5% fetal bovine serum for the appointed stress periods.20 To block the effect of H2O2 at the end of this period, we added catalase from bovine liver (50 U/mL medium, Sigma-Aldrich #C9322-1G)20 and replaced the 0.5 mM H2O2 medium with 250 µL per well of the regular culturing medium.
We quantified the ATP concentration of the fibroblasts per well with the Luminescent ATP detection Assay Kit (Abcam #ab113849) and added different concentrations of the ATP standard (i.e., 0, 10, 100, 1,000, 10,000, 100,000, and 1,000,000 nM) provided by the kit to the blank wells. Luminescence was assessed with Perkin Elmer Multimode Plate Reader, Victor X3. We quantified the ATP concentration per well by creating an ATP standard curve with a corresponding equation based on the luminescence per ATP standard. Using this standard curve equation, we determined the ATP concentration according to the average luminescence of the 3 wells per time point per cell line.
Mitochondrial respiration and glycolysis
Using the Seahorse XF Extracellular Flux Analyzer XF24, we could measure mitochondrial respiration and glycolysis simultaneously and in real time in our fibroblast cell lines. Respiration was measured as the oxygen consumption rate (OCR), and glycolysis was measured as the extracellular acidification rate (ECAR). In addition, the Seahorse allowed for the injection of 4 toxins during the experimental run and could monitor their effects over time. In succession, we injected the following toxins: 1 µM oligomycin (ATP-synthase inhibitor); 1 µM carbonyl, cyanide-4-(trifluoromethoxy) phenylhydrazone (or cabonyl cyanide-p-trifluoromethox-yphenyl-hydrazon [FCCP], oxidative phosphorylation uncoupler); 1M 3-nitropropionic acid (or 3-NP, complex II inhibitor), and 1 µM antimycin A (complex III inhibitor). Fibroblasts were plated 1 day in advance of the experiment at 60,000 cells per well in a XF24 cell culture microplate. This density resulted in confluent cultures in which cell growth was blocked because of contact inhibition, avoiding potential biases because of different growth rates between fibroblast cell lines.21 On the day of the experiment, the regular fibroblast medium was removed, and the cells were washed twice with XF assay medium at 37°C, supplemented with 5 mM glucose and 1 mM sodium pyruvate, and the medium was buffered at pH 7.4. Subsequently, 675 µL of the XF assay medium was added to each well, and the cells were incubated for 60 minutes in a 37°C incubator without CO2 to allow the cells to equilibrate to the new medium.
During the experiment, 4 measurements were taken at baseline for both OCR and ECAR. Afterward, 3 measurements were taken after every toxin injection. From these values, we calculated 6 OCR parameters and 2 ECAR parameters. Basal respiration was defined as the average OCR values at baseline. The average of the 3 OCR values after oligomycin injection was defined as the OCR due to proton leak. Maximal respiration was calculated by taking the average OCR of the 3 measurements after FCCP injection. The average OCR value after injection of 3-NP was defined as “respiration after 3-NP injection,” and the average respiration after antimycin A injection was defined as “non-mitochondrial respiration.” From these OCR parameters, another 3 parameters were calculated: respiration dedicated to ATP production (defined as basal respiration minus proton leak), spare capacity (defined as maximal respiration minus basal respiration), and respiration dependent on complex II activity (defined as maximal respiration minus respiration after 3-NP injection). Basal glycolysis was defined as the average ECAR of the 4 baseline ECAR measurements, and the increase in glycolysis after blocking ATP synthase was calculated by subtracting the basal glycolysis from the average ECAR after FCCP injection. To ensure that differences in the absolute averages were not due to variations in basal respiration and glycolysis, we also calculated the mitochondrial measurements as percentages of basal respiration and glycolysis.
Statistical analysis
To account for both the correlation within matched pairs, as well as the correlation due to serial measurements in time during each trial, we applied generalized linear mixed-effects models to analyze the results of the bioenergetics experiments. We set the calculated average ATP concentrations after oxidative stress of every cell line per time point as the target variable. Because of an exponential association between the ATP concentrations and time, we used the natural logarithmic transform of the index variable as the target variable. Group (i.e., earlier age at onset vs later age at onset), disease duration, calendar age at the time of biopsy, and time of exposure to H2O2 were included as fixed effects. Furthermore, we included a random intercept for each patient pair, as well as a random slope for time of exposure to H2O2, to adequately account for both the matching between each pair of patients and the correlated measurements on each individual during the experiments. For mitochondrial respiration and glycolysis, we analyzed every calculated functional index separately. For each functional index, we defined the absolute OCR and ECAR or the relative OCR and ECAR as the target variables and constructed models as described above. For all models, we used an unstructured random effect covariance matrix and robust estimations of covariance, which result in consistent parameter estimates even if model assumptions are violated. All models were checked both graphically and analytically. All tests were 2-tailed, and the threshold for statistical significance (i.e., α) was set at 0.05. All analyses were performed in SPSS version 23.0 (IBM SPSS Statistics for Windows, IBM Corp).
Data availability
Additional data will be made available at the request of other investigators.
Results
Differences in age at onset
The age at onset in the 18 selected patients with Huntington disease ranged between 22 and 73 years, and the average difference in age at onset between patient pairs was 19.2 years. The patient pairs had CAG repeat sequences between 41 and 46 repeats, and 5 of the 9 pairs were men. Furthermore, the symptoms at onset of disease varied per patient between motor, psychiatric, and cognitive symptoms (table 1).
ATP concentrations were lower in the skin fibroblasts of patients with Huntington disease with an earlier age at onset
In all fibroblast cell lines, the ATP concentration decreased over the time exposed to 0.5 mM H2O2. The decrease was exponential (figure 1 and figure e-1, links.lww.com/NXG/A84). Therefore, we used the natural logarithmic transform of the ATP concentration (lnATP) as the target variable in the analysis. We found that lnATP was significantly lower in patients with an earlier age at onset compared with the matched patients with a later age at onset (figure 1 and table 2). This difference was present at baseline in 7 of the 9 couples and continued to be evident throughout the period the cells were subjected to oxidative stress in 7 couples (figure e-1). Because the difference in age at onset as estimated by the rater was missing in couple number 2, causing the data of this couple to be less reliable (table 1), we performed a sensitivity analysis by excluding these patients. Exclusion of these patients from the analysis did not materially alter our results (table 2).
Figure 1. Average ATP concentration in the skin fibroblasts of patients with Huntington disease exposed to oxidative stress over time.
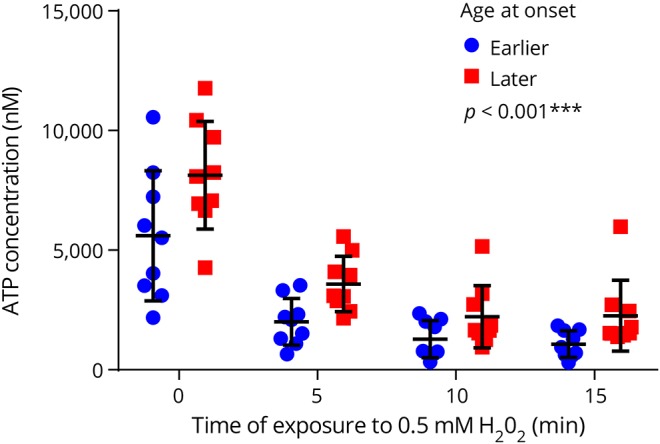
In all fibroblast cell lines, the average ATP concentration decreased exponentially as the time exposed to oxidative stress increased. At every time point, the group of patients with an earlier age at onset had a lower ATP concentration compared with the group of patients with a later age at onset. p < 0.001*** indicates the significant effect of group (i.e., earlier or later age at onset) on the ATP concentration determined using linear mixed-effects models. Error bars indicate ±SD.
Table 2.
Association between the logarithmic transform of the average ATP concentration and earlier vs later age at onset in HD

Mitochondrial indices were lower in the skin fibroblasts of patients with Huntington disease with an earlier age at onset
The average OCR and the ECAR per Huntington disease patient group during the experiment are presented in figure e-2, links.lww.com/NXG/A84. Neither the absolute mitochondrial parameters nor the parameters expressed as percentage of basal respiration differed significantly per group before correction for disease duration and calendar age (figure e-3). However, after correction, we found the absolute OCR averages to be significantly different between the 2 groups in 4 of the 8 estimated parameters (table 3). The average maximal respiration was lower in the group of patients with Huntington disease with an earlier age at onset. All 9 patients with an earlier age at onset had a lower maximal respiration compared with the matched patients with a later age at onset. Similarly, in 8 of the 9 couples, the spare capacity was lower in the earlier age at onset group. Furthermore, the respiration dedicated to ATP production (in 6 couples) and the respiration dependent on complex II activity (in 8 couples) were significantly lower in the group of patients with Huntington disease with an earlier age at onset. The average basal respiration was not markedly different between the 2 groups. Average OCR as a percentage of the basal respiration differed in 3 parameters between the groups, including maximal respiration (in 8 couples), spare capacity (in 8 couples), and respiration dependent on complex II activity (in 9 couples). No difference in average respiration dedicated to ATP production was found when this variable was defined as a percentage of basal respiration. The glycolysis parameters did not differ between the groups, although the difference in basal glycolysis showed a trend toward statistical significance in which the group of patients with Huntington disease with an earlier age at onset had a lower average basal ECAR. To illustrate these effects, we plotted the unadjusted values, as well as the predicted values, calculated using the model estimates after adjustment for disease duration and calendar age (figure 2A-I, and figure e-3). The effects did not change after excluding couple 2 in the sensitivity analysis (table 4).
Table 3.
Association between the calculated mitochondrial stress test parameters and age at onset in Huntington disease


Figure 2. Adjusted estimates of the functional indices calculated from the mitochondrial stress test per age group at symptom onset in Huntington disease.

There was no difference in basal respiration between the 2 groups (A). The absolute mitochondrial respiration dedicated to ATP production was significantly lower in the group of patients with Huntington disease with an earlier age at onset (B). Both the absolute maximal respiration and the maximal respiration as a percentage of the basal respiration were significantly lower in the group of patients with Huntington disease with an earlier age at onset (C and D). The absolute spare capacity and the spare capacity as a percentage of the basal respiration were significantly lower in the group of patients with Huntington disease with an earlier age at onset (E and F). The absolute respiration dependent on complex II activity and the respiration dependent on complex II activity as a percentage of the basal respiration were also significantly lower in the group of patients with Huntington disease with an earlier age at onset (G and H). The basal glycolysis did not differ significantly between the 2 groups (I). Adjusted estimates = values adjusted for disease duration and calendar age at the time of biopsy. Error bars indicate ±SD. *p-value < 0.05. **p-value < 0.01. ***p-value < 0.001. ECAR = extracellular acidification rate; OCR = oxygen consumption rate.
Table 4.
Sensitivity analysis of the association between the mitochondrial stress test parameters and age at onset in Huntington disease

Of interest, disease duration and calendar age were also significantly associated with several indices of mitochondrial bioenergetics. Longer disease duration was accompanied by lower absolute values of basal respiration, maximal respiration, respiration after 3-NP injection, respiration dependent on complex II activity, and glycolysis after blocking ATP synthase. Furthermore, patients with a higher calendar age had significantly lower levels of maximal respiration, respiration dedicated to ATP production, spare capacity, respiration dependent on complex II activity, basal glycolysis, and glycolysis after blocking ATP synthase. In addition, the parameters maximal respiration, spare capacity, and respiration dependent on complex II activity were also significantly lower with a higher calendar age when expressed as percentages of the basal respiration (table 3).
Discussion
We found lower ATP concentrations in the skin fibroblasts of patients with Huntington disease with an earlier age at onset compared with those with a later age at onset, independent of CAG repeat number, sex, calendar age, and disease duration. In addition, we demonstrated that the fibroblasts of patients with Huntington disease with an earlier age at onset exhibited lower mitochondrial respiration indices, including lower maximal respiration, spare capacity, and respiration dependent on complex II activity in an absolute sense, as well as relative to their basal respiration levels. Furthermore, we found that disease duration and age at biopsy were also significantly associated with several parameters of mitochondrial bioenergetics. Although mitochondrial defects have been extensively documented before in Huntington disease, to our knowledge, we are the first to demonstrate that differences in bioenergetics were associated with age at onset among patients with Huntington disease, and thereby, could be an important target for future therapeutic interventions.
The fact that the ATP concentration was lower in patients with Huntington disease with an earlier age at onset suggests that the production of ATP in the cells of these patients may be impaired to a greater extent compared with their counterparts or that the cells of patients with a later age at onset carry a mechanism that protects their ATP metabolism. The presence of a potential problem with ATP production was supported by comparable differences in various indices of mitochondrial respiration. Maximal respiration and mitochondrial spare capacity are measures of the ability of mitochondria to react to increased energy demands and are critical for neuronal survival.22–24 These measures were lower in patients with an earlier age at onset, suggesting that neurons in these patients could be more vulnerable to damage and death, thus causing Huntington disease symptoms to start at a younger age. Of interest, a previous study showed a correlation between decreased levels of spare capacity and increased reactive oxygen species and mitochondrial DNA lesions in mutant Huntington disease striatal immortalized neuronal cells.15 Respiration dependent on complex II activity was also lower in patients with Huntington disease with an earlier age at onset. Different studies showed that in postmortem samples of the striatum and cerebral cortex of patients with Huntington disease, complex II of the mitochondrial electron transport chain displayed reduced activity, which was associated with diminished expression of 2 complex II subunits.25–29 The exact mechanism as to how the mutated huntingtin protein (HTT) causes the loss of complex II is unknown. Possible hypotheses include the direct association of mutant HTT with the mitochondrial membrane, causing decreased import of subunits into the mitochondria, increased degradation, or abnormal assembly.13,29,30 Furthermore, mutant HTT has been shown to increase cellular oxidative stress, which was associated with impaired activity of complex II in a yeast model of Huntington disease.31–33 Our results here suggest that the reduced expression of complex II is more pronounced in patients with an earlier age at onset independent of CAG repeat length, which could perhaps be due to a more potent association of mutant HTT with the mitochondrial membrane or a higher vulnerability to oxidative stress.
Candidate studies found that a single nucleotide polymorphism in PPARGC1A, encoding the mitochondrial regulator peroxisome proliferator-activated receptor gamma coactivator-1α (PGC-1α), affected the age at onset in 3 European Huntington disease cohorts, suggesting that our findings might have a genetic origin.34,35 Moreover, in striata from patients with Huntington disease and different rodent Huntington disease models, the expression of PGC-1α mRNA was reduced, and upregulating this expression caused prevention of striatal neuronal atrophy, improvement of motor deficits, and protection against mitochondrial dysfunction and cell death, implying that PGC-1α expression may modify mutant HTT–induced mitochondrial toxicity.36–38 Together with our results, these findings indicate that differences in PGC-1α expression and the consequential variations in bioenergetics profile may result in additional variation in age at onset in Huntington disease.
We found that as patients suffered from Huntington disease for a longer period of time, several indices of mitochondrial respiration were significantly lower, independent of calendar age. This finding is in line with the fact that Huntington disease pathogenesis is known to involve mitochondrial deficits.39,40 Therefore, a reasonable derivative is that these mitochondrial deficits become worse as the disease progresses. Mitochondrial function is also known to decrease with age.41 In accordance, we found that as the calendar age of patients increased, several parameters of mitochondrial function decreased. The aging process itself is unlikely to have accounted for our main finding (i.e., a better bioenergetics profile in late-onset patients), given that patients with a later age at onset were older at the time of biopsy (table 1).
Our study had several limitations. First, although Huntington disease is primarily a neurologic disease, we were able to find differences between patients with Huntington disease with an early onset and late onset of symptoms in skin fibroblasts. The fact that we could acquire relevant results in these relatively easy to obtain cells is intriguing and supports the use of fibroblast cell models in the research of neurodegenerative diseases. However, these cells are not the primary cells of interest. Therefore, repeating similar experiments in a neuronal cell model, such as striatal neurons derived from induced pluripotent stem cells, is warranted. Second, establishing the age at onset in Huntington disease is subjective.42 In order to achieve additional certainty concerning this age, we noted the age at Huntington disease diagnosis, as well as the age estimated by the rater, which were analogous in 8 of the 9 couples. In addition, our results did not change after excluding the couple in which the rater-estimated age at onset was missing for 1 patient, illustrating that our results are robust and reliable. Last, we performed several statistical analyses without applying a correction for multiple testing, given our relatively small sample size, as well as the fact that bioenergetics assessments are highly correlated and, thus, cannot be regarded as independent. Nevertheless, to obtain results with a higher level of confidence, future research investigating similar parameters should include a larger number of patients.
We demonstrated an association between age at onset and the bioenergetics profile in patients with Huntington disease. Thus, differences in bioenergetics could explain part of the residual variation in age at onset among patients with Huntington disease, whereas therapies aimed at enhancing mitochondrial function may delay disease onset. However, further research into the mechanisms mediating the association between bioenergetics and age at onset are needed to develop novel therapies aimed at delaying symptom onset and disease progression in Huntington disease.
Acknowledgment
The authors cordially thank all patients and caregivers involved in the experiments for their valuable time and efforts. The authors also thank Ms. L.J. Schipper, M.D., for assistance in taking skin biopsies.
Glossary
- CAG
cytosine-adenine-guanine
- ECAR
extracellular acidification rate
- OCR
oxygen consumption rate
Author contributions
S.L. Gardiner and N.A. Aziz contributed to the conception and design of the study and to the acquisition and analysis of data. C. Milanese and W.M.C. van Roon-Mom offered crucial supervision during the experiments. All authors contributed to drafting the text and preparing the figures.
Study funding
NAA is supported by a VENI-grant (#91615080) from the Netherlands Organization for Scientific Research and a Marie Sklodowska-Curie Individual Fellowship grant from the European Union (Horizon 2020, #701130).
Disclosure
S. Gardiner, C. Milanese, M.W. Boogaard, R.A.M. Buijsen, and M. Hogenboom report no disclosures. R.A.C. Roos is a consultant for uniQure. P.G. Mastroberardino serves on the editorial boards of Neurobiology of Disease, Frontiers in Cellular Neuroscience, and Cell Death and Disease; is a consultant for the Neurological Institute; and has received research support from Dorpmans-Wigmans Stichting. W.M.C. van Roon-Mom has served on the medical advisory board of the ADCA Patients' Association; is the main inventor on 2 published patent applications (WO2012/018257 and WO2015/053624) regarding exon skipping approaches for neurodegenerative diseases; and is coinventor on 1 published patent application on exon skipping approaches for SCA3 (WO 2017/053781). N.A. Aziz has received research support from the Netherlands Organization for Scientific Research (NWO), the European Union, and the European Huntington's Disease Network. Full disclosure form information provided by the authors is available with the full text of this article at Neurology.org/NG.
References
- 1.Bruyn G. Huntington's chorea: historical, clinical and laboratory synopsis. In: Vinken P, Bruyn G, editors. Handbook of Clinical Neurology. Amsterdam: Elsevier; 1968:298–378. [Google Scholar]
- 2.A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. The Huntington's Disease Collaborative Research Group. Cell 1993;72:971–983. [DOI] [PubMed] [Google Scholar]
- 3.Langbehn DR, Hayden MR, Paulsen JS. CAG-repeat length and the age of onset in Huntington disease (HD): a review and validation study of statistical approaches. Am J Med Genet B Neuropsychiatr Genet 2010;153B:397–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wexler NS, Lorimer J, Porter J, et al. Venezuelan kindreds reveal that genetic and environmental factors modulate Huntington's disease age of onset. Proc Natl Acad Sci USA 2004;101:3498–3503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gusella JF, MacDonald ME, Lee JM. Genetic modifiers of Huntington's disease. Mov Disord 2014;29:1359–1365. [DOI] [PubMed] [Google Scholar]
- 6.Aziz NA, van der Burg JM, Landwehrmeyer GB, Brundin P, Stijnen T, Roos RA. Weight loss in Huntington disease increases with higher CAG repeat number. Neurology 2008;71:1506–1513. [DOI] [PubMed] [Google Scholar]
- 7.Mochel F, Charles P, Seguin F, et al. Early energy deficit in Huntington disease: identification of a plasma biomarker traceable during disease progression. PLoS One 2007;2:e647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.van der Burg JMM, Gardiner SL, Ludolph AC, Landwehrmeyer GB, Roos RAC, Aziz NA. Body weight is a robust predictor of clinical progression in Huntington disease. Ann Neurol 2017;82:479–483. [DOI] [PubMed] [Google Scholar]
- 9.Feigin A, Leenders KL, Moeller JR, et al. Metabolic network abnormalities in early Huntington's disease: an [(18)F]FDG PET study. J Nucl Med 2001;42:1591–1595. [PubMed] [Google Scholar]
- 10.Ciarmiello A, Cannella M, Lastoria S, et al. Brain white-matter volume loss and glucose hypometabolism precede the clinical symptoms of Huntington's disease. J Nucl Med 2006;47:215–222. [PubMed] [Google Scholar]
- 11.Koroshetz WJ, Jenkins BG, Rosen BR, Beal MF. Energy metabolism defects in Huntington's disease and effects of coenzyme Q10. Ann Neurol 1997;41:160–165. [DOI] [PubMed] [Google Scholar]
- 12.Sawa A, Wiegand GW, Cooper J, et al. Increased apoptosis of Huntington disease lymphoblasts associated with repeat length-dependent mitochondrial depolarization. Nat Med 1999;5:1194–1198. [DOI] [PubMed] [Google Scholar]
- 13.Panov AV, Gutekunst CA, Leavitt BR, et al. Early mitochondrial calcium defects in Huntington's disease are a direct effect of polyglutamines. Nat Neurosci 2002;5:731–736. [DOI] [PubMed] [Google Scholar]
- 14.Seong IS, Ivanova E, Lee JM, et al. HD CAG repeat implicates a dominant property of huntingtin in mitochondrial energy metabolism. Hum Mol Genet 2005;14:2871–2880. [DOI] [PubMed] [Google Scholar]
- 15.Siddiqui A, Rivera-Sanchez S, Castro Mdel R, et al. Mitochondrial DNA damage is associated with reduced mitochondrial bioenergetics in Huntington's disease. Free Radic Biol Med 2012;53:1478–1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.He F, Zhang S, Qian F, Zhang C. Delayed dystonia with striatal CT lucencies induced by a mycotoxin (3-nitropropionic acid). Neurology 1995;45:2178–2183. [DOI] [PubMed] [Google Scholar]
- 17.Ludolph AC, He F, Spencer PS, Hammerstad J, Sabri M. 3-Nitropropionic acid-exogenous animal neurotoxin and possible human striatal toxin. Can J Neurol Sci 1991;18:492–498. [DOI] [PubMed] [Google Scholar]
- 18.Brouillet E, Jenkins BG, Hyman BT, et al. Age-dependent vulnerability of the striatum to the mitochondrial toxin 3-nitropropionic acid. J Neurochem 1993;60:356–359. [DOI] [PubMed] [Google Scholar]
- 19.Brouillet E, Conde F, Beal MF, Hantraye P. Replicating Huntington's disease phenotype in experimental animals. Prog Neurobiol 1999;59:427–468. [DOI] [PubMed] [Google Scholar]
- 20.Mocali A, Caldini R, Chevanne M, Paoletti F. Induction, effects, and quantification of sublethal oxidative stress by hydrogen peroxide on cultured human fibroblasts. Exp Cel Res 1995;216:388–395. [DOI] [PubMed] [Google Scholar]
- 21.Ambrosi G, Ghezzi C, Sepe S, et al. Bioenergetic and proteolytic defects in fibroblasts from patients with sporadic Parkinson's disease. Biochim Biophys Acta 2014;1842:1385–1394. [DOI] [PubMed] [Google Scholar]
- 22.Kumar MJ, Nicholls DG, Andersen JK. Oxidative alpha-ketoglutarate dehydrogenase inhibition via subtle elevations in monoamine oxidase B levels results in loss of spare respiratory capacity: implications for Parkinson's disease. J Biol Chem 2003;278:46432–46439. [DOI] [PubMed] [Google Scholar]
- 23.Vesce S, Jekabsons MB, Johnson-Cadwell LI, Nicholls DG. Acute glutathione depletion restricts mitochondrial ATP export in cerebellar granule neurons. J Biol Chem 2005;280:38720–38728. [DOI] [PubMed] [Google Scholar]
- 24.Yadava N, Nicholls DG. Spare respiratory capacity rather than oxidative stress regulates glutamate excitotoxicity after partial respiratory inhibition of mitochondrial complex I with rotenone. J Neurosci 2007;27:7310–7317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gu M, Gash MT, Mann VM, Javoy-Agid F, Cooper JM, Schapira AH. Mitochondrial defect in Huntington's disease caudate nucleus. Ann Neurol 1996;39:385–389. [DOI] [PubMed] [Google Scholar]
- 26.Brennan WA Jr, Bird ED, Aprille JR. Regional mitochondrial respiratory activity in Huntington's disease brain. J Neurochem 1985;44:1948–1950. [DOI] [PubMed] [Google Scholar]
- 27.Browne SE, Bowling AC, MacGarvey U, et al. Oxidative damage and metabolic dysfunction in Huntington's disease: selective vulnerability of the basal ganglia. Ann Neurol 1997;41:646–653. [DOI] [PubMed] [Google Scholar]
- 28.Tabrizi SJ, Cleeter MW, Xuereb J, Taanman JW, Cooper JM, Schapira AH. Biochemical abnormalities and excitotoxicity in Huntington's disease brain. Ann Neurol 1999;45:25–32. [DOI] [PubMed] [Google Scholar]
- 29.Benchoua A, Trioulier Y, Zala D, et al. Involvement of mitochondrial complex II defects in neuronal death produced by N-terminus fragment of mutated huntingtin. Mol Biol Cell 2006;17:1652–1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Choo YS, Johnson GV, MacDonald M, Detloff PJ, Lesort M. Mutant huntingtin directly increases susceptibility of mitochondria to the calcium-induced permeability transition and cytochrome c release. Hum Mol Genet 2004;13:1407–1420. [DOI] [PubMed] [Google Scholar]
- 31.Browne SE, Ferrante RJ, Beal MF. Oxidative stress in Huntington's disease. Brain Pathol 1999;9:147–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bogdanov MB, Andreassen OA, Dedeoglu A, Ferrante RJ, Beal MF. Increased oxidative damage to DNA in a transgenic mouse model of Huntington's disease. J Neurochem 2001;79:1246–1249. [DOI] [PubMed] [Google Scholar]
- 33.Solans A, Zambrano A, Rodriguez M, Barrientos A. Cytotoxicity of a mutant huntingtin fragment in yeast involves early alterations in mitochondrial OXPHOS complexes II and III. Hum Mol Genet 2006;15:3063–3081. [DOI] [PubMed] [Google Scholar]
- 34.Che HV, Metzger S, Portal E, Deyle C, Riess O, Nguyen HP. Localization of sequence variations in PGC-1alpha influence their modifying effect in Huntington disease. Mol Neurodegener 2011;6:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Taherzadeh-Fard E, Saft C, Andrich J, Wieczorek S, Arning L. PGC-1alpha as modifier of onset age in Huntington disease. Mol Neurodegener 2009;4:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cui L, Jeong H, Borovecki F, Parkhurst CN, Tanese N, Krainc D. Transcriptional repression of PGC-1alpha by mutant huntingtin leads to mitochondrial dysfunction and neurodegeneration. Cell 2006;127:59–69. [DOI] [PubMed] [Google Scholar]
- 37.Chaturvedi RK, Calingasan NY, Yang L, Hennessey T, Johri A, Beal MF. Impairment of PGC-1alpha expression, neuropathology and hepatic steatosis in a transgenic mouse model of Huntington's disease following chronic energy deprivation. Hum Mol Genet 2010;19:3190–3205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hathorn T, Snyder-Keller A, Messer A. Nicotinamide improves motor deficits and upregulates PGC-1alpha and BDNF gene expression in a mouse model of Huntington's disease. Neurobiol Dis 2011;41:43–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bossy-Wetzel E, Petrilli A, Knott AB. Mutant huntingtin and mitochondrial dysfunction. Trends Neurosci 2008;31:609–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen CM. Mitochondrial dysfunction, metabolic deficits, and increased oxidative stress in Huntington's disease. Chang Gung Med J 2011;34:135–152. [PubMed] [Google Scholar]
- 41.Bratic A, Larsson NG. The role of mitochondria in aging. J Clin Invest 2013;123:951–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Orth M, Schwenke C. Age-at-onset in Huntington disease. PLoS Curr 2011;3:RRN1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Additional data will be made available at the request of other investigators.