

Abstract
Traumatic brain injury (TBI) is the major cause of physical disability and emotional vulnerability. Treatment of TBI is lacking due to its multi-mechanistic etiology, including derailed mitochondrial and cellular energy metabolism. Previous studies from our laboratory show that an endogenous nitric oxide (NO) metabolite S-nitrosoglutathione (GSNO) provides neuroprotection and improves neurobehavioral function via anti-inflammatory and anti- neurodegenerative mechanisms. To accelerate the rate and enhance the degree of recovery, we investigated combining GSNO with caffeic acid phenethyl ester (CAPE), a potent antioxidant compound, using a male mouse model of TBI, controlled cortical impact in mice. The combination therapy accelerated improvement of cognitive and depresssive-like behavior compared with GSNO or CAPE monotherapy. Separately, both GSNO and CAPE improved mitochondrial integrity/function and decreased oxidative damage; however, the combination therapy had greater effects on Drp1 and MnSOD. Additionally, while CAPE alone activated AMPK, this activation was heightened in combination with GSNO. CAPE treatment of normal animals also significantly increased the expression levels of pAMPK, pACC (activation of AMPK substrate ACC) and pLKB1 (activation of upstream to AMPK kinase LKB1), indicating that CAPE activates AMPK via LKB1. These results show that while GSNO and CAPE provide neuroprotection and improve functional recovery separately, the combination treatment invokes greater recovery by significantly improving mitochondrial functions and activating the AMPK enzyme. Both GSNO and CAPE are in human consumption without any known adverse effects; therefore, a combination therapy-based multi-mechanistic approach is worthy of investigation in human TBI.
Keywords: TBI, S-nitrosoglutathione, Caffeic acid phenethyl ester, Mitochondria quality, Antioxidant activity, Combination therapy, Functional recovery, RRIDs
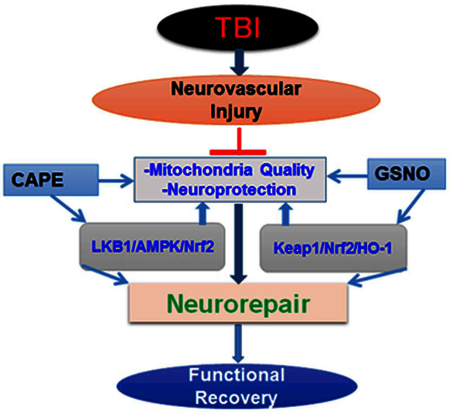
Legend of graphical Abstract:
Combining GSNO with CAPE is more effective in TBI than CAPE or GSNO monotherapy. Both GSNO and CAPE are naturally occurring potent antioxidant and they have been used in human for other indications.
Introduction
Traumatic brain injury (TBI) affects more than 1.7 million people each year in the United States (Gardner and Zafonte 2016). According to the U.S. Centers for Disease Control and Prevention (CDC), TBI is currently associated with direct and indirect annual costs of approximately $76.5 billion (https://www.cdc.gov/traumaticbraininjury/data/rates.html), causing an enormous socioeconomic burden. TBI is a leading cause of morbidity and disability (Gardner and Zafonte 2016). The observed motor and cognitive changes that follow TBI include decreased mental flexibility, depression, impaired attention, poor planning, impaired judgment, deficits in verbal fluency, and problems with working memory (Levin and Kraus 1994). TBI patients are also susceptible to dementia, epilepsy, and Alzheimer’s disease (Johnson et al. 2010). Unfortunately, treatment options for TBI are limited, and a proven FDA-approved therapy for TBI is presently not available, mainly because of limited mechanistic understanding.
Following the primary injury of a TBI event, secondary injury follows from neuroinflammation, oxidative stress, and mitochondrial dysfunction. This secondary injury hampers extant mechanisms of neuroprotection and neurorepair, thus delaying and limiting neurobehavioral recovery. Recently, we documented that GSNO reduces secondary injury, protects the brain, and improves neurological function both in cerebral ischemia reperfusion (IR) injury (Khan et al. 2006; Khan et al. 2005) and in TBI (Khan et al. 2009). We have also observed that GSNO promotes functional recovery in experimental SCI (Chou et al. 2011; Shunmugavel et al. 2012). GSNO is a natural component of the human body (Singh et al. 1996), present in the brain and other organs (Kluge et al. 1997). It invokes its cellular signaling via S-nitrosylation of target proteins, including NF-κB, STAT3, COX-2, caspase-3, calpains, inducible nitric oxide synthase (iNOS), and endothelial NOS (Jaffrey et al. 2001; Khan et al. 2012; Khan et al. 2005; Kim et al. 2013b). Exogenous administration of GSNO (Rassaf et al. 2006) also protects against cardiac ischemic injury (Konorev et al. 1995; Lima et al. 2009), supporting the therapeutic potential of GSNO. Pharmacological inhibition of GSNO reductase (GSNOR), a GSNO degrading enzyme, has also been shown to improve endothelial functions (Chen et al. 2013), indicating a protective role of GSNO in neurovascular dysfunction-related diseases. Studies have also reported that GSNO inhibits platelet activation in humans (Radomski et al. 1992) and protects both BBB integrity and epithelial permeability (Khan et al. 2009; Savidge et al. 2007), indicating GSNO’s clinical potential. The effects of GSNO are mediated mainly by the mechanism of S-nitrosylation, and S-nitrosylation-based mechanistic ability is not embedded in conventional NO donors, making GSNO a unique candidate for TBI therapy. However, GSNO’s effects on mitochondrial efficiency/quality and redox are less understood.
Like GSNO, CAPE is a potent neuroprotective agent; it improves neurobehavioral function mainly by its antioxidant activity (Tolba et al. 2013). Because redox is implicated in neuroinflammation, neuronal cell death and functional deficits (Avila et al. 2008; Besson 2009; Deng-Bryant et al. 2008; Hall et al. 1999; Singh et al. 2007), we investigated the efficacy of CAPE in TBI. CAPE is a plant-originated potent lipophilic compound (Son and Lewis 2002; Zheng and Yenari 2004) and is well-tolerated in humans. It is rapidly absorbed and metabolized by plasmatic esterases to caffeic acid (Celli et al. 2004), which is also an effective antioxidant and inhibits the activity of inflammatory 5-lipoxygenase (Teixeira et al. 2015). A post-TBI administration of CAPE shows reduced cortical tissue loss (Zhao et al. 2012). In another TBI study, CAPE decreased oxidative stress, reduced neuronal loss and protected tissue structure, effects associated with its antioxidant functions (Kerman et al. 2012). Because the bulk of reactive oxygen species are generated by several metabolic enzymes residing in mitochondria, the efficiency of mitochondrial function is significant to detoxify secondary injury-induced exacerbations following TBI. Recent studies from our (Jatana et al. 2006; Khan et al. 2007; Singh et al. 2013) and other laboratories (Lee et al. 2007; Liao et al. 2013; Natarajan et al. 1996; Tsuda et al. 2012) suggest that CAPE invokes its protective effects mainly by maintaining mitochondrial integrity through the activation of AMPK and down regulation of inflammation via the inhibition of NF-ΚB. Recently, studies have shown down regulation of inflammation in microglial cells by CAPE via the inhibition of iNOS and COX-2, activation of AMPK, and the induction of HO-1 (Choi et al. 2015; Tsai et al. 2015). These findings support the notion that mitochondrial integrity is well protected by CAPE, likely through induction of Nrf2 (Kim et al. 2013a) and activation of AMPK (Feng et al. 2008). Based on improved mitochondrial activity/function and the proven efficacy of CAPE in animal models of TBI (Kerman et al. 2012; Tsuda et al. 2012; Zhao et al. 2012) and stroke (Suuronen et al. 2003), we investigated the efficacy of CAPE alone and tested whether CAPE accelerates the efficacy of GSNO treatment in TBI. Greater efficacy of combination therapy compared with monotherapy in TBI has been discussed recently (Kline et al. 2016).
Animal models of TBI using the focal controlled cortical impact (CCI) technique are recognized as physiologically relevant to TBI in humans (Bondi et al. 2015; Osier and Dixon 2016). CCI provides an opportunity to determine both primary as well as secondary injuries and to evaluate functional deficits. We opted for a subacute phase injury (7 days) study over the acute (24–96 h) and chronic phases (weeks and months) to avoid the contribution of contusion- induced oxidative exacerbations and the stimulation of profound neurorepair processes, respectively. It is recognized that very severe injury involves several pathways, thus making it difficult to delineate specific ones. Moreover, 75% of TBI patients in the United States are diagnosed as suffering from mild TBI. Therefore, we used a mild/moderate 7-day TBI mouse model as described (Deng et al. 2007; Khan et al. 2016) to investigate whether the combination (GSNO+CAPE) therapy shows greater efficacy compared with GSNO or CAPE alone in mitigating multi-mechanistic pathology of TBI.
Experimental Procedure
Reagents
GSNO was purchased from World Precision Instruments (Sarasota, FL, USA) and caffeic acid phenethyl ester (CAPE) from Cayman Chemical (Ann Arbor, MI, USA), respectively. All other chemicals and reagents used were purchased from Sigma-Aldrich (St. Louis, MO), unless stated otherwise.
Animals
Animals were young adult male (~3–4 months old) wild type C57BL/6 mice (The Jackson Laboratory, ME: Stock # 00064) weighing between 25 and 30 g at the time of surgery. Animals were group-housed (4/cage) before surgery and individually housed after surgery under specific pathogen-free conditions in static micro-isolator cages. Animals were fed with irradiated rodent chow and tap water ad lib. Temperature of the vivarium was maintained at 25±°C with a 12:12 h light-dark cycle. All animals received humane care in compliance with the Medical University of South Carolina’s (MUSC) guidance and the National Research Council’s criteria for humane care. Animal procedures were approved by the institutional animal care and use committee (IACUC) of MUSC and VA medical center. Animals were euthanized at the endpoints using isoflurane (2.5%).
Experimental groups, drugs and dose
Animal groups were randomized and experiments were blinded. The animals (n=48) were divided into seven groups: 1) TBI animals treated with vehicle (TBI; n=8); 2) TBI+GSNO treatment (GSNO; n=8); 3) TBI+CAPE treatment (CAPE; n=8); 4) TBI+GSNO+CAPE (GSNO+CAPE; n=8); 5) sham-operated treated with 50% DMSO diluted with sterile saline (Sham; n=8); 6) normal control animals treated with CAPE (Norm+CAPE, n=4); and 7) normal control animals treated with DMSO (Norm+DMSO, n=4). The number of animals used in each experiment is also indicated in figure legends. In the GSNO-treated group, the mice were administered freshly prepared GSNO (0.05 mg/kg body weight), which was dissolved in sterile saline (~25μl) and administered iv at 2 h after CCI. CAPE (5 mg/kg, iv), dissolved in sterile 50% DMSO (~25 μl), was also administered at 2 h after CCI. In the combination group, CAPE (iv) was administered first followed by GSNO (iv) initially at 2 h after CCI. Daily thereafter, the same dose of CAPE (orally, in 50% DMSO) was administered first followed by the same dose of GSNO (orally in saline). The dose of GSNO treatment was based on our previously reported dose response curve study in a rat and mouse model of TBI (Khan et al. 2009; Khan et al. 2011). The effective dose of CAPE was determined based on the reported dose study from our laboratory using an animal model of cerebral IR (Khan et al. 2007). The selected dose of both GSNO and CAPE did not alter physiologic parameters, including blood pressure, heart rate, and temperature, measured at 1 hour following GSNO or CAPE treatment. Details of the study on physiologic parameters in rats have been published previously (Khan et al. 2007; Khan et al. 2006; Khan et al. 2011).
Controlled cortical impact (CCI) mouse model of TBI
Surgical anesthesia was induced by ketamine (90 mg/kg body weight) and xylazine (10 mg/kg body weight) administered intraperitoneally (ip). Analgesic buprenorphine was administered pre- emptively to alleviate pain following surgery. The animal was then placed onto a heated pad, with core body temperature maintained at 37.0+1°C. The animals were secured in a stereotaxic frame and ventilated mechanically. A dab of sterile ophthalmic ointment was placed on each eye to compensate for the decrease in lacrimation during anesthesia. A midline scalp incision was made, and the skin and fascia were reflected to expose the skull. A craniotomy (4 mm) was made in the right hemisphere encompassing bregma and lambda and between the sagittal suture and the coronal ridge with a handheld Michele trephine. Care was taken to avoid injury to the underlying dura mater, which was continuously bathed in sterile normal saline warmed to 37.5°C, as described (Fox et al. 1998). The resulting bone flap was removed, and the craniotomy was enlarged further with cranial rongeurs. CCI injury was produced in mice as previously described (Fox et al. 1998; Smith et al. 1995; Wang et al. 2013). Briefly, a cortical contusion was produced on the exposed cortex using a controlled impactor device described by Bilgen (Bilgen 2005) and used in our laboratory (Shunmugavel et al. 2010) and described in our TBI study (Khan et al. 2018; Khan et al. 2016; Khan et al. 2009). Briefly, the impacting shaft was extended, and the impact tip was centered and lowered over the craniotomy site until it touched the dura mater. Then, the rod was retracted and the impact tip was advanced farther to produce a brain injury of moderate severity for mice (tip diameter, 1 mm; cortical contusion depth, 1 mm; impact duration 85 msec; impact velocity, 1.5 m/sec). The impact tip was wiped clean with sterile alcohol after each impact and cleaned/disinfected further with cidex after surgery. Core temperature was maintained at 37± 0.1°C with a heating pad during surgery and recorded with a rectal probe. Immediately after injury, the skin incision was closed with nylon sutures, and 2% lidocaine jelly was applied to the lesion site to minimize discomfort. Sham animals underwent the same procedure, excepting the cortical impact. Exclusion criterion included the brains with inconsistent trauma or deeper damage due to heavy inflammation following the CCI.
Evaluation of motor behavior, sensory function, novel object recognition (NOR) and forced swim test (FST)
Animals in each group were evaluated for beam walking, foot faults, NOR, and forced swim test before and after CCI. All the tasks described below were performed by investigators blinded to the study groups and conditions.
In the motor function test, the mice were examined for fine motor function using a beam- walking task with an elevated narrow beam (80 cm long × 2.5 cm wide) raised 10 cm above ground level. The time to traverse the beam was recorded and analyzed after three trials (60 seconds allotted time). The beam test was selected over other motor function tests to detect motor deficits because the test is suitable for aged as well as young rodents. Animals were pre- trained for 3 days before CCI. We routinely use this task, as published for IR and TBI studies (Khan et al. 2016; Khan et al. 2015a; Khan et al. 2009; Sakakima et al. 2012).
The forelimb foot faults or grid walking task, a motor impairment test, was used to determine forelimb movement dysfunction while walking. Mice were placed on elevated horizontal wire grids. Mice place their paws on the wire while moving along the grid. With each step, the paw may fall or slip between wires. These events were recorded as foot faults. The total number of steps that the mouse used to cross the grid was counted, and the total number of foot faults was recorded as described (Zhang et al. 2002). Mice were trained for 3 days before CCI, and the number of foot faults following CCI at day 7 was recorded.
Non-spatial memory was determined by the NOR test as described (Huang et al. 2014). The NOR test evaluates the ability of the animal to recognize a novel object in the environment. The NOR test wooden box (36cm x 22cm x 18 cm) was located in an isolated and illuminated animal testing room. The animals were allowed to explore the test box for 15 min per day before the actual experiments. The NOR test box was devoid of any object during the habituation trial. The test was performed on day 7 following CCI, as described (Khan et al. 2016). During session I, two objects, A1 and A2, with identical texture, color, and size were presented to the test animal for 10 min. After a 24 h delay in the home cage, the mice were again exposed to the same area with one novel object of different texture, color, and size included (A1 and B) for 4 min. Both the objects and the box were cleaned with alcohol and dried after each trial to remove olfactory cues. Latency (% preference for a novel object) was recorded using a video camera. The advantage of the test is that, unlike the Morris water-maze task, it is independent of motor function (Dhawan et al. 2011).
The forced swim test (FST) has been used to measure depressive-like behavior in mice following trauma. The test was performed as previously described (Can et al. 2012). A cylindrical tank (30 cm height x 20 cm diameter) made of Plexiglass was used in the study. Water level was maintained 15 cm from the bottom uniformly for all the mice tested. During the test, the mice were unable to touch the bottom of the tank either with their hind limbs or tail. The entire experiment was recorded with a video camera mounted on a tripod. Tests were performed individually in sequence, avoiding the use of dividers between the tanks. The mice were forced to swim for 8 min 24 h before the actual test. On the day of the test (7th day after CCI), each mouse was given a forced swim period of 6 min. The last 4 min of the total 6 min was used for final calculation. Periods of mobility and immobility were calculated as described by Can et al (Can et al. 2012).
4.5. Histology and immunohistochemistry
NeuN immunostaining indicates number of neurons. The degree of neuronal damage (loss of viable neurons) was evaluated by cresyl violet (Nissl) staining. Cellular infiltration and tissue damage were assessed by hematoxylin and eosin (H&E) staining as previously described in a rat TBI brain (Khan et al. 2011). Nissl staining highlights important structural features of neurons. Paraffin-embedded brain sections from the formalin-fixed brain tissues, processed at 7 days after CCI, were used to stain with H&E and Nissl, and the staining was performed according to classical histology methods as previously described from our laboratory (Khan et al. 2015a; Khan et al. 2011; Sakakima et al. 2012). Cells that contained Nissl substance were considered to be viable neurons. Condensed, fragmented staining shows neuronal degeneration.
Paraffin-embedded brain sections (6 μm thick) from the formalin-fixed brain tissues, processed at 7 days after CCI, were used to immunostain with NeuN, and the staining was performed using a NeuN-specific antibody, RRID:AB_2298772 (1:250 dilution) from Millipore (Billerica, MA) as described (Liu et al. 2009). The percent of NeuN positive area was determined as previously described from our laboratory (Sakakima et al. 2012), and viable neurons from Nissl staining were quantified by manually counting cells as previously described from our and other laboratories (Khan et al. 2015a; Zhu et al. 2014) in the traumatic penumbra area using an Olympus microscope. Histological analysis was performed by an observer blinded to the identity of groups, as previously described from our laboratory (Khan et al. 2015a; Sakakima et al. 2012).
Western blot analysis
In the traumatic penumbra area from the ipsilateral injured brain tissue, western blot was performed using antibodies against PGC-1 α (RRID:AB_2268435), Drp1 (RRID:AB_11178938), Fis1(RRID:AB_11204021), Opa1 (RRID:AB_944550), AMPKα1 (RRID:AB_310542), pAMPK (Thr172) (RRID:AB_1977022), MnSOD (RRID:AB_525598), HO-1 (RRID:AB_764541), and β-actin (RRID:AB_303668) as described in our publications (Khan et al. 2016; Khan et al. 2015,b; Khan et al. 2012). Detail of antibodies is described in Table 1. After washing, the membranes were incubated with 1:10,000 diluted horseradish Peroxidase- AffiniPure Goat Anti-Rabbit IgG secondary antibody (RRID:AB_2337938) for 1h at room temperature, again washed, reacted with ECL reagent from Amersham Life Science (Pittsbrugh, PA), and subjected to autoradiography. β-actin expression level was used as an internal control for equal protein loading. Protein concentrations were determined using protein assay dye from Bio-Rad Laboratories (Hercules, CA). Blots were scanned for densitometry using an Epson perfection 1200 U scanner, transferred to Adobe Photoshop CS2, and the images were analyzed by NIH Image J software.
Table 1.
Description of antibodies
| Antibody | Company Name | Cat # | Host organism/Clonality | RRID |
|---|---|---|---|---|
| NeuN | Millipore | MAB377 | Mouse/monoclonal | RRID:AB_2298772 |
| PGC1α | Cell Signaling | 4259S | Rabbit/polyclonal | RRID:AB_2268435 |
| Drp1 | Cell Signaling | 5391S | Rabbit/monoclonal | RRID:AB_11178938 |
| Fis1 | Millipore | ABC67 | Rabbit/polyclonal | RRID:AB_11204021 |
| Opa1 | Abcam | ab55772 | Mouse/monoclonal | RRID:AB_944550 |
| AMPKα1 | Millipore | 07–350 | Rabbit/polyclonal | RRID:AB_310542 |
| pAMPK (Thr172) | Millipore | 09–290 | Rabbit/polyclonal | RRID:AB_1977022 |
| MnSOD | Novus | NB 100–931 | Goat/polyclonal | RRID:AB_525598 |
| ACC | Cell Signaling | 3662 | Chicken/polyclonal | RRID:AB_2219400 |
| pACC (Ser79) | Cell Signaling | 11818 | Rabbit/monoclonal | RRID:AB_2687505 |
| LKB1 | Cell Signaling | 3050S | Rabbit/monoclonal | RRID:AB_823559 |
| pLKB1 (Thr189) | Cell Signaling | 3054S | Rabbit/polyclonal | RRID:AB_330068 |
| HO-1 | Abcam | 1922–1 | Rabbit/monoclonal | RRID:AB_764541 |
| β-actin | Abcam | ab3280 | Mouse/monoclonal | RRID:AB_303668 |
| Peroxidase- AffiniPure Goat Anti-Rabbit IgG | Jackson ImmunoResearch Labs | 111–035-045 | Rabbit/polyclonal | RRID:AB_2337938 |
Activation of AMPK by CAPE in mice brain
We determined the expression of activated AMPK and pAMPK (phosphorylated AMPK) in the cortical/striatal brain region from normal control animals. The expression of AMPK substrate acetyl co-A carboxylase (ACC) as pACC and AMPK upstream kinase LKB1 as pLKB1 were determined using specific antibodies to support AMPK activation. Mice were injected iv with 5 mg/kg CAPE in 50% DMSO (25 μl) or DMSO alone. Animals were sacrificed 2 h after the injection, and the expression of pAMPK (RRID:AB_1977022)/AMPK (RRID:AB_310542), pACC (RRID:AB_2687505)/ACC (RRID:AB_2219400), pLKB1 (RRID:AB_330068)/LKB1 (RRID:AB_823559) were determined using specific antibodies in cortical brain region using western analysis and densitometry. Detail of antibodies is described in Table 1.
Statistical evaluation
Statistical analysis was performed using Graph pad Prism 5.01 software as described (Khan et al. 2015a). The results are presented as the mean±SD. Statistical significance was analyzed by one- way or two-way (ANOVA) with repeated measures with time, and the Bonferroni post-hoc test was used for multiple comparisons. Student’s t-test was used to compare values between two groups as described in figure 6. A p value<0.05 was considered significant.
Figure 6.
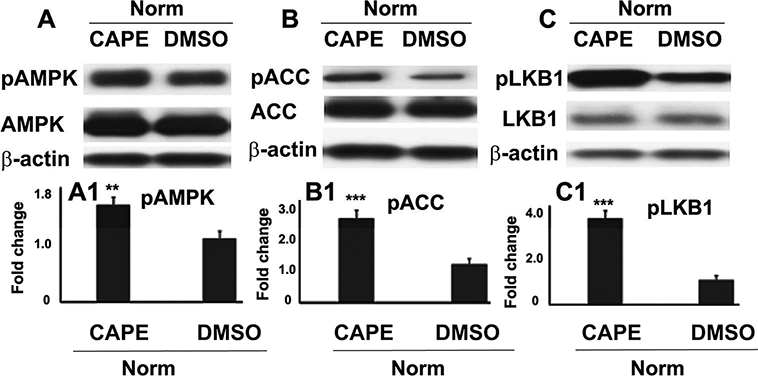
Effect of CAPE on activation and expression of AMPK, ACC, and LKB1 in wild type mouse brain. Normal control mice were treated iv with either 5mg/kg CAPE in 50% DMSO or 50% DMSO alone. The animals were sacrificed 2 h after the injection and the expressions of pAMPK and AMPK with densitometry (A, A1); pACC and ACC with densitometry (B, B1); and pLKB1 and LKB1 with densitometry (C, C1) in cortical area were determined using western blot analysis. Data are presented as mean ± SD (n=4). ***p<0.001, **p<0.01 vs. DMSO.
Results
GSNO or CAPE treatment for 7 days improved motor, NOR and FST functions, and combination GSNO and CAPE treatment accelerated NOR and FST functions following TBI
Using GSNO or CAPE, we tested the hypothesis that TBI-mediated compromised neurobehavioral functions, especially motor coordination and balance, cognition and depressive- like behaviors are improved following TBI. We also determined whether the combination of GSNO+CAPE accelerated functional recovery. GSNO or CAPE treatment significantly improved beam walking latency (Fig 1A) and reduced the number of foot faults (Fig 1B) compared with the TBI group measured at the 7th day after TBI. However, the combination treatment did not show additional improvement compared with the GSNO or CAPE group. The treatment with GSNO or CAPE also significantly improved NOR (Fig 1C) and forced swim test (FST) functions (Fig 1D) at the 7th day after TBI. The combination (GSNO+CAPE) treatment remarkably accelerated both NOR and FST functions and showed significantly greater improvement compared with either the GSNO or the CAPE group (Fig 1C, D).
Figure 1.
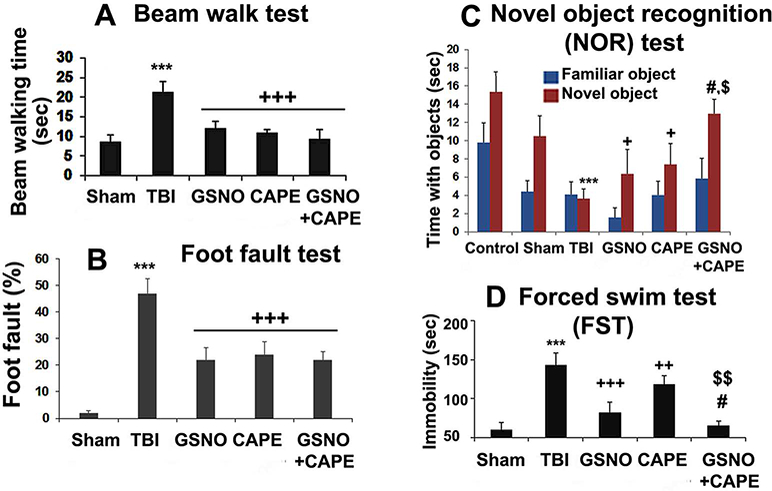
Effect of GSNO, CAPE, and combination GSNO+CAPE for 7 days on neurobehavioral functions. Treatment of TBI animals with GSNO, CAPE, and GSNO+CAPE improved motor functions (Beam walk; A and foot fault: B), cognitive (NOR, C) and depressive- like behavior (FST; D). Sham animals showed deficits neither in motor nor in cognition and depressive-like behavior. The combination therapy of GSNO+CAPE did not show additional improvement in motor function. However, both cognition and depressive-like behavior were significantly accelerated and greatly improved in the combination group compared with GSNO- or CAPE-treated group. Data are presented as mean+SD (n=8). ***p<0.001 vs. Sham, +++p<0.001, ++p<0.01, +p<0.05 vs. TBI, #p<0.05 vs. GSNO, $p<0.05 and $ $p<0.01 vs. CAPE.
GSNO or CAPE treatment for 7 days reduced neuron loss/neurodegeneration and protected brain tissue integrity, and the combination treatment showed additive effects following TBI
The therapeutic efficacy of GSNO, CAPE and CAPE+GSNO was determined in reducing neuronal loss, increasing number of viable neurons and the integrity of brain tissue. Neuronal loss and neurodegeneration were evaluated by neuronal nuclear protein (NeuN, Fig 2A) immunostaining and cresyl violet (Nissl, Fig 2B) staining, respectively. The stainings highlight the neuron population size (NeuN) and structural features of neurons (Nissl). Condensed fragmented Nissl staining in TBI (Fig 2B) shows neuron degeneration. Brain tissue structure and cellular infiltration were examined by histology using H&E staining (Fig 2C). Although treatment with GSNO or CAPE significantly reduced neuron loss and neurodegeneration, combination treatment with GSNO+CAPE yielded a greater number (Fig 2A, Ai) and improved structure of neurons (Fig 2B, Bi) compared with the GSNO or CAPE group. As with NeuN and Nissl, the combination group had similar H&E staining, showing lesser infiltration as well as damage compared with the GSNO or CAPE group (Fig C).
Figure 2.
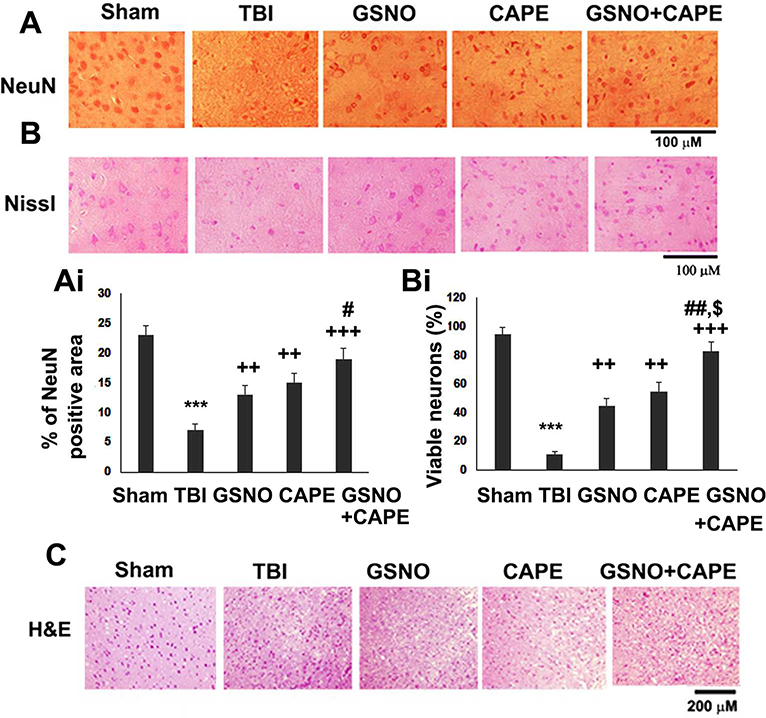
Effect of GSNO, CAPE, and combination GSNO+CAPE for 7 days on neuronal survival/degeneration and tissue histology. Photomicrographs of DAB staining of immunoreactivity and % of area of NeuN (A, Ai), neuronal viability staining, viable neuron count (Nissl; B, Bi), and brain tissue H&E staining (C) showing enhanced inflammatory infiltration in the traumatic penumbra region of TBI group. Data are presented as mean+SD (n=4). ***p<0.001 vs. Sham, +++p<0.001 and ++p<0.01 vs. TBI, #p<0.05 and ##p<0.01 vs. GSNO, $p<0.05 vs. CAPE.
GSNO or CAPE treatment for 7 days reduced the expression of mitochondrial fission mediators (Drp1, Fis1) and increased the expression of the mitochondrial fusion mediator (OPa1), and the combination treatment showed synergistic effects on Drp1 following TBI
Because mitochondrial function is essentially associated with neuroprotection and functional recovery, we investigated the impact of GSNO, CAPE and GSNO+CAPE on mitochondrial integrity/function. Mitochondrial quality control is regulated by the antagonistic forces of fission and fusion. Therefore, we determined the expression of fission (Drp1 and Fis 1)- and fusion (Opa1)- associated proteins using western blot analysis (Fig 3A) and densitometry (Fig 3B). While the expression of Drp1 and Fis1 was increased, Opa1 was decreased in TBI compared with the Sham group (Fig 3A, B). Treatment with GSNO or CAPE decreased the expression of both Drp1 and Fis1 and treatment with the combination had greater effects on Drp1. GSNO or CAPE alone or in combination increased the expression of Opa1. Unlike the effect on the expression of Drp1, the combination group showed no greater effects on Opa1 (Fig 3A, B).
Figure 3.
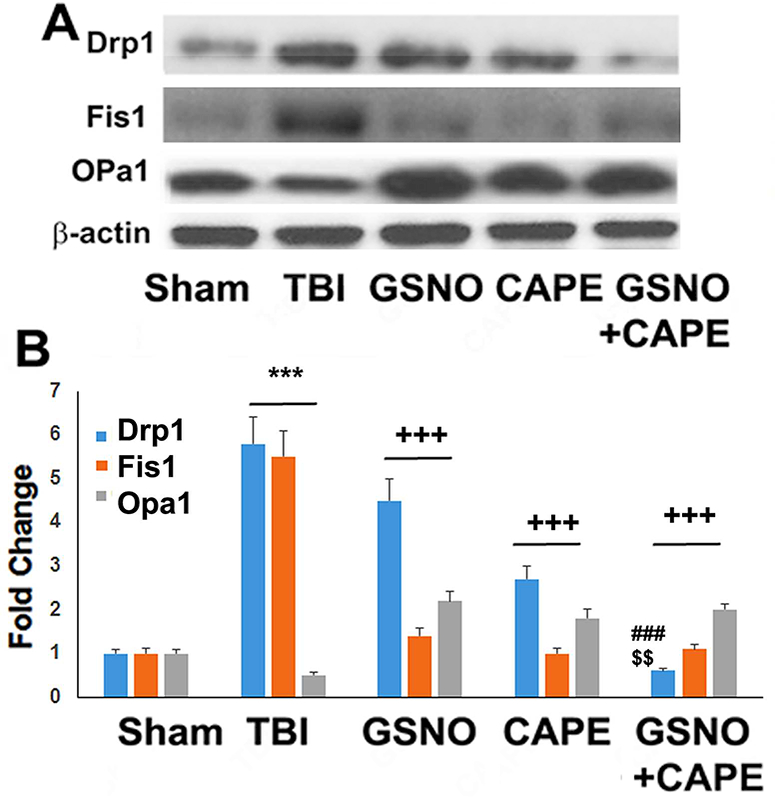
Effect of GSNO, CAPE, and combination GSNO+CAPE for 7 days on expression of Drp1, Fis1, and OPa1. The expression of Drp1, Fis1, and OPa1 was measured in the traumatic penumbra region using western analysis (A) and its densitometry (B). TBI had an increased expression of Drp1 and Fis1 and a decreased expression of OPa1. The levels of both Drp1 and Fis1 were decreased in both GSNO and CAPE groups compared with TBI group. Treatment with GSNO+CAPE further decreased the levels of Drp1. All treatment groups also had increased expression of Opa1. Data are presented as mean ± SD (n=4). ***p<0.001 vs. Sham, +++p<0.001 vs. TBI, ###p<0.001 vs. GSNO, $ $p<0.01 vs. CAPE.
GSNO or CAPE treatment for 7 days increased expression of mitochondrial biogenesis factor PGC1α and antioxidant enzymes HO-1 and MnSOD, and combination treatment showed additive effects following TBI
Mitochondrial function (PGC1α, MnSOD) and antioxidant activity (HO-1) were determined using western blot analysis (Fig 4A) and densitometry (Fig 4B). The expression PGC1α was decreased whereas HO-1 and MnSOD increased in the TBI group. GSNO treatment had no effect on the expression levels of PGC1α (Fig 4A, B); however, GSNO increased the expression of both HO-1 and MnSOD compared with the TBI group. CAPE treatment increased the expression of PGC1α as well as HO-1 and MnSOD (fig 4A, B). Treatment with combined GSNO and CAPE had synergistic effects on the increased expression of HO-1 and MnSOD whereas the combination treatment had no additive effect on PGC1α levels (Fig 4A, B).
Figure 4.
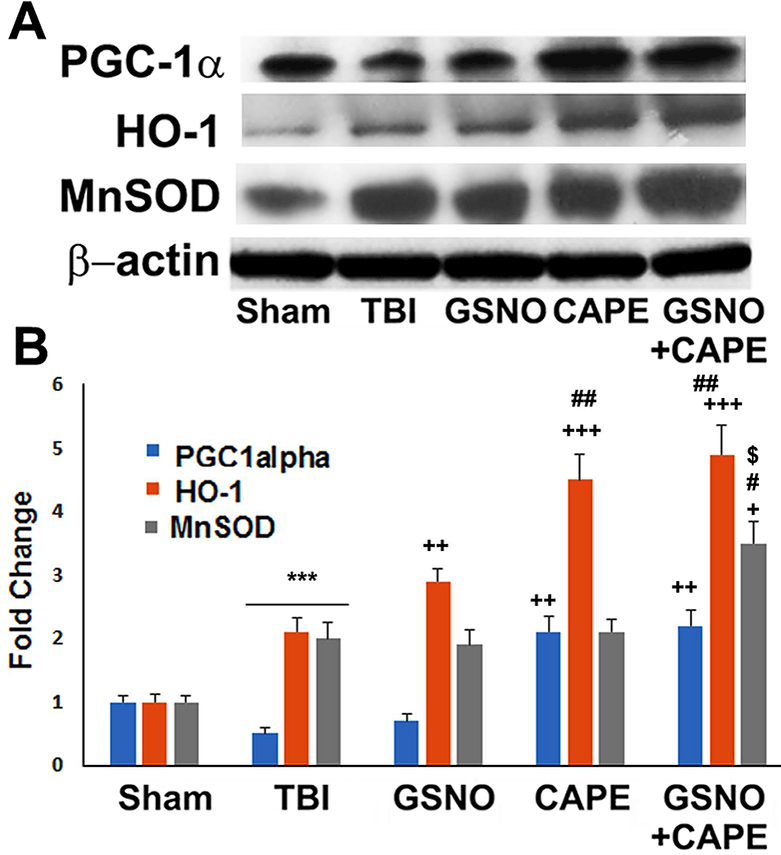
Effect of GSNO, CAPE, and combination GSNO+CAPE for 7 days on the expression of PGC1α, HO-1, and MnSOD. The expression of PGC1α, HO-1, and MnSOD was measured in the traumatic penumbra region using western analysis (A) and its densitometry (B). TBI brain had reduced expression of PGC1α, HO-1 and increased expression of MnSOD. All treated groups had increased expression of PGC1α, HO-1, and MnSOD compared with TBI group. However, GSNO+CAPE group had increased expression of HO-1 and MnSOD compared with GSNO or CAPE alone. Data are presented as mean ± SD (n=4). ***p<0.001 vs. Sham, +++p<0.001, ++p<0.01, +p<0.05 vs. TBI, ##p<0.01, #p<0.05 vs. GSNO, $p<0.05 vs. CAPE.
Effect of GSNO or CAPE or GSNO+CAPE treatment for 7 days on the activation of AMPK (pAMPK) following TBI
AMPK activation is directly associated with bioenergetics and mitochondrial activity. The effects of GSNO, CAPE and GSNO+CAPE on the activation of AMPK (evaluated as pAMPK) were determined using western blot analysis (Fig 5A) and densitometry (Fig 5B). AMPK was more activated in the TBI compared with the Sham group. While GSNO treatment had no effect on the expression of pAMPK, CAPE treatment increased the expression compared with the TBI group. Interestingly, treatment with combined GSNO+CAPE had significantly increased AMPK activation (pAMPK) compared with GSNO or CAPE alone (Fig 5A, B).
Figure 5.
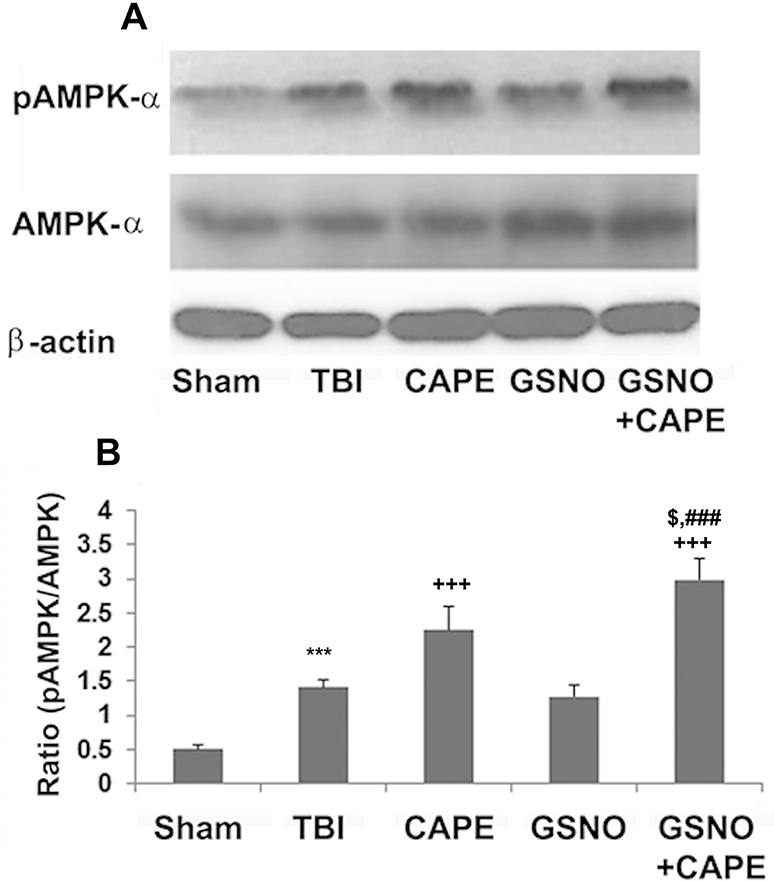
Effect of GSNO, CAPE, and combination GSNO+CAPE for 7 days on activation and expression of AMPK. Activation of AMPK (measured as increased levels of AMPK phosphorylation) and expression of total AMPK (AMPKα) were measured in traumatic penumbra region using western analysis (A) and its densitometry (B). While CAPE treatment increased AMPK activation levels, GSNO treatment had no significant effects on AMPK activation. Treatment with GSNO+CAPE had synergistic effects. Data are presented as mean ± SD (n=4). ***p<0.001 vs. Sham, +++p<0.001 vs. TBI, ###p<0.001 vs. GSNO, $p<0.05 vs. CAPE.
Effect of CAPE treatment of wild type normal mice for 2 h on the activation of AMPK, ACC and LKB1 in brain tissue
The effects of CAPE on the activation of AMPK (Fig 6A), its substrate ACC (Fig 6B) and upstream AMPK kinase LKB1 (Fig 6C) were determined using western blot analysis (Fig 6A-C) and densitometry (Fig 6 A1-C1). Treatment with CAPE increased the expression levels pAMPK (Fig 6A, A1), pACC (Fig 6B, B1) and pLKB1 (Fig 6C, C1) in cortical brain tissue compared with the vehicle (DMSO)-treated brain tissue from the very same cortical region. The expression of AMPK, ACC and LKB1 remained unchanged between CAPE and DMSO-treated animals. Because GSNO did not change pAMPK expression levels in TBI brain (Fig 5A, B), we did not evaluate GSNO’s effect on the activation of AMPK in the normal mouse brain.
Discussion
Both GSNO and CAPE have an acceptable mechanistic rationale, good safety record and favorable pharmacokinetic profile. We designed this study to investigate, first, the efficacy of these neuroprotective compounds alone and then extended the study to examine the greater efficacy, if any, of GSNO+CAPE combination therapy. Combination therapies have been documented better for optimal neurobehavioral and cognitive recovery after experimental TBI (Kline et al. 2016). Our previous studies showing the efficacy of GSNO in a rat/mouse model of TBI (Khan et al. 2018; Khan et al. 2016; Khan et al. 2009; Khan et al. 2011) and the neuroprotective effect of CAPE in a rat model of cerebral IR (Khan et al. 2007) provided a rationale to combine them for potentially greater efficacy and accelerated functional recovery.
As expected, all three treated groups showed significant improvements with respect to improved beam walk latency (Fig 1A) and foot faults (Fig 1B). However, the anticipated greater balance and coordination recovery in the combination group compared with the GSNO or the CAPE alone group was not observed (Fig 1A, B). The beam walk test evaluates balance and coordination of movement, and it is sensitive to degree of injury and therapeutic manipulation (Fujimoto et al. 2004). Normalization of beam walk and fault functions at 7th day after TBI (Fig 1A, B) in all groups may be due to the mild/moderate degree of injury in our model. In contrast to motor function, non-spatial memory (evaluated by NOR test, Fig 1C) and depressive-like behavior (evaluated by FST, Fig 1D) were significantly improved in the combination (GSNO+CAPE) group compared with the GSNO or the CAPE alone group (Fig 1C, D).
Although depressive-like behavior (FST) is a significant consequence and common to mild, moderate and severe TBI (Washington et al. 2012), only one study has investigated its pharmacological regulation in TBI via an inhibition of the 5-lipoxygenase-induced oxidative stress pathway (Higashi et al. 2014). Both CAPE (Feng et al. 2008; Khan et al. 2007) and GSNO (Chiueh and Rauhala 1999; Khan et al. 2011; Rauhala et al. 1998) are potent antioxidants. Thus, the greatly improved FST in the combination group compared with the GSNO or the CAPE alone indicates the potential of the combination therapy to ameliorate depression and emotion- associated deficits following TBI. Cognitive deficit is another major issue associated with TBI. We used a NOR test to evaluate cognition as described (Huang et al. 2014). The advantage of this test is that it can be performed well irrespective of the age of mice or rats. Unlike the Morris water-maze task, its outcomes are independent of motor function (Dhawan et al. 2011). Like FST function (Fig 1D), NOR function was also significantly improved in the combination group compared with the GSNO or the CAPE group alone. These results clearly support the notion that the combination treatment is more effective to reduce cognitive and emotional deficits. Because both mitochondrial function and cellular energy metabolism play key roles in neuroprotection and functional recovery, we investigated the effect of GSNO, CAPE and GSNO+CAPE on improving mitochondrial function and cellular energy metabolism.
Treated groups had reduced infiltration and improved tissue structure as shown by H&E staining (Fig 2C). In parallel, they provided significant neuroprotection as evident from a significantly increased number of viable neurons (NeuN and Nissl staining); however, the combination group had additional neuroprotection compared with the GSNO- or CAPE-alone group (Fig 2A, B). To examine potential correlates of neuroprotection (Fig 2) and functional recovery (Fig 1) with mitochondrial function, we determined the quality and efficiency of mitochondria. Altered mitochondrial dynamics are salient features of TBI (Fischer et al. 2016), and mitochondrial quality control is regulated by antagonistic fission and fusion, which determine mitochondrial quality, integrity and functions (Di Pietro et al. 2017). The fission of mitochondria has been reported to be regulated by AMPK (Wang and Youle 2016). Therefore, we determined the expression of fission (Drp1 and Fis 1)- and fusion (Opa1)-associated proteins (Fig 3A, B). These proteins were correlated with mitochondrial biogenesis (PGC1α), mitochondrial antioxidant enzyme expression (MnSOD), and the overall cellular antioxidant activity measured as the expression of HO-1 (Fig 4A, B). While the process of fission was activated (up regulation of Drp1 and Fis1, Fig 3A, B), fusion was inhibited (down regulation of Opa1, Fig 3A, B). In the TBI group, both GSNO and CAPE significantly inhibited fission; the combination treatment had a synergistic effect on the expression of Drp1. On the other hand, fusion was inhibited in all treated groups. Collectively, regulation of fission and fusion processes (Fig 3) shows that both GSNO and CAPE maintained mitochondrial quality, correlating well with neuroprotection and functional recovery. Furthermore, controlling mitochondrial integrity translated into functional activities of mitochondria as shown by increased expression of PGC1α (a mediator of mitochondrial biogenesis) and MnSOD (a regulator of mitochondrial antioxidant activity) (Fig 4A, B). Combined, GSNO+CAPE increased expression of MnSOD (Fig 4A, B). Like MnSOD, both GSNO and CAPE increased the expression levels of HO-1, and the combination resulted in higher levels of HO-1 expression (Fig 4A, B), indicating that the combination is more effective in reducing the overall oxidative damage. HO-1 is transcriptionally regulated by Nrf2, and the activity of Nrf2 is regulated by both GSNO and CAPE. These linkages indicate that they provide neuroprotection and stimulate functional recovery, at least in part, by inducing the activity of Nrf2.
Inappropriate heme metabolism contributes to the development of pathology, including increased susceptibility to TBI. The heme oxygenases (HO) are metabolic enzymes that utilize NADPH and oxygen to break apart the heme moiety, liberating biliverdin (BV), carbon monoxide (CO), and iron. Heme that is derived from hemoproteins can be toxic to the cells and and causes apoptosis and local inflammation if not broken and removed immediately. HO-1 plays a significant role in brain heme metabolism (Wegiel et al. 2014). Therefore, HO-1 plays an important role in the regulation of cellular redox. Increased expression of HO-1 by GSNO and CAPE (Fig 4A, B) indicates that both invoke antioxidant effects and, in combination, they do so greatly.
In TBI, the NO metabolome is derailed due to oxidative stress (Khan et al. 2016; Khan et al. 2017). After injury, NO released by nitric oxide synthases (NOS) is consumed by superoxide to form peroxynitrite, a highly potent oxidant in biological systems (Pacher et al. 2007). In such an environment, the levels of endogenous redox modulator GSNO are depleted, causing activation of calpains and caspases, leading to neuroinflammation, cell death and functional deficits (Annamalai et al. 2015; Corti et al. 2014; Khan et al. 2016; Khan et al. 2011). Supplementation of GSNO in a number of neurodegenerative diseases provides neuroprotection and functional recovery (Khan et al. 2016; Khan et al. 2012; Khan et al. 2011; Khan et al. 2005; Won et al. 2015). These neuroprotective effects of GSNO may be associated with its reported inhibition of the activity of caspase-3 (Khan et al. 2005; Mohr et al. 1997) as well as calpains (Annamalai et al. 2015; Khan et al. 2016). The same is anticipated in TBI because conventional NO-donors or NO gas itself may immediately be inactivated by superoxide, forming peroxynitrite. This disadvantage of inactivation is not associated with the S-nitrosylating agent GSNO. In addition, S-nitrosylation of cysteine residue (a reversible post translational modification) prevents it from further oxidation to sulfinic and sulfonic acids (an irreversible modification), thereby preventing inactivation of both NO and proteins. Neurorepair from GSNO may be mediated by two different mechanisms: 1) S-nitrosylation and 2) maintaining redox and protecting mitochondrial functions by mechanistically reducing the production of oxidants, including peroxynitrite. This multi- mechanistic functional and therapeutic potential is not embedded in conventional NO donors (Khan et al. 2006), making GSNO a unique candidate to be investigated for the stimulation of functional recovery following TBI.
Like GSNO, CAPE confers neuroprotection in animal models of TBI (Kerman et al. 2012) and cerebral ischemia/reperfusion (Khan et al. 2007). It also protects against neonatal brain hypoxic-ischemic injury (Wei et al. 2004). In experimental TBI, it protects BBB (Zhao et al. 2012). However, the molecular mechanisms underlying CAPE-mediated reductions in brain injury and improvements of neurological functions are not clear. Recent studies from our (Singh et al. 2013) and other laboratories (Aladag et al. 2006; Cengiz et al. 2007; Fontanilla et al. 2012; Ilhan et al. 2004; Natarajan et al. 1996; Tsai et al. 2005) suggest that CAPE invokes its protective effects mainly by maintaining mitochondrial integrity and activating AMPK. These findings support the notion that CAPE is a suitable agent to be used as a potent antioxidant therapy in TBI. In view of these properties in neurodegenerative animal models, we used CAPE as a therapeutic agent to enhance the efficacy of GSNO by improving redox and mitochondrial structure/function (Feng et al. 2008). CAPE is a plant-originated naturally occurring lipophilic antioxidant without any apparent toxicity in humans. In this study, CAPE was found to improve significantly mitochondrial efficiency by regulating the fission/fusion process and increasing antioxidant levels for accelerated recovery in a mouse model of experimental TBI. The treatment also improved cognitive function alone and accelerated recovery when combined with GSNO. In addition to improving the mitochondrial function and reducing the oxidative damage, CAPE likely contributed an additional benefit by activating AMPK. AMPK activation has been documented to ameliorate neurodegeneration and improve neurological functions in several neurodegenerative diseases, including TBI (Tsai et al. 2015).
To provide evidence that CAPE is a potent AMPK activator, normal animals were treated with CAPE for 2 h. An increased level of pLKB1, pAMPK and pACC in brain tissue (Fig 6) indicates that CAPE activates AMPK via LKB1. Several flavonoid-based phytochemicals are reported to activate AMPK. While the exact sequence of molecular events leading to CAPE- mediated activation of AMPK has yet to be clarified, our evidence suggests that CAPE directly activates AMPK (Fig 6). Based on our data and discussions herein, it is clear that combining antioxidant/Nrf2 with AMPK pathway results in greater neuroprotection and accelerated functional recovery. Supporting this notion, a dual AMPK/Nrf2 activator has been reported to ameliorate stroke injury (Wang et al. 2017).
Limitations:
The most affected population from TBI is recognized to be young adult males. Therefore, we used young adult males in this study. At the same time, the differential effects of therapeutic compounds such as GSNO are well established (Wu et al. 2007). Therefore, in follow-up longer time study, the efficacy of GSNO, CAPE and GSNO+CAPE will be investigated using both male and female young adult mice to determine sex-based mechanistic differences and functional recovery, if any.
Our study shows that a combined treatment with GSNO and CAPE mitigates TBI pathology. The combination reduces cognitive and emotional deficits through additive mechanisms, improving mitochondrial efficiency and stimulating the cellular energy switch AMPK. Therefore, combination GSNO+CAPE treatment represents the focus of our next level research as a promising therapy for human TBI.
Significance Statement:
TBI is a common and devastating disorder, which causes neurobehavioral deficits in young adults, the elderly and children. These deficits are often the cause of significant disability and morbidity. TBI patients are also susceptible to dementia, epilepsy, and Alzheimer’s disease. Although several therapeutic compounds provide some degree of improvement by targeting single mechanistic pathways, they are not sufficient in rehabilitating patients with TBI due to the multi-mechanistic nature of the injury. Therefore, the goal of this study is to determine whether a combination therapy targeting two different mechanistic pathways results in greater efficacy compared to monotherapy following TBI.
Acknowledgments
We thank Ms. Joyce Bryan for her technical help and secretarial assistance. We are grateful to Ms. Danielle Lowe (MD/PhD student at the MUSC) for statistical analysis. We also acknowledge Dr. Tom Smith from the MUSC Writing Center for his valuable editing of the manuscript.
Funding information: This work was supported by grants from VA merit award (BX003401). This work was also supported by the VA grant (RX002090) and the NIH, Grants C06 RR018823 and No C06 RR015455 from the Extramural Research Facilities Program of the National Center for Research Resources.
Footnotes
Conflict of interests
The author(s) declare that they have no conflict of interests.
Authors’ Contributions
This study is based on an original idea of MK, AKS and IS. MK wrote the manuscript and all authors reviewed the manuscript. AS and TSD carried out animal and biochemical studies. MK, AKS, TSD, HK, and AS critically examined animal and biochemical studies. MK and HK performed statistical analysis. AS and TSD performed histological, immunoblotting and IHC studies. All authors have approved the manuscript.
Declaration of Transparency
The authors affirm that in accordance to the policies set by the Journal of Neuroscience Research this manuscript presents an accurate and transparent account of the study being reported and that all critical details describing the methods and results are present.
Data Accessibility
All data generated or analyzed during this study are included in this manuscript.
References
- Aladag MA, Turkoz Y, Ozcan C, Sahna E, Parlakpinar H, Akpolat N, Cigremis Y. 2006. Caffeic acid phenethyl ester (CAPE) attenuates cerebral vasospasm after experimental subarachnoidal haemorrhage by increasing brain nitric oxide levels. Int J Dev Neurosci 24(1):9–14. [DOI] [PubMed] [Google Scholar]
- Annamalai B, Won JS, Choi S, Singh I, Singh AK. 2015. Role of S-nitrosoglutathione mediated mechanisms in tau hyper-phosphorylation. Biochem Biophys Res Commun 458(1):214–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avila MA, Sell SL, Kadoi Y, Prough DS, Hellmich HL, Velasco M, Dewitt DS. 2008. L- Arginine decreases fluid-percussion injury-induced neuronal nitrotyrosine immunoreactivity in rats. J Cereb Blood Flow Metab 28(10):1733–1741. [DOI] [PubMed] [Google Scholar]
- Besson VC. 2009. Drug targets for traumatic brain injury from poly(ADP-ribose)polymerase pathway modulation. Br J Pharmacol 157(5):695–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilgen M 2005. A new device for experimental modeling of central nervous system injuries. Neurorehabil Neural Repair 19(3):219–226. [DOI] [PubMed] [Google Scholar]
- Bondi CO, Semple BD, Noble-Haeusslein LJ, Osier ND, Carlson SW, Dixon CE, Giza CC, Kline AE. 2015. Found in translation: Understanding the biology and behavior of experimental traumatic brain injury. Neurosci Biobehav Rev 58:123–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Can A, Dao DT, Arad M, Terrillion CE, Piantadosi SC, Gould TD. 2012. The mouse forced swim test. J Vis Exp(59):e3638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celli N, Mariani B, Dragani LK, Murzilli S, Rossi C, Rotilio D. 2004. Development and validation of a liquid chromatographic-tandem mass spectrometric method for the determination of caffeic acid phenethyl ester in rat plasma and urine. J Chromatogr B Analyt Technol Biomed Life Sci 810(1): 129–136. [DOI] [PubMed] [Google Scholar]
- Cengiz N, Colakoglu N, Kavakli A, Sahna E, Parlakpinar H, Acet A. 2007. Effects of caffeic acid phenethyl ester on cerebral cortex: structural changes resulting from middle cerebral artery ischemia reperfusion. Clin Neuropathol 26(2):80–84. [DOI] [PubMed] [Google Scholar]
- Chen Q, Sievers RE, Varga M, Kharait S, Haddad DJ, Patton AK, Delany CS, Mutka SC, Blonder JP, Dube GP, Rosenthal GJ, Springer ML. 2013. Pharmacological inhibition of S-nitrosoglutathione reductase improves endothelial vasodilatory function in rats in vivo. J Appl Physiol (1985) 114(6):752–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiueh CC, Rauhala P. 1999. The redox pathway of S-nitrosoglutathione, glutathione and nitric oxide in cell to neuron communications. Free Radic Res 31(6):641–650. [DOI] [PubMed] [Google Scholar]
- Choi EY, Choe SH, Hyeon JY, Choi JI, Choi IS, Kim SJ. 2015. Effect of caffeic acid phenethyl ester on Prevotella intermedia lipopolysaccharide-induced production of proinflammatory mediators in murine macrophages. J Periodontal Res. [DOI] [PubMed] [Google Scholar]
- Chou PC, Shunmugavel A, Sayed HE, Desouki MM, Nguyen SA, Khan M, Singh I, Bilgen M. 2011. Preclinical use of longitudinal MRI for screening the efficacy of s- nitrosoglutathione in treating spinal cord injury. J Magn Reson Imaging 33(6): 1301–1311. [DOI] [PubMed] [Google Scholar]
- Corti A, Franzini M, Scataglini I, Pompella A. 2014. Mechanisms and targets of the modulatory action of S-nitrosoglutathione (GSNO) on inflammatory cytokines expression. Arch Biochem Biophys 562:80–91. [DOI] [PubMed] [Google Scholar]
- Deng-Bryant Y, Singh IN, Carrico KM, Hall ED. 2008. Neuroprotective effects of tempol, a catalytic scavenger of peroxynitrite-derived free radicals, in a mouse traumatic brain injury model. J Cereb Blood Flow Metab 28(6): 1114–1126. [DOI] [PubMed] [Google Scholar]
- Deng Y, Thompson BM, Gao X, Hall ED. 2007. Temporal relationship of peroxynitrite-induced oxidative damage, calpain-mediated cytoskeletal degradation and neurodegeneration after traumatic brain injury. Exp Neurol 205(1): 154–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhawan J, Benveniste H, Luo Z, Nawrocky M, Smith SD, Biegon A. 2011. A new look at glutamate and ischemia: NMDA agonist improves long-term functional outcome in a rat model of stroke. Future Neurol 6(6):823–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Pietro V, Lazzarino G, Amorini AM, Signoretti S, Hill LJ, Porto E, Tavazzi B, Lazzarino G, Belli A. 2017. Fusion or Fission: The Destiny of Mitochondria In Traumatic Brain Injury of Different Severities. Sci Rep 7(1):9189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Y, Lu YW, Xu PH, Long Y, Wu WM, Li W, Wang R. 2008. Caffeic acid phenethyl ester and its related compounds limit the functional alterations of the isolated mouse brain and liver mitochondria submitted to in vitro anoxia-reoxygenation: relationship to their antioxidant activities. Biochim Biophys Acta 1780(4):659–672. [DOI] [PubMed] [Google Scholar]
- Fischer TD, Hylin MJ, Zhao J, Moore AN, Waxham MN, Dash PK. 2016. Altered Mitochondrial Dynamics and TBI Pathophysiology. Front Syst Neurosci 10:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontanilla CV, Wei X, Zhao L, Johnstone B, Pascuzzi RM, Farlow MR, Du Y. 2012. Caffeic acid phenethyl ester extends survival of a mouse model of amyotrophic lateral sclerosis. Neuroscience 205:185–193. [DOI] [PubMed] [Google Scholar]
- Fox GB, Fan L, Levasseur RA, Faden AI. 1998. Sustained sensory/motor and cognitive deficits with neuronal apoptosis following controlled cortical impact brain injury in the mouse. J Neurotrauma 15(8):599–614. [DOI] [PubMed] [Google Scholar]
- Fujimoto ST, Longhi L, Saatman KE, Conte V, Stocchetti N, McIntosh TK. 2004. Motor and cognitive function evaluation following experimental traumatic brain injury. Neurosci Biobehav Rev 28(4):365–378. [DOI] [PubMed] [Google Scholar]
- Gardner AJ, Zafonte R. 2016. Neuroepidemiology of traumatic brain injury. Handb Clin Neurol 138:207–223. [DOI] [PubMed] [Google Scholar]
- Hall ED, Kupina NC, Althaus JS. 1999. Peroxynitrite scavengers for the acute treatment of traumatic brain injury. Ann N Y Acad Sci 890:462–468. [DOI] [PubMed] [Google Scholar]
- Higashi Y, Hoshijima M, Yawata T, Nobumoto A, Tsuda M, Shimizu T, Saito M, Ueba T. 2014. Suppression of oxidative stress and 5-lipoxygenase activation by edaravone improves depressive-like behavior after concussion. J Neurotrauma 31(20):1689–1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang TN, Chuang HC, Chou WH, Chen CY, Wang HF, Chou SJ, Hsueh YP. 2014. Tbr1 haploinsufficiency impairs amygdalar axonal projections and results in cognitive abnormality. Nat Neurosci 17(2):240–247. [DOI] [PubMed] [Google Scholar]
- Ilhan A, Akyol O, Gurel A, Armutcu F, Iraz M, Oztas E. 2004. Protective effects of caffeic acid phenethyl ester against experimental allergic encephalomyelitis-induced oxidative stress in rats. Free Radic Biol Med 37(3):386–394. [DOI] [PubMed] [Google Scholar]
- Jaffrey SR, Erdjument-Bromage H, Ferris CD, Tempst P, Snyder SH. 2001. Protein S- nitrosylation: a physiological signal for neuronal nitric oxide. Nat Cell Biol 3(2):193–197. [DOI] [PubMed] [Google Scholar]
- Jatana M, Giri S, Ansari MA, Elango C, Singh AK, Singh I, Khan M. 2006. Inhibition of NF-kB activation by 5-lipoxygenase inhibitors protects brain against injury in a rat model of focal cerebral ischemia. J Neuroinflammation 3(1): 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson VE, Stewart W, Smith DH. 2010. Traumatic brain injury and amyloid-beta pathology: a link to Alzheimer’s disease? Nat Rev Neurosci 11:361–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerman M, Kanter M, Coskun KK, Erboga M, Gurel A. 2012. Neuroprotective effects of caffeic acid phenethyl ester on experimental traumatic brain injury in rats. J Mol Histol 43(1):49–57. [DOI] [PubMed] [Google Scholar]
- Khan M, Dhammu TS, Baarine M, Kim J, Paintlia MK, Singh I, Singh AK. 2018. GSNO promotes functional recovery in experimental TBI by stabilizing HIF-1alpha. Behav Brain Res 340:63–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan M, Dhammu TS, Matsuda F, Annamalai B, Dhindsa TS, Singh I, Singh AK. 2016. Targeting the nNOS/peroxynitrite/calpain system to confer neuroprotection and aid functional recovery in a mouse model of TBI. Brain Res 1630:159–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan M, Dhammu TS, Matsuda F, Baarine M, Dhindsa TS, Singh I, Singh AK. 2015a. Promoting endothelial function by S-nitrosoglutathione through the HIF-1alpha/VEGF pathway stimulates neurorepair and functional recovery following experimental stroke in rats. Drug Des Devel Ther 9:2233–2247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan M, Dhammu TS, Matsuda F, Singh AK, Singh I. 2015. b. Blocking a vicious cycle nNOS/peroxynitrite/AMPK by S-nitrosoglutathione: implication for stroke therapy. BMC Neurosci 16:42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan M, Dhammu TS, Sakakima H, Shunmugavel A, Gilg AG, Singh AK, Singh I. 2012. The inhibitory effect of S-nitrosoglutathione on blood-brain barrier disruption and peroxynitrite formation in a rat model of experimental stroke. J Neurochem 123 Suppl 2:86–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan M, Elango C, Ansari MA, Singh I, Singh AK. 2007. Caffeic acid phenethyl ester reduces neurovascular inflammation and protects rat brain following transient focal cerebral ischemia. J Neurochem 102(2):365–377. [DOI] [PubMed] [Google Scholar]
- Khan M, Im YB, Shunmugavel A, Gilg AG, Dhindsa RK, Singh AK, Singh I. 2009. Administration of S-nitrosoglutathione after traumatic brain injury protects the neurovascular unit and reduces secondary injury in a rat model of controlled cortical impact. J Neuroinflammation 6:32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan M, Jatana M, Elango C, Paintlia AS, Singh AK, Singh I. 2006. Cerebrovascular protection by various nitric oxide donors in rats after experimental stroke. Nitric Oxide 15(2): 114124. [DOI] [PubMed] [Google Scholar]
- Khan M, Khan H, Singh I, Singh AK. 2017. Hypoxia inducible factor-1 alpha stabilization for regenerative therapy in traumatic brain injury. Neural Regen Res 12(5):696–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan M, Sakakima H, Dhammu TS, Shunmugavel A, Im YB, Gilg AG, Singh AK, Singh I. 2011. S-Nitrosoglutathione reduces oxidative injury and promotes mechanisms of neurorepair following traumatic brain injury in rats. J Neuroinflammation 8(1):78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan M, Sekhon B, Giri S, Jatana M, Gilg AG, Ayasolla K, Elango C, Singh AK, Singh I. 2005. S-Nitrosoglutathione reduces inflammation and protects brain against focal cerebral ischemia in a rat model of experimental stroke. J Cereb Blood Flow Metab 25(2):177–192. [DOI] [PubMed] [Google Scholar]
- Kim H, Kim W, Yum S, Hong S, Oh JE, Lee JW, Kwak MK, Park EJ, Na DH, Jung Y. 2013a. Caffeic acid phenethyl ester activation of Nrf2 pathway is enhanced under oxidative state: structural analysis and potential as a pathologically targeted therapeutic agent in treatment of colonic inflammation. Free Radic Biol Med 65:552–562. [DOI] [PubMed] [Google Scholar]
- Kim J, Won JS, Singh AK, Sharma AK, Singh I. 2013b. STAT3 Regulation by S-Nitrosylation: Implication for Inflammatory Disease. Antioxid Redox Signal. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kline AE, Leary JB, Radabaugh HL, Cheng JP, Bondi CO. 2016. Combination therapies for neurobehavioral and cognitive recovery after experimental traumatic brain injury: Is more better? Prog Neurobiol 142:45–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kluge I, Gutteck-Amsler U, Zollinger M, Do KQ. 1997. S-Nitrosoglutathione in Rat Cerebellum: Identification and Quantification by Liquid Chromatography-Mass Spectrometry. J Neurochem 69(6):2599–2607. [DOI] [PubMed] [Google Scholar]
- Konorev EA, Tarpey MM, Joseph J, Baker JE, Kalyanaraman B. 1995. S-nitrosoglutathione improves functional recovery in the isolated rat heart after cardioplegic ischemic arrest- evidence for a cardioprotective effect of nitric oxide. J Pharmacol Exp Ther 274(1):200–206. [PubMed] [Google Scholar]
- Lee ES, Uhm KO, Lee YM, Han M, Lee M, Park JM, Suh PG, Park SH, Kim HS. 2007. CAPE (caffeic acid phenethyl ester) stimulates glucose uptake through AMPK (AMP-activated protein kinase) activation in skeletal muscle cells. Biochem Biophys Res Commun 361(4):854–858. [DOI] [PubMed] [Google Scholar]
- Levin H, Kraus MF. 1994. The frontal lobes and traumatic brain injury. J Neuropsychiatry Clin Neurosci 6(4):443–454. [DOI] [PubMed] [Google Scholar]
- Liao CC, Ou TT, Huang HP, Wang CJ. 2013. The inhibition of oleic acid induced hepatic lipogenesis and the promotion of lipolysis by caffeic acid via up-regulation of AMP- activated kinase. J Sci Food Agric. [DOI] [PubMed] [Google Scholar]
- Lima B, Lam GK, Xie L, Diesen DL, Villamizar N, Nienaber J, Messina E, Bowles D, Kontos CD, Hare JM, Stamler JS, Rockman HA. 2009. Endogenous S-nitrosothiols protect against myocardial injury. Proc Natl Acad Sci U S A 106(15):6297–6302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F, Schafer DP, McCullough LD. 2009. TTC, fluoro-Jade B and NeuN staining confirm evolving phases of infarction induced by middle cerebral artery occlusion. J Neurosci Methods 179(1):1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohr S, Zech B, Lapetina EG, Brune B. 1997. Inhibition of caspase-3 by S-nitrosation and oxidation caused by nitric oxide. Biochem Biophys Res Commun 238(2):387–391. [DOI] [PubMed] [Google Scholar]
- Natarajan K, Singh S, Burke TR Jr., Grunberger D, Aggarwal BB 1996. Caffeic acid phenethyl ester is a potent and specific inhibitor of activation of nuclear transcription factor NF- kappa B. Proc Natl Acad Sci U S A 93(17):9090–9095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osier ND, Dixon CE. 2016. The Controlled Cortical Impact Model: Applications, Considerations for Researchers, and Future Directions. Front Neurol 7:134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacher P, Beckman JS, Liaudet L. 2007. Nitric oxide and peroxynitrite in health and disease. Physiol Rev 87(1):315–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radomski MW, Rees DD, Dutra A, Moncada S. 1992. S-nitroso-glutathione inhibits platelet activation in vitro and in vivo. Br J Pharmacol 107(3):745–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rassaf T, Poll LW, Brouzos P, Lauer T, Totzeck M, Kleinbongard P, Gharini P, Andersen K, Schulz R, Heusch G, Modder U, Kelm M. 2006. Positive effects of nitric oxide on left ventricular function in humans. Eur Heart J 27(14):1699–1705. [DOI] [PubMed] [Google Scholar]
- Rauhala P, Lin AM, Chiueh CC. 1998. Neuroprotection by S-nitrosoglutathione of brain dopamine neurons from oxidative stress. Faseb J 12(2): 165–173. [DOI] [PubMed] [Google Scholar]
- Sakakima H, Khan M, Dhammu TS, Shunmugavel A, Yoshida Y, Singh I, Singh AK. 2012. Stimulation of functional recovery via the mechanisms of neurorepair by S- nitrosoglutathione and motor exercise in a rat model of transient cerebral ischemia and reperfusion. Restor Neurol Neurosci 30(5):383–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savidge TC, Newman P, Pothoulakis C, Ruhl A, Neunlist M, Bourreille A, Hurst R, Sofroniew MV. 2007. Enteric glia regulate intestinal barrier function and inflammation via release of S- nitrosoglutathione. Gastroenterology 132(4): 1344–1358. [DOI] [PubMed] [Google Scholar]
- Shunmugavel A, Khan M, Chou PC, Dhindsa RK, Marcus M, Copay AG, Subach BR, Schuler TC, Bilgen M, Orak JK, Singh I. 2010. Simvastatin protects bladder and renal functions following spinal cord injury in rats. J Inflamm (Lond) 7(1): 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shunmugavel A, Khan M, Chou PC, Singh I. 2012. Spinal Cord Injury Induced Arrest in Estrous Cycle of Rats Is Ameliorated by S-nitrosoglutathione: Novel Therapeutic Agent to Treat Amenorrhea. J Sex Med 9(1): 148–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh IN, Sullivan PG, Hall ED. 2007. Peroxynitrite-mediated oxidative damage to brain mitochondria: Protective effects of peroxynitrite scavengers. J Neurosci Res 85(10):2216–2223. [DOI] [PubMed] [Google Scholar]
- Singh J, Khan M, Singh I. 2013. Caffeic acid phenethyl ester induces adrenoleukodystrophy (Abcd2) gene in human X-ALD fibroblasts and inhibits the proinflammatory response in Abcd1/2 silenced mouse primary astrocytes. Biochim Biophys Acta 1831(4):747–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh SP, Wishnok JS, Keshive M, Deen WM, Tannenbaum SR. 1996. The chemistry of the S- nitrosoglutathione/glutathione system. Proc Natl Acad Sci U S A 93(25):14428–14433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith DH, Soares HD, Pierce JS, Perlman KG, Saatman KE, Meaney DF, Dixon CE, McIntosh TK. 1995. A model of parasagittal controlled cortical impact in the mouse: cognitive and histopathologic effects. J Neurotrauma 12(2):169–178. [DOI] [PubMed] [Google Scholar]
- Son S, Lewis BA. 2002. Free radical scavenging and antioxidative activity of caffeic acid amide and ester analogues: structure-activity relationship. J Agric Food Chem 50(3):468–472. [DOI] [PubMed] [Google Scholar]
- Suuronen T, Huuskonen J, Pihlaja R, Kyrylenko S, Salminen A. 2003. Regulation of microglial inflammatory response by histone deacetylase inhibitors. J Neurochem 87(2):407–416. [DOI] [PubMed] [Google Scholar]
- Teixeira J, Soares P, Benfeito S, Murphy MP, Oliveira PJ, Borges F. 2015. Bridging the gap between nature and antioxidant setbacks: delivering caffeic acid to mitochondria. Methods Mol Biol 1265:73–83. [DOI] [PubMed] [Google Scholar]
- Tolba MF, Azab SS, Khalifa AE, Abdel-Rahman SZ, Abdel-Naim AB. 2013. Caffeic acid phenethyl ester, a promising component of propolis with a plethora of biological activities: a review on its anti-inflammatory, neuroprotective, hepatoprotective, and cardioprotective effects. IUBMB Life 65(8):699–709. [DOI] [PubMed] [Google Scholar]
- Tsai CF, Kuo YH, Yeh WL, Wu CY, Lin HY, Lai SW, Liu YS, Wu LH, Lu JK, Lu DY. 2015. Regulatory effects of caffeic Acid phenethyl ester on neuroinflammation in microglial cells. Int J Mol Sci 16(3):5572–5589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai SK, Lin MJ, Liao PH, Yang CY, Lin SM, Liu SM, Lin RH, Chih CL, Huang SS. 2005. Caffeic acid phenethyl ester ameliorates cerebral infarction in rats subjected to focal cerebral ischemia. Life Sci. [DOI] [PubMed] [Google Scholar]
- Tsuda S, Egawa T, Ma X, Oshima R, Kurogi E, Hayashi T. 2012. Coffee polyphenol caffeic acid but not chlorogenic acid increases 5’AMP-activated protein kinase and insulin- independent glucose transport in rat skeletal muscle. J Nutr Biochem 23(11): 1403–1409. [DOI] [PubMed] [Google Scholar]
- Wang C, Youle R. 2016. Cell biology: Form follows function for mitochondria. Nature 530(7590):288–289. [DOI] [PubMed] [Google Scholar]
- Wang G, Zhang J, Hu X, Zhang L, Mao L, Jiang X, Liou AK, Leak RK, Gao Y, Chen J. 2013. Microglia/macrophage polarization dynamics in white matter after traumatic brain injury. J Cereb Blood Flow Metab 33(12):1864–1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Huang Y, Xu Y, Ruan W, Wang H, Zhang Y, Saavedra JM, Zhang L, Huang Z, Pang T. 2017. A Dual AMPK/Nrf2 Activator Reduces Brain Inflammation After Stroke by Enhancing Microglia M2 Polarization. Antioxid Redox Signal. [DOI] [PubMed] [Google Scholar]
- Washington PM, Forcelli PA, Wilkins T, Zapple DN, Parsadanian M, Burns MP. 2012. The effect of injury severity on behavior: a phenotypic study of cognitive and emotional deficits after mild, moderate, and severe controlled cortical impact injury in mice. J Neurotrauma 29(13):2283–2296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wegiel B, Nemeth Z, Correa-Costa M, Bulmer AC, Otterbein LE. 2014. Heme oxygenase-1: a metabolic nike. Antioxid Redox Signal 20(11):1709–1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei X, Zhao L, Ma Z, Holtzman DM, Yan C, Dodel RC, Hampel H, Oertel W, Farlow MR, Du Y. 2004. Caffeic acid phenethyl ester prevents neonatal hypoxic-ischaemic brain injury. Brain 127(Pt 12):2629–2635. [DOI] [PubMed] [Google Scholar]
- Won JS, Annamalai B, Choi S, Singh I, Singh AK. 2015. S-nitrosoglutathione reduces tau hyper- phosphorylation and provides neuroprotection in rat model of chronic cerebral hypoperfusion. Brain Res 1624:359–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, Romieu I, Sienra-Monge JJ, Estela Del Rio-Navarro B, Anderson DM, Jenchura CA, Li H, Ramirez-Aguilar M, Del Carmen Lara-Sanchez I, London SJ. 2007. Genetic variation in S-nitrosoglutathione reductase (GSNOR) and childhood asthma. J Allergy Clin Immunol 120(2):322–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Schallert T, Zhang ZG, Jiang Q, Arniego P, Li Q, Lu M, Chopp M. 2002. A test for detecting long-term sensorimotor dysfunction in the mouse after focal cerebral ischemia. J Neurosci Methods 117(2):207–214. [DOI] [PubMed] [Google Scholar]
- Zhao J, Pati S, Redell JB, Zhang M, Moore AN, Dash PK. 2012. Caffeic Acid phenethyl ester protects blood-brain barrier integrity and reduces contusion volume in rodent models of traumatic brain injury. J Neurotrauma 29(6): 1209–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Z, Yenari MA. 2004. Post-ischemic inflammation: molecular mechanisms and therapeutic implications. Neurol Res 26(8):884–892. [DOI] [PubMed] [Google Scholar]
- Zhu Y, Bu Q, Liu X, Hu W, Wang Y. 2014. Neuroprotective effect of TAT-14–3-3epsilon fusion protein against cerebral ischemia/reperfusion injury in rats. PLoS One 9(3):e93334. [DOI] [PMC free article] [PubMed] [Google Scholar]