

Abstract
Objective
To identify the genetic basis of a patient with symptoms of normokalemic sporadic periodic paralysis (PP) and to study the effect of KCNJ18 mutations.
Methods
A candidate gene approach was used to identify causative gene mutations, using Sanger sequencing. KCNJ18 promoter activity was analyzed in transfected HEK293 cells with a luciferase assay, and functional analysis of Kir2.6 channels was performed with the two-electrode voltage-clamp technique.
Results
Although we did not identify harmful mutations in SCN4A, CACNA1S, KCNJ2 and KCNE3, we detected a monoallelic four-fold variant in KCNJ18 (R39Q/R40H/A56E/I249V), together with a variant in the respective promoter of this channel (c.-542T/A). The exonic variants in Kir2.6 did not alter the channel function; however, luciferase assays revealed a 10-fold higher promoter activity of the c.-542A promoter construct, which is likely to cause a gain-of-function by increased expression of Kir2.6. We found that reducing extracellular K+ levels causes a paradoxical reduction in outward currents, similar to that described for other inward rectifying K+ channels. Thus, reducing the extracellular K+ levels might be a therapeutic strategy to antagonize the transcriptionally increased KCNJ18 currents. Consistently, treatment of the patient with K+ reducing drugs dramatically improved the health situation and prevented PP attacks.
Conclusions
We show that a promoter defect in the KCNJ18 gene is likely to cause periodic paralysis, as the observed transcriptional upregulation will be linked to increased Kir2.6 function. This concept is further supported by our observation that most of the PP attacks in our patient disappeared on medical treatment with K+ reducing drugs.
Familiar forms of periodic paralysis (PP) belong to a group of skeletal muscle disorders characterized by episodes of muscle paralysis, accompanied by decreased (hypoPP) or increased (hyperPP) serum potassium levels, which are associated with mutations in the genes encoding the skeletal muscle isoforms of the voltage-gated sodium channel SCN4A or the respective calcium channel CACNA1S.1–3 Andersen-Tawil-Syndrome (ATS) that results from loss-of-function mutations in the inward rectifying Kir2.1 channel is characterized by a unique triad of symptoms, which include PP, cardiac arrhythmias, as well as facial and skeletal malformations. In the skeletal muscle, the loss-of-function mutations in Kir2.1 are believed to cause a depolarization of the membrane potential.4
The most common form of sporadic PP (SPP) is the thyrotoxic PP (TPP), which is clinically similar to hypoPP or ATS. TPP can result from mutations in the KCNJ18 gene, encoding Kir2.6 channels5–7 that are under transcriptional regulation of thyroid hormones.8 TPP-causing mutations in Kir2.6 include loss-of-function mutations, similar to that in Kir2.1 and ATS, but also gain-of-function mutations8 that are believed to hyperpolarize the skeletal muscle, leading to a reduced excitability, as the membrane potential is too far from the action potential threshold.3
While analyzing a case of sporadic normokalemic PP, we identified a novel disease-causing mechanism, with a heterozygous variant in the KCNJ18 promoter that generates a strong increase in KCNJ18 transcription, which is likely to cause a hyperpolarization of the skeletal muscle membrane potentials, leading to inexcitability under normokalemic conditions.
Methods
Standard protocol approvals, registrations, and patient consents
Informed consent was provided by the patient and his relatives for genetic screening, as well as for publication of the data in its present form. Controls (n = 100) without any neurologic signs of paralysis were tested for the identified single-nucleotide variations (SNVs) of our index patient.9 The work was approved by the local ethics committee of the Philipps-University Marburg (study 10/03), and all procedures were in accordance with the Declaration of Helsinki of 1975, as revised in 1996.
Screen for genomic mutations
For mutation analysis, we Sanger sequenced the genomic DNA of the patient for mutations in all relevant candidate genes of PP, as recommended by the guidelines of the German Neurological Society (DGN). These included the following: SCN4A (NM_000334), CACNA1S (NM_000069), KCNJ2 (NM_000891,) KCNE3 (NM_005472), and KCNJ18 (NM_001194958).10–12 Novel detected SNVs were further assessed for their appearance in a cohort of 100 controls.
Functional analysis of detected mutations
Measurements of promoter activity in luciferase assays
We analyzed a 766-bp fragment of the KCNJ18 promoter upstream of exon 1 for promoter activity. Wild-type or mutant KCNJ18 promoter constructs were generated by cloning a 766-bp polymerase chain reaction (PCR) fragment from the patient's genomic DNA containing KpnI/NcoI linkers into the corresponding sites of pGL3-Basic (Promega). Transient transfection assays in HEK293 cells were performed in six-fold reactions using HeLafect (OZ Biosciences) with 0.5 µg KCNJ18 pGL3 construct and 0.1 µg internal control plasmid pcDNA3.1 CMV-LacZ (Invitrogen). Cells were maintained in Dulbecco modified Eagle medium supplemented with 10% fetal bovine serum and penicillin/streptomycin (100 µg/mL). Forty-eight hours after transfection, the cells were harvested and cellular luciferase expression was measured using the luciferase assay system (Promega) on a Glow-Max—instrument (Promega). The assay was replicated 8 times in independent transfection experiments. Site-directed mutagenesis using the Gene Tailor Site-Directed Mutagenesis System (Invitrogen) was used to validate functional mutations from luciferase assays. For this purpose, the deleterious effect of the mutation was restored by introduction of the corresponding normal base into the mutant KCNJ18 promoter construct.
Two-electrode voltage-clamp recordings of Kir2.6 channels in Xenopus oocytes
Ovarian lobes were dissected from mature Xenopus laevis toads anesthetized with tricaine and treated with collagenase (1 mg/mL, Worthington, type II) in OR2 solution (NaCl 82.5 mM, KCl 2 mM, MgCl2 1 mM, HEPES 5 mM, pH 7.4) for 120 minutes. Isolated oocytes were stored at 18°C in ND96 recording solution (NaCl 96 mM, KCl 2 mM, CaCl2 1.8 mM, MgCl2 1 mM, HEPES 5 mM, pH 7.5) supplemented with Na-pyruvate (275 mg/L), theophylline (90 mg/L), and gentamicin (50 mg/L). Wild-type KCNJ18 and mutant channels were subcloned into an oocyte expression vector and cDNA was linearized. cRNA was synthesized with mMESSAGE mMACHINE-Kit (Ambion). The quality of cRNA was tested using gel electrophoresis. Oocytes were injected with 50 nL (5 ng) of the wild-type KCNJ18 or a cRNA containing the single-nucleotide variants (SNVs) of the patient. Standard two-electrode voltage-clamp (TEVC) experiments were performed at room temperature (21–22°C) with an Axoclamp 900A amplifier, a Digidata 1440A, and pClamp10 software (Axon Instruments) 2 days after injection of oocytes with cRNA. Microelectrodes were fabricated from glass pipettes filled with 3-M KCl and had a resistance of 0.3–1.0 MΩ. To examine the effect of furosemide (10 µM, Sigma), currents were elicited by a voltage step from a holding potential of −100 to −150 mV, followed by a ramp to +40 mV in 0.8 seconds (and a final voltage step to 0 mV, not illustrated). The blocker was applied in the ND96 recording solution.
Statistical analysis
SigmaPlot/SigmaStat software (Systat Software) was used for statistical analyses. For luciferase expression analysis, the continuous variables were expressed as mean and standard deviations. In TEVC experiments, data were expressed as mean and SEM. Continuous variables were compared with the unpaired Student t test if they were normally distributed or the Mann-Whitney U test otherwise. For all tests, a p value ≤ 0.05 (*) was considered statistically significant and p values ≤ 0.001 (***) as highly significant.
Results
Clinical case description
The patient described here is a 69-year-old man with more than a 50-year-long history of severe attacks of weakness in both, legs and arms, speech disturbances, as well as recurring extreme muscle tiredness mostly in the late afternoon. These attacks may lead to a tetraplegia sometimes associated with fear of death. In these situations, the patient was always clear minded during these attacks. He was fully aware of the situation but was unable to express himself verbally because of severe motoric dysarthria. Impairment of the muscles responsible for speech is atypical for PP. Numerous EEGs, as well as EMGs, were without signs for epilepsy or myotonia, respectively. The family history was negative for any neurologic disorders. The mother died at the age of 70 years from stomach cancer, and the father died at the age of 77 years. The brother of the patient died at the age of 61 years from stomach and bone cancer. His children are both noncarriers of the c.-542T/A mutation and showed no clinical symptoms.
Despite various examinations by neurologists, psychiatrists, internists, and general practitioners in the past decades, no conclusive diagnosis was made. Repeated measurements of serum K+, Na+, Ca++, Cl−, and glucose before and during these attacks were found to be within the normal range. Treatment trials with antidepressants, neuroleptic and anticonvulsant therapy, as well as psychotherapeutic treatment, were unsuccessful.
Detection of nonsynonymous SNVs in the SCN4A, CACNA1S, and KCNJ18 genes
We consecutively screened the genes of the voltage-gated sodium channel type IV (SCN4A), the voltage-dependent L-type calcium channel 1S (CACNA1S), the potassium voltage-gated channel subfamily E regulatory subunit 3 (KCNE3), and the inwardly rectifying potassium channels J2 (KCNJ2) and J18 (KCNJ18) in the patient. DNA sequencing detected nine coding variants in the following genes: SCN4A, CACNA1S, and KCNJ18 (table 1). Variation analysis at the Exome Variant Server (EVS) revealed a high minor allele frequency (MAF) >5% and no deleterious effects of the variants as predicted by SIFT and Polyphen2, indicating that these variants are naturally occurring polymorphisms. No EVS data were available for variants of KCNJ18 (table 1). However, the four-fold SNV in Kir2.6, which is located on a single allele of our patient, has been recently identified as a natural variant, which apparently has no functional difference to wild-type channels.13
Table 1.
Detected coding variants in the following genes: SCN4A, CACNA1S, and KCNJ18

The four-fold nonsynonymous SNV in the patient's KCNJ18 gene does not alter the electrophysiologic characteristics of Kir2.6
The four-fold SNV in Kir2.6 R39Q/R40H/A56E/I249V was previously studied under high extracellular potassium concentration (20 mM), so only inward currents could be analyzed and not the physiologically relevant outward conductance and physiologic rectification pattern.13 Therefore, we recorded the variant under physiologic saline conditions by injecting similar amounts of cRNA in X laevis oocytes and recording currents 48 hours after injection. In TEVC recordings, the current rectification was indistinguishable between the two KCNJ18 variants (figure 1, A and B). A representative current trace is illustrated for the wild-type KCNJ18 (black) and the KCNJ18 SNVs (gray), respectively (figure 1A). The SNVs did not alter the rectification pattern or current amplitudes (figure 1, B and C), and the mean inward current amplitude analyzed at −140 mV was similar for the wild-type KCNJ18 and the SNVs (figure 1C), but no substantial differences in physiologically relevant outward currents were observed (figure 1B).
Figure 1. Voltage-clamp recording of wild-type KCNJ18 and the KCNJ18 SNVs expressed in Xenopus laevis oocytes.

(A) Representative current traces of wild-type KCNJ18 channels (upper panel) or KCNJ18 channels containing the SNVs R39Q/R40H/A56E/I249V (lower panel). For these experiments, 5 ng of the respective cRNA was injected and the indicated voltage protocol was applied. Currents were elicited by a voltage step from a holding potential of −100 to −150 mV, followed by a ramp to +50 mV in 0.8 seconds. (B) Mean current-voltage relationship of wild-type KCNJ18 (black) or the KCNJ18 construct containing the SNVs of the patient (gray) n = 13. (C) Relative current amplitudes at −140 mV normalized to wild-type KCNJ18. Values are expressed as mean ± SEM. n.s. = no of significant difference; SNV = single-nucleotide variation.
Allelic variation in the KCNJ18 promoter
Because the variants in the KCNJ18 channel did not affect mean current amplitudes, we assessed the promoter regions of SCN4A, CACNA1S, KCNJ2, and KCNE3 for SNVs based on published promoter sequence data from GeneCards.14 However, no sequence information for the KCNJ18 promoter was available. Therefore, we designed and sequenced a 766-bp PCR fragment according to the mapped and published promoter sequence of the KCNJ18 gene.8 No SNVs were found in the promoters of SCN4A, CACNA1S, KCNJ2, and KCNE3. Sequencing analysis of the KCNJ18 promoter detected 13 heterozygous SNVs. To elucidate the overall frequencies of these SNVs, we sequenced the DNA of 200 control alleles and detected a total of 14 polymorphic sites within the 766-bp KCNJ18 promoter fragment. However, the variant c.-542T>A was detected only in the genomic DNA of our patient (table 2). A search for population diversity of detected variants in Ensembl and gnomAD revealed no frequency data. However, we found entries by searching for small variations in dbSNP based on data from NHLBI TOPMed (Trans-Omics for Precision Medicine [TOPMed]) including >54,000 whole genomes.15 Thirteen of our detected SNPs were common with high MAFs, whereas the c.-542T>A SNP was not listed.
Table 2.
Detected single-nucleotide variations (SNVs) in the KCNJ18 promoter (n = 100 controls)a

Promoter variant c.-542A strongly increases KCNJ18 transcription
We determined the functional relevance of the c.-542T>A KCNJ18 promoter SNV with luciferase assays in transfected HEK293 cells. Compared with the wild-type promoter, the c.-542T/A promoter variant produced a tenfold higher luciferase activity (p < 0.001). This effect was completely restored to normal wild-type promoter activity, when we introduced the corresponding wild-type base T at position c.-542 in the mutant promoter (figure 2). These results show that the c.-542A KCNJ18 promoter SNV increases KCNJ18 transcription tremendously and thus might be the key to the phenotype of SPP in our patient.
Figure 2. Luciferase assays of transiently transfected HEK 293 cells with reporter constructs containing the 766-bp wild-type KCNJ18 or c.-542 T/A mutant promoter.
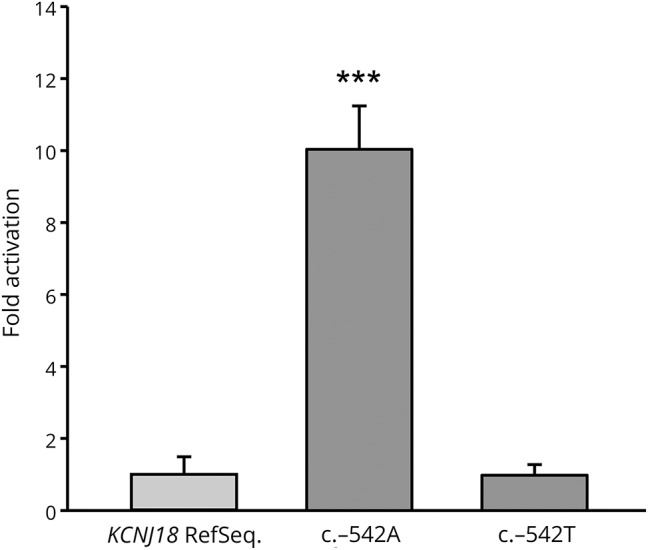
Activation of the promoter-driven luciferase in HEK293 cells by wild-type and mutated constructs, as well as in mutated constructs after introduction of wild-type base T at position c.-542 in the mutant construct. An approximately 10-fold increased luciferase activation could be detected only in the c.-542A construct. Experiments were performed in a six-fold approach, and mean and standard deviations are shown. *** indicates t = 16.983, p < 0.001 vs wild-type KCNJ18 and t = 17.855, p < 0.001 vs c.-542T, respectively.
KCNJ18 currents have an extracellular potassium dependency
Inward rectifier K+ (Kir) channels play an important role in maintaining stable resting membrane potentials, thus controlling excitability, shaping the initial depolarization, and the final repolarization of action potentials in many cell types. It has been known for several decades that Kir-mediated outward currents increase paradoxically with increasing extracellular potassium (K+o) concentration.16 However, to our knowledge, it is not known whether KCNJ18 also has this paradoxical activation by extracellular potassium. Thus, we studied the extracellular potassium dependency of wild-type KCNJ18 and the SNVs using TEVC experiments and systematically changing the K+o concentration. Note that in the oocyte expression system, because of a lower overall osmolarity, the physiologic intracellular K+ is 96 mM and the extracellular K+ is reflected by 2 mM K+o. When K+o was gradually increased between 0 and 4 mM, the outward current increased, despite a reduction in the electrochemical gradient (figure 3A). This effect was similarly observed for wild-type KCNJ18 (left) and the KCNJ18 containing four-fold SNV (right). In the physiologic range of the resting membrane potential of the skeletal muscle, the outward current was clearly reduced under hypokalemic compared with normokalemic conditions (figure 3B). Figure 3 illustrates a strong reduction of the outward currents at −80 and −70 mV for the wild-type KCNJ18 (left) and the SNVs (right) when extracellular K+ was reduced. The current reduction at 50% hypokalemic conditions (K+o = 1 mM instead of 2 mM) and no extracellular potassium (K+o = 0 mM) conditions was 23% and 46% for wild-type KCNJ18 or 22% and 44% for the KCNJ18 containing the SNVs, respectively. Thus, reducing the K+o concentration can be used to antagonize the increased KCNJ18 currents caused by the gain-of-function in the promoter of the channel.
Figure 3. Voltage-clamp recording of the wild-type KCNJ18 (left) and the KCNJ18 carrying the patient's SNVs (right) expressed in Xenopus laevis oocytes.

(A) Different extracellular K+ concentrations of 0–4 mM and (B) under normokalemic, hypokalemic (50%), and no potassium conditions, analyzed in the voltage range of −40 to −80 mV. Data are presented as mean ± SEM. *** = p < 0.001; ns = not significant; SNV = single-nucleotide variation.
Clinical treatment of the patient in relation to experimental findings
We studied the Na+ and K+ levels in more detail, and our patient recorded the electrolyte content of his food closely. From this information, in agreement with our KCNJ18 promoter findings, we decided to test the effect of lowering potassium in vivo by the administration of furosemide (starting with 20 mg per day) during a short inpatient stay in our hospital. Our patient was admitted with a potassium level of 4.1 mmol/L. A few hours later, he had a paralysis attack and the potassium level was at this point 4.2 mmol/L. Three days after the initiation of furosemide 20 mg per day, the patient felt much better, had no attacks, and was able to walk long distances. The potassium level in the following days was 3.4, 3.1, and 3.0 mmol/L, respectively. The chloride levels were within the normal range. Since the diagnosis, the patient was able to lower his potassium level under tight control to a mean level of 3.5 mmol/L (SD ±0.46; n = 234). He felt clearly better on days with potassium levels below normal and had no severe paralysis attacks for more than 2 years.
Furosemide does not alter KCNJ18 currents
Since the treatment of the patient with furosemide ameliorated symptoms of PP, we analyzed whether this drug may directly antagonize the KCNJ18 gain-of-function resulting from the increased transcription. Furosemide 10 µM was applied to oocytes expressing either wild-type KCNJ18 or the SNVs of our patient. In TEVC recordings, neither wild-type KCNJ18 nor the KCNJ18 containing the SNVs was blocked by furosemide (figure 4), supporting an indirect effect of furosemide by reducing serum potassium levels, which antagonizes the Kir2.6 gain-of-function in our patient.
Figure 4. Voltage-clamp recordings of KCNJ18 carrying the patient's SNVs expressed in Xenopus laevis oocytes under control conditions and after application of 10 µM furosemide.
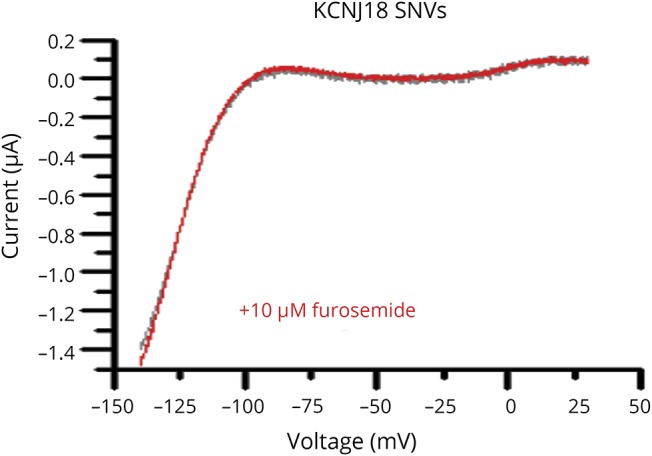
SNV = single-nucleotide variation.
Discussion
In this report, we describe a novel pathophysiologic mechanism for sporadic normokalemic PP in a patient by a gain-of-function variant in the promoter of the KCNJ18 gene. We identified a variation in position c.-542 of the KCNJ18 promoter, which dramatically (>tenfold) increases KCNJ18 transcription. The c.-542T>A KCNJ18 SNV was not found in 200 control alleles of healthy individuals and is also not reported in several databases.15,17 We detected no other putative genetic defects in PP-associated ion channels or their subunits, indicating that the KCNJ18 promoter variant is the most likely disease-relevant mutation. However, transfection experiments were performed only in HEK293 cells; it would be beneficial to confirm KCNJ18 promoter activity in a muscle biopsy of the patient. But because of the strong results of the cell culture experiments and because the patient is stable and feels much better under the current medication, we have refrained from a biopsy. KCNJ18 was described as an inwardly rectifying potassium channel in the skeletal muscle, and mutations in this gene were identified in patients with TPP.8 This study also reported that the KCNJ18 gene contains a 5′-upstream thyroid hormone response element at position c.-594, which enhances transcription by thyroid hormones. The authors proposed that mutations in the gene of Kir2.6 in combination with transcriptional upregulation of the KCNJ18 gene by T3 thyroid hormones can cause TPP, predisposing patients to muscle weakness and paralysis. The authors reported that TPP might involve as a secondary cause of increased KCNJ18 promoter activity; our study describes for the first time a mutation in the KCNJ18 promotor as a primary cause of an atypical form of PP.8 Considering the effects of thyroid hormone–dependent KCNJ18 transcription in TPP, it becomes evident that KCNJ18 promoter variants may cause PP by influencing Kir2.6 current levels in the skeletal muscle. Kir channels play a key role in the pathogenesis of PP, as they set the resting membrane potentials, thus controlling excitability. Increased Kir2.6 expression is likely to hyperpolarize the membrane potentials in the skeletal muscle cells, leading to a reduced excitability. It is well known that the outward currents of Kir channels are paradoxically increased when extracellular Ko+ concentration is elevated.18 However, it was not known whether Kir2.6 channels also have this paradoxical activation by extracellular potassium. Both gain-of-function mutations and loss-of-function mutations can cause TPP. For gain-of-function mutations, it was proposed that a hyperpolarization by increased IKir currents leads to a hyperpolarization and difficulties to reach the action potential threshold, ultimately resulting in an altered muscle excitability, weakness, and/or paralyses.8 Therefore, we tested the extracellular potassium dependence of Kir2.6 with and without the four-fold nonsynonymous SNVs. Our data obtained in vitro (Kir2.6 measurements) and in vivo (effects of furosemide by lowering plasma K+ and dietary control of potassium intake) show that reducing IKir currents is in our patient protective against further attacks, as it should antagonize the hyperpolarization caused by increased Kir2.6 transcription and IKir currents. Kir2.6 channels are believed to act as a “brake” or suppressor of inward rectifier currents. Kir2.6 is largely retained in the ER where it can bind and sequester Kir2.1 and Kir2.2 to reduce the surface expression of inward rectifier K+ channels. Therefore, the increased expression of Kir2.6 might reduce Kir2.x currents.19 This model would be consistent with the loss-of-function defects of Kir2.x channels in Andersen-Tawil syndrome. However, low extracellular K+ levels are believed to reduce Kir2.x outward currents in muscle cells. Thus, our approach of keeping plasma K+ levels low would rather aggravate symptoms than being beneficial, if the disease-causing mechanism would already include a Kir2.x reduction–mediated depolarization. Moreover, the phenotype of our patient is rather atypical and does not match with an ATS-like PP, making a similar disease-causing mechanism less likely. An alternative explanation why the patient improved on furosemide would be the fact that inhibition of the NKCC cotransporter prevents Cl− influx into muscle cells and thereby lowers myoplasmic [Cl−], which is protective against attacks of PP.20 However, although this is clearly a beneficial mechanism for PP attacks with depolarized membrane potentials, the therapeutical effect is in our case more likely related to the furosemide-mediated lowering of the plasma K+ level, which antagonizes the Kir2.6 gain-of-function. This is supported by our observation that the K+ plasma levels, which we monitored, correlate with the occurrence of PP attacks and the fact that the incidence of the attacks can be titrated not only by furosemide but also by the dietary of potassium intake.
Perhaps the most important regulatory mechanism that is involved in the pathogenesis of TPP is the transcriptional regulation of the KCNJ18 gene through the action of thyroid hormones via a thyroid hormone response element at position c.-594 in the KCNJ18 promoter. Because our c.-542T>A mutation is not localized in this thyroid response element, we searched for other transcription factor binding sites in the KCNJ18 promoter. In silico analysis for potential transcription factor binding sites21 revealed that the c.-542T>A variation replaces an androgen receptor binding site (AR) by an estrogen receptor element (ER). In PP, the attacks increase in frequency and severity during puberty, adolescence, and early adulthood. This may be caused by a change in hormone levels. Regulatory variants that affect clinical phenotypes have been identified in more than 700 genes so far, and most of these variations are located in proximal and distal promoter elements that map within 1 kb of the transcription start site.22,23 The impact of SNVs in promoters and enhancer elements on voltage-gated channel expression has been already shown for the SCN5A/SCN10A locus in arrhythmia susceptibility and Brugada syndrome.24–26 Therefore, our findings support that in PP, promoter mutations can be disease causative by altering the current densities of ion channels.
There has been a controversial debate to consider KCNJ18 as a PP gene because of its large homology with KCNJ12, and conflicting results about the pathogenicity of rare variants in these genes for the diagnosis of PP exist, as shown in a recently published work.27 We detected several promoter SNVs and functionally tested their impact in transfection experiments. Consistent with our findings, a recently published article has shown that RYR-1 mutations can cause atypical PP phenotypes in patients in whom mutations in SCN4A, CACNA1S, and KCNJ2 have been excluded.28 Here, we did not screen the RYR-1 gene for mutations. Nevertheless, from the clinical symptoms, our patient can be distinguished from the RYR-1 phenotype as he lacks symptoms of exertional myalgia, cramps, or ophthalmoplegia. Given the increasing use of whole-exome sequencing approaches to identify disease-causing genes and mechanisms, this first description of promoter-induced dysregulation of KCNJ18 in PP highlights the need to analyze nonexonic genomic patients' DNA with atypical phenotypes of PP. However, because we report on a unique case so far, further studies are needed to proof our—as we think—intriguing concept.
Acknowledgment
The authors are grateful for the participation and support of the patient and his family to perform this study.
Glossary
- ATS
Andersen-Tawil-Syndrome
- EVS
Exome Variant Server
- MAF
minor allele frequency
- PP
periodic paralysis
- PCR
polymerase chain reaction
- SNV
single-nucleotide variation
- SPP
sporadic PP
- TEVC
two-electrode voltage-clamp
- TPP
thyrotoxic PP
Author contributions
M. Soufi and V. Ruppert: genetic analysis and cell culture experiments, writing and drafting the manuscript, and study concept. S. Rinné: voltage clamp experiment data collection and analysis, revising the manuscript for content, and data interpretation. T. Mueller, B. Kurt, and A. Maieron: clinical examination of the patient, revising the manuscript for content, and data interpretation. G. Pilz: mathematical analysis, revising the manuscript for content, and data interpretation. R. Dodel: clinical examination of the patient, writing and revising the manuscript for content, and data interpretation. N. Decher: voltage clamp data collection and analysis, writing, drafting and revising the manuscript for intellectual content, and study concept. J.R. Schaefer: study concept and supervision, writing, drafting, and revising the manuscript for intellectual content.
Study funding
Dr. Reinfried Pohl Foundation provided financial support.
Disclosure
M. Soufi, V. Ruppert, and S. Rinné report no disclosures. T. Mueller is an employee of Rhön Klinikum AG. B. Kurt, G. Pilz, and A. Maieron report no disclosures. R. Dodel has served on scientific advisory boards of Octapharma and Lilly; has received travel funding/speaker honoraria from Pfizer, Lundbeck, Merz, Solvay, AstraZeneca, CSL Behring, Octapharma, Orion Pharma, UCB, Teva, and BMBF; receives publishing royalties from Thieme Verlag Stuttgart; has been a consultant for Octapharma and Lilly; has received research support from BMBF, JPND (BMBF), UKGM Inhouse grants, Behring Röntgen Stiftung, the Michael J. Fox Foundation, Deutsche Parkinson-Vereinigung, the International Parkinson Foundation, Faber Stiftung, the Movement Disorders Society, Hector-Stiftung, and Alzheimer Forschungsinitiative; receives revenue from patents (which are held by the University of Marburg); and holds the following patents: Method of detecting progression of a neurodementing disease; Method of treatment of neurodementing diseases using isolated, monoclonal, human, anti-b-amyloid antibody; Use of tetracyclines as neuro-protective agents and for the treatment of Parkinson's disease and related disorders; Human beta-amyloid antibody and use thereof for treatment of Alzheimer's disease; Human monoclonal anti-amyloid-beta antibodies; Synthetische Liganden für humane anti-aß-Antikörper; Diagnosis, prophylaxis and therapy of Alzheimer's disease and other neurodementing disorders; Naturally occurring autoantibodies against alpha-synuclein that inhibit the aggregation and cytotoxicity of alpha-synuclein; and Verfahren, insbesondere enzyme-linked immunosorbent assay (ELISA), zum in vitro Nachweis von Amyloid beta Autoantikörpern, Mikrotiterplatte und Testkit. N. Decher reports no disclosures. J.R. Schaefer serves as a scientific advisor for MSD, Sanofi-Genzyme, and Amgen; received lecture fees from MSD, Sanofi-Genzyme, Novartis Academy, Synlab Academy, and Berlin-Chemie; serves on the editorial board of Der Internist (Springer Nature Verlag); and has received research support from the Dr. Reinfried Pohl Foundation. Full disclosure form information provided by the authors is available with the full text of this article at Neurology.org/NG.
References
- 1.Lehmann-Horn F, Jurkat-Rott K. Voltage-gated ion channels and hereditary disease. Physiol Rev 1999;79:1317–1372. [DOI] [PubMed] [Google Scholar]
- 2.Burge JA, Hanna MG. Novel insights into the pathomechanisms of skeletal muscle channelopathies. Curr Neurol Neurosci Rep 2012;12:62–69. [DOI] [PubMed] [Google Scholar]
- 3.Tricarico D, Camerino DC. Recent advances in the pathogenesis and drug action in periodic paralyses and related channelopathies. Front Pharmacol 2011;2:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Plaster NM, Tawil R, Tristani-Firouzi M, et al. Mutations in Kir2.1 cause the developmental and episodic electrical phenotypes of Andersen's syndrome. Cell 2001;105:511–519. [DOI] [PubMed] [Google Scholar]
- 5.Falhammar H, Thoren M, Calissendorff J. Thyrotoxic periodic paralysis: clinical and molecular aspects. Endocrine 2013;43:274–284. [DOI] [PubMed] [Google Scholar]
- 6.Cheng CJ, Lin SH, Lo YF, et al. Identification and functional characterization of Kir2.6 mutations associated with non-familial hypokalemic periodic paralysis. J Biol Chem 2011;286:27425–27435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zheng J, Liang Z, Hou Y, et al. A novel Kir2.6 mutation associated with hypokalemic periodic paralysis. Clin Neurophysiol 2016;127:2503–2508. [DOI] [PubMed] [Google Scholar]
- 8.Ryan DP, da Silva MR, Soong TW, et al. Mutations in potassium channel Kir2.6 cause susceptibility to thyrotoxic hypokalemic periodic paralysis. Cell 2010;140:88–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Soufi M, Sattler AM, Maerz W, et al. A new but frequent mutation of apoB-100-apoB His3543Tyr. Atherosclerosis 2004;174:11–16. [DOI] [PubMed] [Google Scholar]
- 10.DGN-Leitlinien. Available at: dgn.org/leitlinien/3457-030-055-myotone-dystrophien-nicht-dystrophe-myotonien-und-periodische-paralysen-2017#definition. Accessed April 23, 2018.
- 11.Jurkat-Rott K, Lehmann-Horn F. Genotype-phenotype correlation and therapeutic rationale in hyperkalemic periodic paralysis. Neurotherapeutics 2007;4:216–224. [DOI] [PubMed] [Google Scholar]
- 12.GHR. Available at: ghr.nlm.nih.gov/search?query=periodic+paralysis. Accessed April 23, 2018.
- 13.Paninka RM, Mazzotti DR, Kizys MM, et al. Whole genome and exome sequencing realignment supports the assignment of KCNJ12, KCNJ17, and KCNJ18 paralogous genes in thyrotoxic periodic paralysis locus: functional characterization of two polymorphic Kir2.6 isoforms. Mol Genet Genomics 2016;291:1535–1544. [DOI] [PubMed] [Google Scholar]
- 14.GeneCards. Available at: genecards.org/cgi-bin/carddisp.pl?gene=KCNJ18. Accessed April 23, 2018.
- 15.dbSNP. Available at: ncbi.nlm.nih.gov/snp/?term=kcnj18. Accessed April 23, 2018.
- 16.Leech CA, Stanfield PR. Inward rectification in frog skeletal muscle fibres and its dependence on membrane potential and external potassium. J Physiol 1981;319:295–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ensembl. Available at: ensembl.org/Homo_sapiens/Gene/Summary?db=core;g=ENSG00000260458;r=17:21692523-21704612;t=ENST00000567955 Accessed April 23, 2018.
- 18.Jurkat-Rott K, Weber MA, Fauler M, et al. K+-dependent paradoxical membrane depolarization and Na+ overload, major and reversible contributors to weakness by ion channel leaks. Proc Natl Acad Sci U S A 2009;106:4036–4041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dassau L, Conti LR, Radeke CM, et al. Kir2.6 regulates the surface expression of Kir2.x inward rectifier potassium channels. J Biol Chem 2011;286:9526–9541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wu F, Mi W, Cannon SC. Beneficial effects of bumetanide in a CaV1.1-R528H mouse model of hypokalaemic periodic paralysis. Brain 2013;136:3766–3774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Messeguer X, Escudero R, Farré D, Núñez O, Martínez J, Albà MM. PROMO: detection of known transcription regulatory elements using species-tailored searches. Bioinformatics 2002;18:333–334. [DOI] [PubMed] [Google Scholar]
- 22.Epstein DJ. Cis-regulatory mutations in human disease. Brief Funct Genomic Proteomic 2009;8:310–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.De GM, Viprakasit V, Hughes JR, et al. A regulatory SNP causes a human genetic disease by creating a new transcriptional promoter. Science 2006;312:1215–1217. [DOI] [PubMed] [Google Scholar]
- 24.Bezzina CR, Shimizu W, Yang P, et al. Common sodium channel promoter haplotype in Asian subjects underlies variability in cardiac conduction. Circulation 2008;113:338–344. [DOI] [PubMed] [Google Scholar]
- 25.van den Boogaard M, Barnett P, Christoffels VM. From GWAS to function: genetic variation in sodium channel gene enhancer influences electrical patterning. Trends Cardiovasc Med 2014;24:99–104. [DOI] [PubMed] [Google Scholar]
- 26.Park JK, Martin LJ, Zhang X, Jegga AG, Benson DW. Genetic variants in SCN5A promoter are associated with arrhythmia phenotype severity in patients with heterozygous loss-of-function mutation. Heart Rhythm 2012;9:1090–1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kuhn M, Jurkat-Rott K, Lehmann-Horn F. Rare KCNJ18 variants do not explain hypokalaemic periodic paralysis in 263 unrelated patients. J Neurol Neurosurg Psychiatry 2016;87:49–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Matthews E, Neuwirth C, Jaffer F, et al. Atypical periodic paralysis and myalgia: a novel RYR1 phenotype. Neurology 2018;90:e412–e418. [DOI] [PMC free article] [PubMed] [Google Scholar]