

Abstract
Background
Fibronectin (FN) polymerization is necessary for collagen matrix deposition and is a key contributor to increased abundance of cardiac myofibroblasts (MF) following cardiac injury. We hypothesized that interfering with FN polymerization or its genetic ablation in fibroblasts would attenuate MF, fibrosis and improve cardiac function following ischemia/reperfusion (I/R)-injury.
Methods
Mouse and human MF were utilized to assess the impact of the FN polymerization inhibitor (pUR4) in attenuating pathologic cellular features such as proliferation, migration, extracellular matrix (ECM) deposition and associated mechanisms. To evaluate the therapeutic potential of inhibiting FN polymerization in vivo, wildtype (WT) mice received daily intraperitoneal injections of either pUR4 or control peptide (III-11C) immediately after cardiac surgery, for seven consecutive days. Mice were analyzed seven days post-I/R to assess myofibroblast markers and inflammatory cell infiltration, or four weeks post-I/R, to evaluate long-term effects of FN inhibition on cardiac function and fibrosis. Further, inducible, fibroblast-restricted, FN gene ablated (Tcf21MerCreMer;Fnflox) mice were utilized to evaluate cell specificity of FN expression and polymerization in the heart.
Results
pUR4 administration on activated MF reduced FN and collagen deposition into the ECM and attenuated cell proliferation, likely mediated through decreased c-myc signaling. pUR4 also ameliorated fibroblast migration accompanied by increased β1 integrin internalization and reduced levels of phosphorylated focal adhesion kinase (FAK) protein. In vivo, daily administration of pUR4 for seven days post-I/R significantly reduced MF markers and neutrophil infiltration. This treatment regimen also significantly attenuated myocardial dysfunction, pathologic cardiac remodeling and fibrosis up to 4 weeks post-I/R. Finally, inducible ablation of FN in fibroblasts post-I/R resulted in significant functional cardioprotection with reduced hypertrophy and fibrosis. Interestingly, addition of pUR4 to the FN ablated mice did not confer further cardioprotection, suggesting that the salutary effects of inhibiting FN polymerization may be largely mediated through effects on FN secreted from the CF lineage.
Conclusions
Inhibiting FN polymerization or CF gene expression attenuates pathologic properties of MF in vitro and ameliorates adverse cardiac remodeling and fibrosis in an in vivo model of HF. Interfering with FN polymerization may be a new therapeutic strategy for treating cardiac fibrosis and HF.
Keywords: fibronectin, extracellular matrix, cardiac fibroblast, fibrosis, heart failure
Introduction
Heart failure (HF) is a devastating disease that remains a leading cause of death worldwide as well as a major socioeconomic burden for Western societies. In the U.S. alone, HF affects ≈6.5 million people ≥20 years of age,1 and projections show that by 2030 the prevalence of HF will increase to >8 million people ≥18 years of age with an estimated cost of $70 billion.2 However, despite many advances, there are few options for efficacious treatment of patients with end-stage HF.
Most of the etiologies of HF are associated with progressive fibrosis that reduces myocardial compliance and function. Cardiac fibrosis is characterized by exacerbated accumulation of extracellular matrix (ECM) components in the myocardium. This dynamic meshwork is composed of structural and nonstructural proteins that provide an architectural scaffold, surrounding and connecting various cardiac cell populations. In addition to its function in tissue support, the myocardial ECM acts as a signal transducer for cell–cell and cell-ECM interaction that modulates cell motility, survival and proliferation.3, 4
The cardiac ECM is primarily comprised of collagen types I and III that form an intricate 3D network in which cardiac cell types reside and interact to maintain organ function.5 Cardiac fibroblasts (CF) are thought to be the major producers of ECM proteins and are responsible for ECM homeostasis. However, following cardiac injury and under stress stimuli, quiescent CF transdifferentiate into myofibroblasts (MF) which display exacerbated proliferative, migratory and contractile abilities as well as a greater capacity to produce ECM proteins.6 Although MF are initially thought to be beneficial by promoting tissue healing after injury, their continued activation and propagation results in adverse tissue remodeling and fibrosis, eliciting cardiac systolic and diastolic dysfunction, thus actively contributing to HFprogression.
Although collagen is the most abundant ECM protein in the heart, cellular fibronectin (FN) is likely to play a pivotal role in cardiac fibrosis. This multifunctional glycoprotein is mainly secreted as a soluble protein by different cell types including CF, MF or endothelial cells7, 8, and it is polymerized into the ECM by a cell-dependent process.9 FN levels are increased in humans with ischemic and dilated cardiomyopathy as well as in animal models of HF10–12. Global FN null animals are embryonic lethal13; prior reports suggests that systemic FN genetic ablation may attenuate hypertrophy/dysfunction following pressure overload HF14 and may play a role in stem cell recruitment to the injured heart.15 Further, FN plays an essential role in the development of lung and liver fibrosis16–18. Polymerized FN regulates the deposition, maturation and stabilization of other ECM proteins, including collagen I19; thus, it is recognized that polymerized FN guides aspects of ECM biogenesis.20 Polymerized FN also influences a variety of crucial cellular processes, including adhesion,21 growth,21 proliferation,22, 23 migration,24, 25 survival26 and differentiation.27 Therefore, targeting FN deposition/polymerization and ablating its gene expression in CF populations may be a novel approach to attenuate myocardial fibrosis and HF progression.
pUR4 is a recombinant peptide derived from the bacterial F1 adhesin that exclusively mimics the cell surface binding site for the five N-terminal type I modules of FN28, 29 impeding its polymerization by blocking the binding of soluble FN to the cell surface.30 As previously reported, pUR4 has a specific affinity for FN that is not shared with other ECM components such as Collagen I, laminin, fibrinogen or vitronectin, indicating a relatively specific bond between pUR4 and FN without affecting additional FN properties.31
The potential therapeutic effects of pUR4 in reducing FN accumulation and limiting organ fibrosis in models of liver and flow-induced vascular fibrosis have been recently reported31, 32 raising the possibility that, by interfering with cardiac FN polymerization in a mouse model of HF, pUR4 peptide will elicit a reduction of pathological ECM deposition and preserve cardiac architecture and function. To assess the therapeutic efficacy and specificity of our FN polymerization inhibitors as well as to determine the cardiac cell-specific roles of FN inhibition/ablation, we have also utilized inducible, fibroblast-specific FN-knockout (KO) mice. The basic helix-loop-helix transcription factor Tcf21 is expressed in virtually all resident cardiac fibroblasts; these fibroblasts also constitute the source of activated myofibroblasts in the infarct region of the heart.33, 34 The inducible Tcf21MerCreMer knock in mouse was crossed with Fnflox/flox mice to examine the functional role of cellular FN in a temporal and cell-type specific manner.
In this manuscript, we demonstrate a potential therapeutic role for inhibiting FN polymerization or ablating local cellular expression in a mouse model of HF in attenuating the excess deposition of both FN and collagen that occurs during adverse cardiac remodeling, thereby limiting the pathogenesis of cardiac fibrosis and preserving cardiac function.
Methods
The data, analytic methods, and study materials that support the findings of this study are available from the corresponding author upon reasonable request.
Mouse cardiac fibroblasts and cardiomyocytes isolation and culture
Adult CF were collected from 10- 12-week-old C57BL6/J WT mouse hearts by enzymatic digestion. Briefly, mice were given 100 μl heparin (100 U/ml) via intraperitoneal injection and anesthetized with isoflurane. The heart was quickly excised and retrograde-perfused using a Langendorff apparatus under constant pressure (60 mmHg; 37 °C, 4 min) in Ca2+-free perfusion buffer containing 113 mM NaCl, 4.7 mM KCl, 1.2 mM MgSO4, 5.5 mM glucose, 0.6 mM KH2PO4, 0.6 mM Na2HPO4, 12 mM NaHCO3, 10 mM KHCO3, 10 mM Hepes, 10 mM 2,3-butanedione monoxime, and 30 mM taurine. Digestion was achieved by perfusing for 3 minutes with Ca2+-free perfusion buffer containing collagenase II (units/ml) (600 units/mL of collagenase II in perfusion buffer; Worthington LS004177) followed by 8 minutes of perfusion with digestion buffer containing 12.5 μM CaCl2. Subsequently, it was removed from the apparatus and gently teased into small pieces with fine forceps in the same enzyme solution. Heart tissue was further dissociated mechanically using 2, 1.5, and 1 mm-diameter pipettes, until all large heart tissue pieces were dispersed. The digestion buffer was neutralized with buffer containing 10 % FBS and 12.5 μM CaCl2 and cell suspension was filtered through 200 μm mesh. Cardiomyocytes were pelleted by gravity (20 min) and the supernatant collected and stored on ice. CM were resuspended in perfusion solution containing 5 % FBS and 12.5 μM CaCl2 and subsequently allowed to settle for 20 min. The two supernatants were pooled and both non-myocyte containing CF fractions were centrifuged at 500 xg for 7 min. CF were resuspended and plated on the desired culture plates previously coated with 1% gelatin, in culture media containing Iscove’s modified Dulbecco’s medium (IMDM), 10 % fetal bovine serum (ESCell FBS), 100 U/ml penicillin, 100 mg/ml streptomycin, 103 units ESGRO Supplement (Millipore), 10 ng/ml EGF (epidermal growth factor) and 20 ng/ml FGF (fibroblast growth factor) (37 °C, 5 % CO2).
Human Fibroblast Isolation
Failing human cardiac fibroblasts were isolated from ventricular tissue excised during left ventricular assist device implantation surgery under the IRB protocol 2013-1386. Tissue was washed in HBSS and cut into 1 mm cubed pieces with scissors. Digestion mixture, consisting of 100 mg collagenase (Worthington), 1 mg trypsin (Worthington TLR3) and 15 mg BSA in DMEM (HyClone SH30022.01), was applied to dissected tissue, and mixture was placed in a shaker at 125 rpm for 20 min at 37°C. After incubation, a 10 mL pipette was used to gently dissociate cells, and supernatant was collected and combined with neutralization media containing DMEM+10% FBS; this process was then repeated until the tissue pieces were completely digested. The combined supernatants were centrifuged at 1300 rpm for 7 minutes to collect the non-myocyte pellet, which was then resuspended in growth media (as described above) and plated on 1% gelatin-coated dishes.
Animals
All animal procedures were conducted according to the Department of Laboratory Animal Medicine and the University Committee on Animal Resources at Cincinnati Children’s Hospital Medical Center.
C57BL6/J wild type (WT) 10- 12-week-old male mice (The Jackson Laboratories) were utilized for systemic peptide administration. Fnflox/flox mice were kindly provided by Dr. Reinhard Fässler and crossed with mice expressing tamoxifen-inducible Cre recombinase under the control of the bHLH transcription factor Tcf21 promotor (Tcf21MerCreMer)33 to generate double transgenics (DTG) inducible fibroblast-specific FN knockout mice (Tcf21mERCremERxFnflox/flox; CF-FN-KO). DTG without tamoxifen or tamoxifen treated Fnflox/flox littermates served as controls. Animals were fed tamoxifen chow (400mg/kg; Harlan TD.130860) two weeks before cardiac surgery and 4 weeks following injury to induce FN ablation in Tcf21 lineage fibroblasts (quiescent before surgery and activated after injury).34
Peptides and treatment
pUR4 is a peptide derived from a surface protein of Streptococcus pyogenes termed F1 adhesin30 and the first described inhibitor of FN polymerization. pUR4 mimics the binding site required for soluble FN to anchor into the cell surface inhibiting its adhesion without interfering with the critical FN-integrin cell binding domain. Importantly, pUR4 specifically binds to FN but not bind to other ECM proteins35. Additionally, FN polymerization tightly regulates the assembly of different ECM proteins, including collagen type I.19, 20 pUR4 has been shown to reduce the deposition of endogenous collagen I into matrix fibrils both in vitro an in vivo.32, 35 We have used a control peptide termed III-11C that is based on sequences in FN’s III-11 module36 that has proven not to have biological effects in vitro or in vivo.35
The pUR4 and III-11C peptides were produced as described,35 dialyzed in PBS and endotoxin was removed from peptides using Acrodisc Unit with Mustang E membrane 0.2 μm (VWR MSTG25E3). Subsequently, endotoxin levels were measured with a Limulus amebocyte lysate assay kit (Lonza 50-647U). After purification, endotoxin levels were less than 0.1 endotoxin units per mg of peptide.
Mice received daily i.p. injections of pUR4 or III-11C (25 mg/kg/day) for 7 consecutive days starting the same day of I/R surgery or for 14 days starting 4 weeks after I/R injury. Cells received pUR4 or III-11C (500 nM) for 72h.
Statistical Analysis
Data are reported as mean ± standard error of the mean (SEM). For single biochemical and physiological observations, student t-test was applied. Paired t-test was utilized when different treatments (pUR4 and III-11C) were applied to cells isolated form the same animal. Unpaired t-test was applied when pUR4 and III-11C were administered to different animals. Multiple responses were analyzed by one-way or two-way ANOVA. Post-hoc analysis was performed as indicated if statistical significance (P ≤ 0.05) was achieved. Calculations were performed using Graphpad Prism 6.0.
An expanded Materials and Methods are available in the Data Supplement.
Results
pUR4 attenuates fibronectin deposition in adult mouse cardiac fibroblasts in vitro
A key function of CF is to produce structural proteins that comprise the myocardial ECM and regulate its homeostasis. Polymerized FN regulates the deposition and maturation of several ECM proteins, including collagen I, and actively plays a role in cellular survival, proliferation, migration and differentiation. To explore the functional role and efficacy of pUR4 in reducing FN assembly into the ECM, in vitro studies were performed using primary CF cultures. Primary adult mouse CF from unchallenged hearts were isolated and treated for 72 h with either the FN polymerization inhibitor, pUR4, or the control (III-11C) peptide, which binds FN but does not possess inhibitory activity.31 pUR4-treated cells displayed a robust reduction of ECM network organization compared to cells treated with the control III-11C peptide (Fig. 1A); however, no effects were observed on cellular expression of Fn mRNA following pUR4 administration (Fig. 1B). Further, to explore whether there were cellular or ECM-related changes in FN protein content, the cellular and ECM fractions were isolated separately from CF cultures. Although pUR4 treatment did not alter FN intracellular protein content compared to III-11C (Fig. 1C), pUR4-treated cells exhibited attenuation of FN deposition into the ECM (Fig. 1D). However, the normal stoichiometry of total extracellular FN expression was maintained, as pUR4 increased FN abundance in cell culture media measured by ELISA (Fig. 1E). These data demonstrate that pUR4 efficiently blocks FN polymerization within the ECM of CF with no effect on cellular transcript or protein level.
Figure 1. pUR4 attenuates fibronectin polymerization in unchallenged primary mouse cardiac fibroblasts 72 h post-treatment.
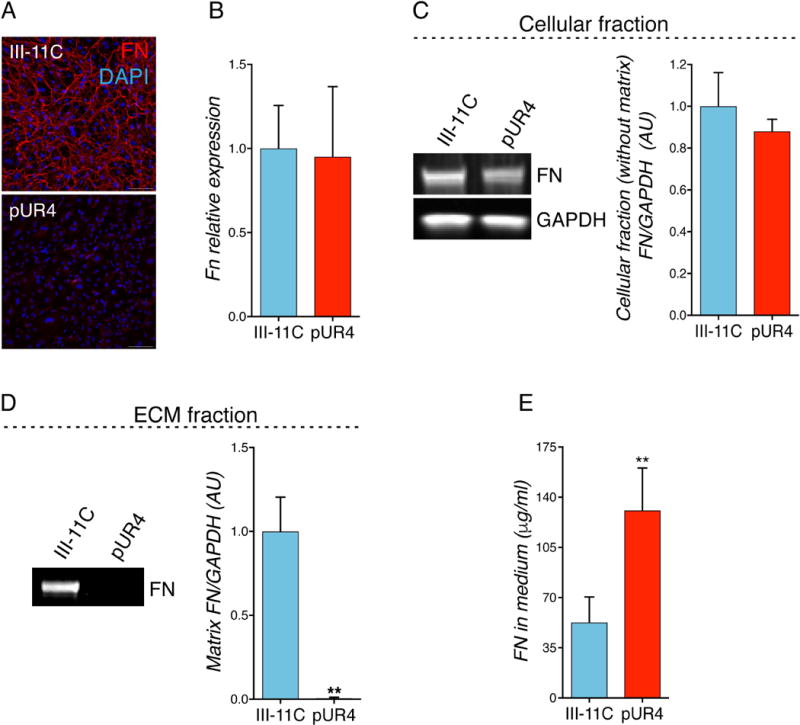
(A) FN immunofluorescence staining in healthy CF. Scale bars, 100 μm. (B) FN transcript level expression. n=5. (C) Cellular FN protein level expression (without matrix) (representative immunoblots, left, and densitometry, right). n=5. (D) Matrix FN deposition in the extracellular space of CF is attenuated with pUR4 (representative immunoblots, left, and densitometry, right). n=5. (E) FN is accumulated in the cultured media measured by ELISA. n=5. Statistical significance was determined with paired t-test: *P<0.05, **P<0.01. Data are represented as mean ± SEM.
pUR4 attenuates pathologic mouse cardiac myofibroblast activation profile in vitro
CF actively participate in the complex and dynamic intercellular crosstalk between the myriad of cardiac cells to maintain cardiac structure and function. After cardiac insult, this crosstalk is dysregulated and elicits the transition of CF to MF, resulting in exacerbated synthesis and deposition of ECM, as well as enhanced proliferation and migration. To explore the impact of inhibiting FN polymerization on cell behavior, primary activated MF were isolated from wild-type (WT) hearts 5 days after myocardial ischemia reperfusion (I/R) injury. First, the activation profile of MF was confirmed by identifying increased fibrotic and inflammatory cytokine gene expression (Suppl. Fig. 1). Subsequently, experiments investigated whether pUR4 reduces FN polymerization in MF ECM to the same extent as observed in unchallenged CF. pUR4 exhibited a robust inhibitory effect on FN polymerization in MF isolated from injured mouse hearts compared to III-11C-treated cells (Suppl. Fig. 2). FN polymerization also tightly regulates the assembly of collagen I; when MF were cultured in the presence of pUR4, deposition of collagen into matrix fibrils was reduced (Fig. 2A and B). Neither intracellular protein content nor collagen I mRNA transcript levels were affected by pUR4 treatment (Fig. 2C and 2D).
Figure 2. pUR4 attenuates mouse pathologic myofibroblast phenotype.
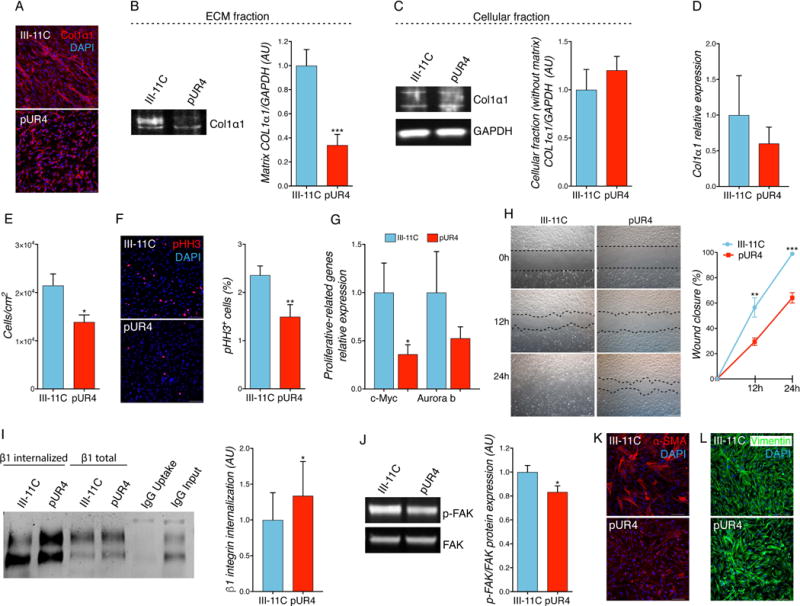
(A) pUR4 decreases collagen I staining in CF isolated after cardiac I/R injury by immunofluorescence. Scale bars, 100 μm. (B) Matrix collagen I deposition in activated MF is attenuated upon pUR4 treatment (representative immunoblots, left, and densitometry, right). n=5. (C) Cellular collagen I protein level expression (without matrix) (representative immunoblots, left, and densitometry, right). n=5. (D) Collagen I transcript level expression. n=6. (E) pUR4 decreases cell proliferation monitored by counting cell number with a hemocytometer. n=6. (F) Phospho-histone H3 (pHH3) staining in mouse MF shows a decrease in pHH3+ cells in pUR4-treated cells (representative pictures, left, and quantification, right). Scale bars, 100 μm. n=3. (G) RT-qPCR of proliferative-related genes in pUR4 and III-11C-treated mouse MF. n=6. (H) Scratch wound-healing assay in mouse CF cell migration; pictures were taken at 0 h, 12 h and 24 h post-scratch. Black dotted lines denote the wound borders (representative photographs, left, and mobility quantification, right). Scale bars, 1000 μm. n=6. (I) Increased β1 integrin internalization was found in MF treated with pUR4 compared to III-11C peptide (representative immunoblots, left, and densitometry, right). n=5. (J) Expression of phosphorylated focal adhesion kinase (p-FAK) was evaluated by Western blotting and quantified relative to total FAK expression (representative immunoblots, left, and densitometry, right). n=4. (K) Alpha-smooth muscle actin (α-SMA) staining in III-11C and pUR4-treated MF. Scale bars, 100 μm. (L) Vimentin staining in peptide-treated cells. Scale bars, 100 μm. Statistical significance was determined with paired t-test: *P<0.05, **P<0.01, ***P<0.001. Data are represented as mean ± SEM.
To examine whether pUR4 attenuates MF activation, MF were treated with pUR4 or III-11C peptides and different cellular functions were analyzed. In vitro MF proliferation was evaluated by counting cell number as well as by the expression of the mitosis marker phospho-histone H3 (pHH3) 72 h after peptide administration. Cell proliferation and pHH3+ cells were significantly reduced after pUR4 treatment in activated cardiac MF (Fig. 2 E and F). Reduced FN polymerization resulted in decreased transcript expression of proliferation-related genes such as c-myc and auroraB kinase (Fig. 2G). In line with these results, a cell cycle analysis was performed in activated MF at different time points after peptide administration (12, 24, 36, 48, 56 and 72 h); a significant decrease was observed in the percentage of cells in S-phase at 24, 36, 48 and 56 h (Suppl. Fig. 3A and B). However, this decrease was not associated with changes in signaling via the ERK or AKT pathways (Suppl. Fig. 3C). Surprisingly, inhibiting FN polymerization did not affect cell proliferation in unchallenged CF isolated from healthy animals (Suppl. Fig. 4A).
Another central property of MF is their enhanced migratory ability following cardiac injury. The effect of pUR4 in MF migratory capacity was evaluated by performing a scratch-wound healing assay. pUR4-treated cells displayed a significant reduction in MF migration (Fig. 2H). This finding was also observed in unchallenged CF (Suppl. Fig. 4B). In this context, integrins contribute to the recruitment and activation of crucial signaling proteins involved in cell migration, including focal adhesion kinases (FAKs). A significant increase of integrin internalization was observed following pUR4 treatment (Fig. 2I). Since FAKs are among the most prominent proteins involved in ECM-integrin signaling pathways crucial for cell migration, abundance of phospho-FAK was assessed after peptide treatment. p-FAK protein content was also reduced in pUR4-treated cells compared to control III-11C-treated cells (Fig. 2J). Further, pUR4 decreased the expression of α-SMA and reduced the complexity of fiber formation (Fig. 2K) without affecting vimentin expression (Fig. 2L). Together, these data suggest that pUR4 exhibits a principal role in diminishing MF proliferation via the c-myc signaling axis, reduces cell migration by promoting integrin internalization and decreasing FAK activation as well as ameliorating the activated MF phenotype.
pUR4 does not alter mitochondria content, oxidative stress or cell metabolism in mouse cardiac myofibroblasts
Our data suggest that it is possible to attenuate the pathologic cardiac MF phenotype by interfering with FN polymerization. We next aimed to determine whether pUR4 treatment affected cell survival or metabolism that may explain the changes overserved in cell behavior. To this end, the impact of inhibiting FN polymerization in cell apoptosis, reactive oxygen species (ROS) production and metabolic state were determined. pUR4 administration did not result in a significant difference in apoptosis measured by flow cytometry (Fig. 3A). Also, the accumulation of total and mitochondrial ROS, known to be increased after cardiac injury, did not differ between control and pUR4-treated cells (Fig. 3B and C). Further, cellular mitochondrial content was examined to evaluate whether the limited proliferative and migratory abilities exhibited by pUR4-treated MF may be explained by an overall reduction of energy production. Flow cytometry analysis utilizing Mitrotracker combined with TOM20 cell staining revealed no differences in total mitochondrial content after inhibiting FN polymerization (Fig. 3D and E). Finally, to evaluate whether pUR4 treatment affects the metabolic state of cardiac MF, cellular ATP levels and oxygen consumption were measured in MF-treated cells. No differences were detected in cells lacking a FN network (Fig. 3F and G). Additionally, cellular glycolysis and fatty acid oxidation were measured with no effect on the aforementioned pathways following pUR4 administration (Fig. 3H and I).
Figure 3. pUR4 does not alter MF cell survival, ROS production, mitochondrial content and metabolism after 72 h of peptide treatment.
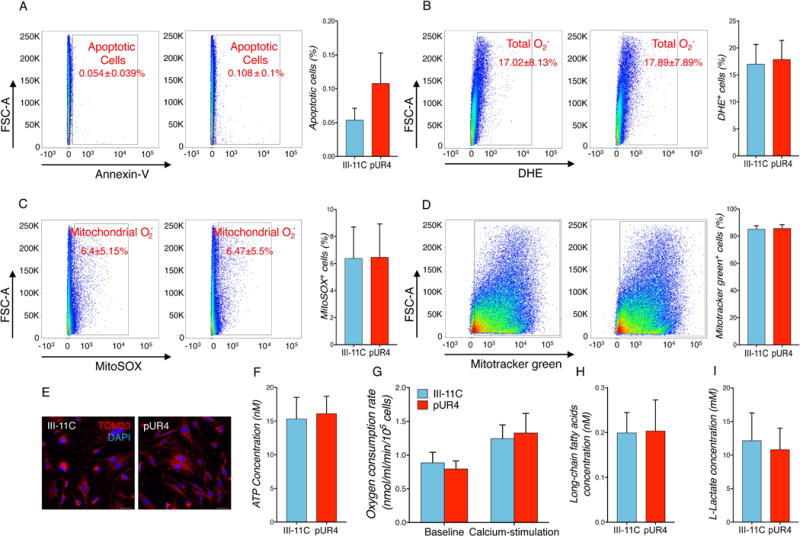
(A) Flow cytometry analysis of annexin-V staining of apoptotic MF following pUR4 or III-11C peptide treatment. n=5. (B) MF treated either with pUR4 or III-11C were stained with superoxide anion-specific dye DHE. Oxidized version of DHE was detected by flow cytometry. n=5. (C) Levels of mitochondrial-associated ROS in peptide-treated MF were analyzed by MitoSOX labeling and analyzed using flow cytometry. n=5. (D) Mitochondria were stained with MitoTracker Green and analyzed by flow cytometry. n=5. (E) Mitochondria immunostaining with TOM20 in pUR4 or III-11C-treated cells. Scale bars, 20 μm. (F) ATP content was measured in peptide-treated MF after 72 h. n=5. (G) Oxygen consumption was determined without or with calcium stimulation in MF treated with III-11C or pUR4. n=7. (H) Mitochondrial fatty acid oxidation was measured by long-chain fatty acid concentration existent in the cellular content. n=5. (I) Glycolysis activity was measured by L-lactate concentration in the cultured media. n=5. Absence of statistical significance was determined with paired t-test. Data are represented as mean ± SEM.
These findings demonstrate that inhibiting FN polymerization does not alter cell survival or cellular metabolic state, highlighting the direct ECM-mediated effect of pUR4 in attenuating MF migration and proliferation.
pUR4 reduces the pathologic activation of human failing cardiac myofibroblasts in vitro
Human CF regulate normal cardiac function and organ homeostasis but also are the main mediators of pathological myocardial remodeling following cardiac injury that contributes to the progression of organ fibrosis and HF. To determine the effects of FN polymerization inhibition on human activated cardiac MF, primary failing human MF were isolated from the hearts of end-stage heart failure patients undergoing left ventricular assist device implantation, characterized (Suppl. Fig. 5) and treated with pUR4. As with murine MF, pUR4 treatment impaired the deposition of FN fibrils into the ECM of human MF (Fig. 4A). pUR4 significantly reduced in vitro cell growth as monitored by total cell counts and pHH3+ cells (Fig. 4B and C). Also, pUR4 reduced expression levels of proliferative and inflammatory-related transcripts in human failing cardiac MF (Fig. 4D and E) and significantly blunted their migratory ability in a scratch-wound healing assay (Fig. 4F). Furthermore, pUR4-treated cells exhibited a reduced p-FAK expression (Fig. 4G and H) as well as a reduction of F-actin patterning expression (Fig. 4I). Additionally, human cardiac MF exhibited reduced expression of α-SMA and complexity of fiber formation (Fig. 4J), without affecting vimentin expression (Fig. 4K) upon inhibition of FN polymerization. Collectively, these data show the potential therapeutic effect of inhibiting FN fibril formation in reducing the pathologic phenotype of human failing cardiac MF.
Figure 4. pUR4 decreases proliferation and migration of human failing cardiac fibroblasts.
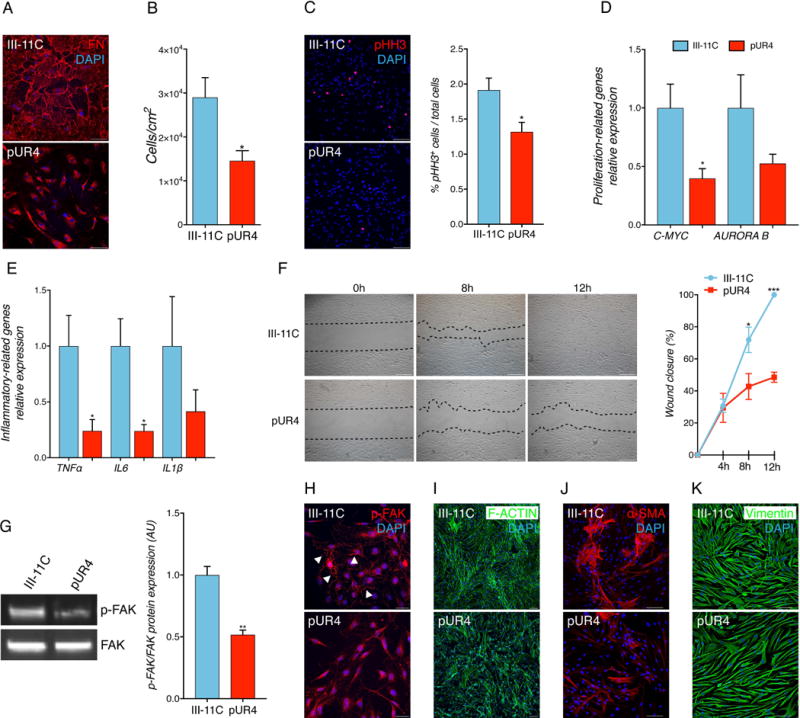
(A) pUR4 reduces FN staining. Scale bars, 100 μm. (B) pUR4 decreases cell proliferation. (C) pUR4 decreases phospho- histone H3 (pHH3)-positive human heart failure CF (representative pictures, left, and quantification, right). Scale bars, 100 μm. (D) pUR4 downregulates proliferation-related genes. (E) Expression levels of pro-inflammatory cytokine transcripts are significantly reduced following pUR4 treatment in failing human cardiac fibroblasts measured by RT-qPCR. Tumor necrosis factor alpha (TNFα), Interleukin 6 (Il6) and Interleukin 1ß (IL1ß). (F) Cardiac fibroblasts were scratch-wounded and recorded at 0 h, 4 h, 8 h and 12 h post-scratch. Black dotted lines denote the wound borders (representative photographs, left, and mobility quantification, right). Scale bars, 500 μm. (G) p-FAK protein level expression (representative immunoblots, left, and densitometry, right). (H) Immunofluorescence analysis of p-FAK in pUR4 and III-11C-treated human MF (arrowheads). Scale bars, 10μm. (I) F-actin staining in pUR4-treated cells indicated a compromised defect in cellular organization compared to control group. Scale bars, 100 μm. (J) Alpha-smooth muscle actin (α-SMA) staining in III-11C and pUR4-treated human MF. Scale bars, 100 μm. (K) Vimentin staining in peptide-treated cells. Scale bars, 100 μm. Statistical significance was determined with paired t-test: *P<0.05, **P<0.01, ***P<0.001. Data are represented as mean ± SEM. n=5.
Neither pUR4 nor III-11C affect baseline cardiac function in unchallenged hearts in vivo or isolated cardiomyocytes in vitro
To explore the effects of pUR4 peptide administration on cardiac function at baseline in vivo, WT mice were injected intraperitoneally with III-11C or pUR4 once daily for 7 consecutive days. A fluorescent dye was covalently bound to the peptides to verify peptide localization in vivo 24 h after the last administration. As shown in Suppl. Fig. 6, the peptide was detected throughout the animal body, in addition to the heart.
No effects on cardiac function or morphometry parameters were detected after 7 days of peptide exposure (Suppl. Fig. 7A and B) (Suppl. Table 1). Further, pUR4 administration did not result in CM hypertrophy in healthy hearts (Suppl. Fig. 7C). To determine whether there was any effect on the cardiac inflammatory milieu following exposure to the peptides, flow cytometry analysis was performed and no significant changes in the inflammatory cell population of the heart were observed (Suppl. Fig. 7D). Also, FN inhibition in healthy animals did not result in a significant decrease of collagen deposition (Suppl. Fig. 7E). To further explore possible effects on cardiac muscle cell contractility, primary cardiomyocytes (CM) were isolated from unchallenged hearts and assessed for contractile function. No basal differences in sarcomeric shortening were observed with either peptide (Suppl. Fig. 7F). Additionally, CM isolated 5 days following I/R and acutely treated with III-11C and pUR4 for 1 h did not show significant alterations in contraction (Suppl. Fig. 7G). Overall, these data suggest that peptide administration has no significant effect on baseline cardiovascular function.
Inhibition of FN polymerization attenuates myofibroblast markers and inflammatory cell infiltration 7 days after injury in vivo.
Our in vitro data suggest that blocking FN deposition into the ECM attenuates the pathologic phenotype exhibited by activated MF isolated from failing mouse and human hearts. Therefore, we sought to examine whether inhibition of FN polymerization by pUR4 treatment may be salutary following myocardial injury. To test this hypothesis, WT mice were subjected to myocardial ischemia followed by 7 days of reperfusion (Fig. 5A). First, FN expression was markedly upregulated 7 days post I/R (Suppl. Fig. 8) as previously described.37, 38 7 days of pUR4 treatment following cardiac injury trended toward reduction of the expression of FN by 30% (Fig. 5B) as well as fibrosis-related transcripts, such as α-SMA and collagen 3 (Fig. 5C). Additionally, pUR4 significantly reduced proliferative fibroblast numbers in both the infarcted and remote areas of the heart 7 days post injury as observed by EdU administration (Fig. 5D).
Figure 5. Inhibition of FN polymerization attenuates fibrotic mediators and inflammatory cell infiltration 7 days post-I/R in vivo.
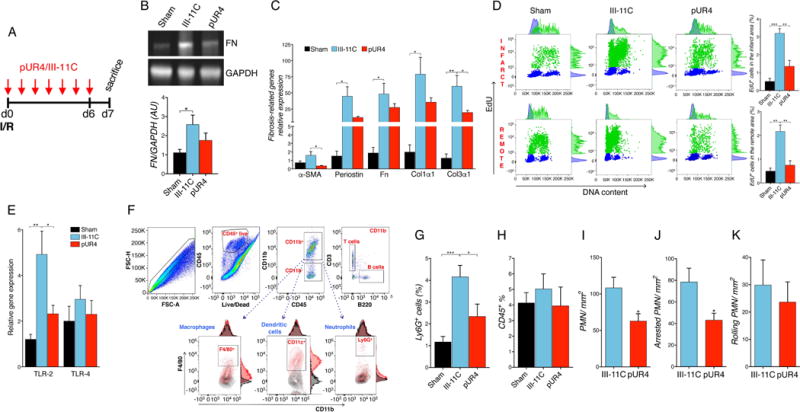
(A) Schematic of the experimental timeline showing initiation of pUR4 or III-11C treatment after the I/R injury. (B) Fibronectin protein expression in the heart (representative immunoblots, top, and densitometry, bottom). n=5. (C) Fibrosis-related gene expression in left ventricular tissue of peptide-treated hearts. α-SMA: alpha-smooth muscle actin, Fn; Fibronectin, Col1α1: Collagen1α1 and Col3α1: Collagen 3α1. n=5-6. (D) Cardiac proliferative cells, S phase, (EdU+ staining, green) detected in the infarct and remote areas by flow cytometry, (representative plots, left, and quantification, right). n=3-4. (E) TLR-2 and TLR-4 expression 7 days after infarction in the specified treatment groups. n=5-6. (F) Flow cytometry strategy workflow to analyze the different immune cell compartments. (G and H) Quantification using flow cytometry of neutrophils (G) and CD45+ cells (H) in the hearts of sham, pUR4 or III-11C-treated animals. n=5-8. (I) Quantification of total accumulation of neutrophils to MHEC. (J) Quantification of firm adhesion of neutrophils to MHEC. (K) Quantification of neutrophil rolling on TNFα-activated MHEC pre-treated with the indicated peptides for 72 h. Neutrophils were perfused at 1 dyne/cm2, and interactions with MHEC were quantified in 10 min videos and additional fields of view. Statistical significance determined by one-way ANOVA with Tukey post-hoc analysis: *P<0.05, **P<0.01, ***P<0.001. Data are represented as mean ± SEM.
ECM homeostasis relies on a tight balance between matrix metalloproteinases (MMPs) and tissue inhibitors of metalloproteinases (TIMPs), which collectively regulate ECM components in the process of cardiac remodeling.39 However, exacerbated MMP activity is detrimental for cardiac output.39 To characterize MMP activity, we studied the expression of MMPs and TIMPs, as well as their enzymatic activity. Analysis in pUR4-treated hearts revealed a trend toward reduction of MMP-9 activity (Suppl. Fig. 9A) as well as its transcript level (Suppl. Fig. 9B). TIMP-1 transcript level was significantly reduced by pUR4 treatment (Suppl. Fig. 9B).
The role of FN in the recruitment of inflammatory cells into different tissues has been extensively described.40, 41 The robust inflammatory response triggered after cardiac insult is initially crucial for cardiac repair, but it is also involved in pathological tissue remodeling and heart failure at later stages.42 Cardiomyocyte necrosis triggers an acute inflammatory response characterized by immune cell recruitment and a rapid increase in cytokine and chemokine levels.43 Toll-like receptors (TLR) play a pivotal role in recognizing endogenous “danger signals” released during cell death.43 Since FN is a possible ligand for TLR-2 and TLR-4, we sought to investigate the role of FN inhibition in TLR expression and inflammatory progression following I/R injury. TLR-2, but not TLR-4, expression exhibited a significant reduction in the left ventricle (LV) of pUR4-treated hearts (Fig. 5E). Further, the different immune subsets residing in the heart were evaluated by flow cytometry 7 days after I/R injury in pUR4- and III-11C-treated animals (Fig. 5F). The recruitment of neutrophils into the heart was significantly attenuated after pUR4 treatment (Fig. 5G), while the total number of CD45+ cells recruited was not significantly altered (Fig. 5H), nor were the number of macrophages, dendritic cells and other compartments affected by pUR4 treatment (Suppl. Fig. 10 A-F). Also, we did not detect a significant decrease in the mRNA expression of relevant cytokines such as Il-1β and Il-6 in the LV of pUR4-treated animals 7 days after organ reperfusion (Suppl. Fig. 10G).
Given the observed decrease in neutrophil infiltration in the heart post-I/R in mice treated with pUR4, we hypothesized that pUR4 would impair neutrophil adhesion to heart endothelial cells as a mechanism for the observed in vivo defect in recruitment. To test whether pUR4 had any effect on neutrophil adhesion, mouse heart endothelial cells (MHEC) were evaluated under shear flow conditions in vitro. Treatment of neutrophils or MHEC with pUR4 did not alter the expression of the neutrophil markers Ly6G or CD11b, the expression of neutrophil adhesion molecules such as PSGL-1, or the MHEC monolayer integrity, and it efficiently inhibited FN polymerization in MHEC (Suppl. Fig. 11). Strikingly, pUR4 treatment of MHEC resulted in significantly reduced total accumulation and firm adhesion of neutrophils to MHEC compared to III-11C control treatment (Fig. 5I and J) without affecting neutrophil rolling on MHEC (Fig. 5K). This decreased adhesion observed in pUR4-treated cells was not related to an alteration in the expression of integrins involved in neutrophil adhesion such as VLA-4 and LFA-144 (Suppl. Fig. 12A and B). Subsequent experiments tested whether FN polymerization regulated the expression of VCAM-1 and ICAM-1, which are the endothelial ligands for neutrophil VLA-4 and LFA-1,45 upon TNFα stimulation. Although flow cytometry data showed that pUR4 does not significantly decrease VCAM-1 and ICAM-1 surface expression in MHEC treated for 72 h (Suppl. Fig. 12C and D), cellular MHEC VCAM-1 and ICAM-1 staining demonstrated overall reduction of protein expression (Suppl. Fig. 12E and F). Taken together, these results suggest that pUR4 reduces neutrophil adhesion and arrest on MHEC under inflammatory conditions by a putative decrease of total ICAM-1 and VCAM-1 protein expression.
Systemic treatment with pUR4 reduces pathologic cardiac fibrosis and preserves cardiac function up to 4 weeks post-I/R
To examine whether pUR4 treatment limits long term cardiac injury and fibrosis in vivo, mice were subjected to I/R injury, treated daily with pUR4 or III-11C for the first 7 days, followed by evaluation of cardiac function and histology 4 weeks post-I/R (Fig. 6A). Interestingly, 7 days of pUR4 treatment significantly preserved myocardial function up to 4 weeks post-I/R as measured by ejection fraction and end systolic volume (Fig. 6B and C) (Suppl. Table 2), suggesting an injury-reducing effect following just acute (7 days) pUR4 administration. Attenuation of systolic dysfunction observed in pUR4-treated animals was possibly due to a reduction in the size of the infarct, either through direct effects of the peptide on cardiomyocyte survival, or through attenuation of inflammatory cardiomyocyte injury. To further evaluate these possibilities, the infarct size was assessed 24 h post-injury with concomitant peptide treatment followed by quantification of percent of infarcted area versus the area at risk (Suppl. Fig. 13A). In addition, terminal deoxynucleotidyl transferase dUTR nick-end labeling (TUNEL) was performed to measure the number of apoptotic cells in the infarct area 24 hours post-treatment and I/R (Suppl. Fig. 13B). No significant differences were observed either in the area of infarct or in the number of apoptotic cells between treatments.
Figure 6. Seven days of pUR4 preserves cardiac function and reduces remodeling up to 4 weeks post-I/R.
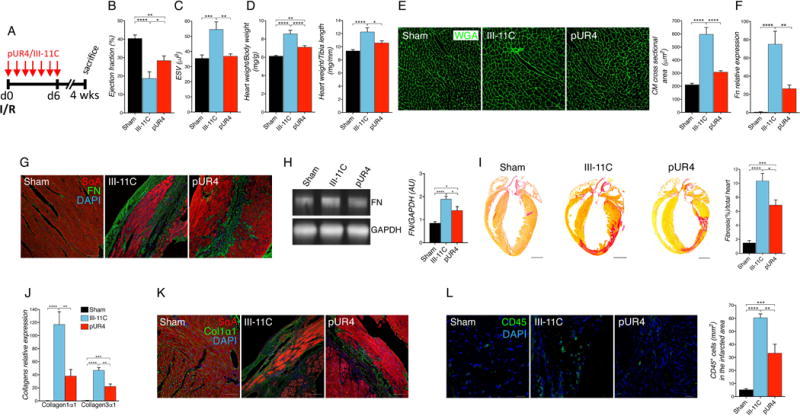
(A) Animals received daily intraperitoneal peptide injection for 7 days immediately after cardiac surgery, and were followed for 4 weeks total. (B) Cardiac function evaluated by echocardiography shown by percent ejection fraction at 4 weeks post-I/R. n=8-10 per group. (C) Cardiac dilation assessed by echocardiography shown by end systolic volume (ESV) 4 weeks post-I/R. n=8-10 per group. (D) Cardiac morphometry is shown as heart weight to body weight ratio (left) and heart weight to tibia length ratio (right). n=11-14 per group. (E) Representative cardiomyocyte cross-sectional images of wheat germ agglutinin (WGA) staining (green) in the infarct border zone of the indicated treatment (left) and quantification (right). Scale bars, 100 μm. n=5. (F) FN transcript expression in the left ventricle of the different treatment groups. n=5-6. (G) Representative images of FN staining obtained in the infarcted area of I/R groups or in the left ventricles of sham animals. Scale bars, 100 μm. (H) FN expression in the left ventricle of the indicated treatments was determined by Western blotting. n=6. (I) Fibrotic scar formation was evaluated by picrosirius red staining (representative pictures, left and quantification, right). n=5. (J) Collagen transcript expression in the left ventricle of the different treatment groups. n=5-6. (K) Representative images of collagen staining obtained from infarcted area of I/R groups or in the left ventricles of sham animals. Scale bars, 100 μm. (L) Inflammatory cells recruited in the infarcted area were detected with CD45 marker (green) (representative pictures, left and quantification, right). Scale bars, 10 μm. n=5-6. Statistical significance was determined with one-way ANOVA with Tukey post-hoc analysis. *P<0.05, **P<0.01, ***P<0.001, ****P<0.001. Data are represented as mean ± SEM.
pUR4-treated animals also exhibited an attenuation in pathologic hypertrophy detected in cardiac morphometry (Fig. 6D). The observed attenuation of cardiac hypertrophy was confirmed by a reduction of CM size following pUR4 administration (Fig. 6E) although pUR4 appeared not to have a direct effect in CM size in response to the pro-hypertrophic signal angiotensin II using primary neonatal rat cardiomyocytes in vitro. (Suppl. Fig. 14).
A reduction of the pathologic fibrotic response was detected in pUR4-treated animals 4 weeks after I/R. FN content in the LV as analyzed by qPCR, immunofluorescence and Western blotting was reduced following peptide administration (Fig. 6F-H). At 4 weeks post-injury, pUR4 administration resulted in decreased fibrosis and collagen I accumulation as evidenced by picrosirius red (Fig. 6I), reduction of transcript level expression (Fig. 6J) and immunostaining (Fig. 6K). Further, second harmonic generation (SHG) represents a powerful tool to image collagen at high resolution, with diffraction-limited resolution (sub-300 nm), that allows the study of collagen architecture, orientation and crystallinity at a microscopic level better than is possible by other microscopy or histologic techniques.46, 47 SHG imaging was performed to further study collagen organization in peptide-treated groups. A notable reduction of collagen deposition was observed in pUR4-treated animals comparted to III-11C peptide. (Suppl. Fig. 15). Additionally, total numbers of CD45+ cells localized in the infarcted area was reduced in the LV of pUR4-treated animals (Fig. 6L). Collectively, these data demonstrate the salutary effects of pUR4 treatment in preserving cardiac function and attenuating myocardial hypertrophy and fibrosis following organ injury.
To investigate whether pUR4 may revert the pre-established scar existing in a later stage of heart failure, animals underwent I/R cardiac surgery and pUR4 or III-11C treatment was started 4 weeks post-I/R for two weeks, followed by two more weeks of survival; hearts were harvested 2 weeks post-peptide administration (Suppl. Fig. 16A). Delayed pUR4-treated hearts exhibited a trend toward reduction of hypertrophy and fibrosis (Suppl. Fig. 16B and C) with no apparent effect on cardiac function (Suppl. Fig. 16D) (Suppl. Table 3). Although we did not observe significant changes in cardiac remodeling or dysfunction, these data suggest that pUR4 may hold potential to attenuate the pre-stablished scar 4 weeks post-I/R with no protection in cardiac function at this time point.
Inducible fibroblast-specific FN ablation is protective following cardiac injury
Based on the salutary effects of systemic pUR4 treatment following I/R, the therapeutic efficacy of attenuating local FN polymerization was further evaluated utilizing fibroblast restricted FN knockout mice. These mice were further treated with III-11C or pUR4 to characterize cell specificity of FN in the heart. Fnflox/flox (FN+/+) animals were crossed with mice expressing inducible Cre recombinase under the control of Tcf21 promoter (Tcf21mERCremER)33. These mice allow conditional ablation of FN gene expression upon tamoxifen (TM) administration in essentially all cardiac resident fibroblasts as well as in their activated myofibroblast progeny34. TM chow diet was initiated two weeks prior to surgery and continued for four weeks to conditionally ablate FN gene expression both in resident and activated fibroblasts (Fig. 7A). To determine gene knockdown, qPCR and immunostaining analysis for FN was performed in freshly isolated cardiac fibroblasts from double transgenic (DTG) mice with or without TM (Suppl. Fig. 17). Remarkably, mice in which FN was ablated in Tcf21+ fibroblast lineage offered a significant preservation of cardiac function post-I/R as measured by improved percent ejection fraction (Fig. 7B) and reduced of cardiac dilation (Fig. 7C) compared to FN+/+ control mice (Suppl. Table 4). Interestingly, administration of the pUR4 FN inhibitor following I/R injury to DTG mice did not further improve cardiac function (Fig. 7B and C). Additionally, restricted CF-FN-KO mice exhibited ameliorated pathologic changes in cardiac morphometry treated with III-11C or pUR4 (Fig. 7D). The observed attenuation in cardiac hypertrophy was confirmed by a reduction in CM size in CF-FN-KO animals (Fig. 7E), however, pUR4 treatment did not further attenuate CM hypertrophy (Fig. 7E).
Figure 7. Effect of FN genetic ablation in cardiac fibroblasts.
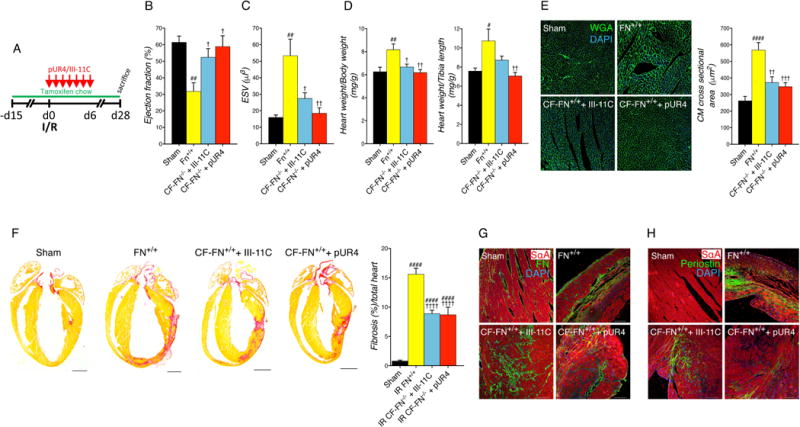
(A) Animals were fed with tamoxifen chow diet 15 days prior to cardiac surgery and continued for 4 weeks after the injury and received daily intraperitoneal peptide injection for 7 days immediately after I/R. Animals were followed for 4 weeks total. (B) Cardiac function evaluated by echocardiography shown by percent ejection fraction at 4 weeks post-I/R. (C) Cardiac dilation assessed by echocardiography shown by end systolic volume (ESV) 4 weeks post-I/R. (D) Cardiac morphometry represented as heart weight to body weight ratio (left) and heart weight to tibia length ratio (right). (E) Representative cardiomyocyte cross-sectional images of wheat germ agglutinin (WGA) staining (green) and DAPI (blue) in the infarct border zone of the indicated treatment (left) and quantification (right). Scale bars, 100 μm. (F) Fibrotic scar formation was evaluated by picrosirius red staining (representative pictures, left and quantification, right). Scale bars, 1000 μm (G) Representative images for FN staining from the infarcted area of the mentioned experimental groups. Scale bars, 100 μm. (H) Representative images for periostin staining from the infarcted area of the mentioned experimental groups. Scale bars, 100 μm. Fn+/+: Fnflox/flox; CF-FN−/−:Tcf21mERCremER;Fnflox/flox. Sarcomeric α-actinin (SαA). Statistical significance was determined with one-way ANOVA with Tukey post-hoc analysis. #P<0.05 versus (vs.) Sham, ##P<0.01 vs. Sham, ####P<0.0001 vs. Sham, †P<0.05 vs. Fn+/+, ††P<0.01 vs. Fn+/+, †††P<0.001 vs. Fn+/+, ††††P<0.0001 vs. Fn+/+. Data are represented as mean ± SEM. n=6-7 per group.
To examine the effects of FN genetic ablation in cardiac fibrosis, hearts of DTG mice +TM treated with III-11C or pUR4 were histologically assessed. Collagen content was significantly reduced in CF-FN-KO mice with no further changes with pUR4 treatment (Fig. 7F). Also, immunofluorescence staining for FN and periostin revealed a decreased FN expression in DTG mice compared to FN+/+ controls following injury and tamoxifen administration (Fig. 7G and H).
Together, these data demonstrate that animals in which FN was ablated in cardiac fibroblast populations exhibited preserved cardiac function and reduced hypertrophy and fibrosis in vivo. These findings may be due to a reduced post-I/R fibrotic scar as a consequence of the attenuation of pathologic FN and collagen deposition derived from cardiac fibroblasts.
Discussion
Myocardial ischemia after coronary artery obstruction triggers the recruitment and activation of resident and quiescent CF. These CF are stimulated by the release of cytokines, chemokines and growth factors, as well as by mechanical forces48, which alter activation patterns in the myocardium, promoting their differentiation to MF.49 MF are a highly proliferative, migratory and secretory cell type that are involved in the pathological remodeling of the myocardium upon injury, promoting cardiac fibrosis that ultimately leads to HF. Although MF initially trigger a reparative wound healing response, prolonged persistence of MF activity is detrimental for organ compliance.
Production and polymerization of FN are both elevated in clinical and experimental models of HF.37, 38 Polymerization of FN tightly regulates the assembly of different ECM proteins, including collagen type I.19, 20, 50 These processes are necessary for normal tissue repair but become exacerbated during fibrosis. However, the impact of inhibiting fibronectin polymerization or targeting its gene expression in cardiac fibrosis and organ function following injury had not been elucidated.
In the present study, the functional role of FN matrix deposition in cardiac remodeling was evaluated using the adhesin-based peptide pUR4, which specifically binds to FN and inhibits its cell-mediated polymerization. Administration of pUR4 for 7 days following cardiac I/R preserved cardiac function, reduced FN and collagen deposition, and attenuated pathological cardiac remodeling, cardiac fibrosis and myocardial neutrophil infiltration up to 4 weeks post-I/R. Similar results were obtained in vitro, where pUR4 attenuated key properties of primary mouse and human failing cardiac MF, such as proliferation and migration. Our data demonstrate that pUR4 blocked FN polymerization in the extracellular space in primary mouse CF isolated from unchallenged hearts. However, pUR4-treated cells did not show a decrease in FN transcript level or intracellular protein content. These observations are in agreement with previous studies31, 32 showing that pUR4 administration does not interfere with FN transcript level expression or intracellular protein content.
The ECM microenvironment modulates cell phenotype and function. FN is known to be required during tissue homeostasis and post-injury wound healing influencing a variety of crucial cellular processes including proliferation and migration. The in vitro data demonstrate that pUR4 blocks the deposition of FN into ECM fibrils in primary cardiac MF isolated from mouse hearts after I/R. Also, decreasing FN polymerization rendered a concomitant reduction of collagen deposition and maturation into the ECM measured by immunocytochemistry and Western blotting. These data are consistent with several studies demonstrating the requirement of an established FN network to nucleate the elaboration of a collagen matrix into the ECM.19, 20, 51 Mouse MF exhibited a significant reduction in cell proliferation, likely mediated through c-myc signaling pathway downregulation. Previous in vitro studies have demonstrated the role of FN in cell proliferation through c-myc axis regulation,23 pinpointing its central role as a master regulator of cellular proliferation and its essential requirement for cell cycle progression.52, 53 Also, FN polymerization promotes migration of a variety of cell types, including myofibroblasts.19, 24, 25 Therefore, our data are consistent with previous reports that recognize a positive effect of FN polymerization on cell migration. Further, depletion of FN deposition promoted an increase of β1 integrin internalization accompanied by a reduction of phosphorylated FAK protein level and a decrease in α-SMA expression. Numerous studies have demonstrated that FN typically binds to α5β1 integrins54 and that cessation of FN polymerization triggers the internalization of β1 integrins in a caveolin-1 dependent manner.55 Also, FAKs plays a critical role in cell migration,56 localizing with integrins at focal contact sites and activated by cell binding to ECM proteins, such as FN.57 Our results suggest that pUR4 treatment of primary mouse CF promotes integrin internalization upon inhibition of FN polymerization with the consequent disruption of p-FAK localization and expression, hence decreasing the mobility of pUR4-treated cells. Similar results were observed in pUR4-treated human failing CF. As with murine CF, FN remarkably attenuated the ability of the failing human CF to proliferate and migrate, exhibiting a downregulation in c-myc and p-FAK pathways as well as α-SMA expression. Collectively, the findings in mouse and human heart failure MF highlight the crucial role of FN in modulating cell behavior and the well-established relationship between FN and cell proliferation and migration. Our in vitro human data in failing MF following pUR4 treatment suggests therapeutic promise for the treatment of HF.
The positive role that FN exerts in cell survival, thus inhibiting apoptosis, has been previously reported.23, 58 Our data demonstrate that inhibiting FN matrix formation does not significantly alter cell survival, suggesting that this process may rely on other mechanisms or that the low levels of FN deposited into the cell surface is sufficient to maintain cell survival. Additionally, no differences in ROS production or total mitochondria content were observed in MF after pUR4 administration, suggesting that inhibiting FN polymerization does not interfere with normal mitochondrial functions. Also, pUR4 did not alter cellular metabolism as evidenced by unaffected ATP production, oxygen consumption, glycolysis and fatty acid oxidation. Together, these data suggest that the attenuation of proliferation and migration detected in MF upon pUR4 administration are not attributed to survival and metabolic state, highlighting that blocking FN polymerization directly affects specific aspects of cellular behavior.
Although FN expression levels are low in unchallenged hearts59, 60, its expression is elevated upon cardiac injury in both mouse and human hearts.60, 61 Daily pUR4 peptide treatment, initiated the same day of I/R injury, decreased fibrotic-related genes including FN and collagens in the LV of 7 days post-I/R hearts. Interestingly, pUR4 was sufficient to decrease FN expression and accumulation in the heart. Concurrently, in vivo EdU pulse-chase experiments revealed a significant decrease of proliferative fibroblasts, both in remote and in infarcted areas 7 days after cardiac insult. These in vivo data are consistent with much of our in vitro results demonstrating the role of pUR4 in attenuating cell proliferation.
The tight balance between matrix metalloproteinases (MMPs) and tissue inhibitors of metalloproteinases (TIMPs) regulates ECM homeostasis.62 However, after cardiac injury, several MMPs and TIMPs are significantly increased in the myocardium63, contributing to the development and progression of pathological ventricular remodeling. pUR4 treatment displayed a downward trend in MMP-9 activity and expression and a significant downregulation of TIMP-1, suggesting that blocking FN deposition may attenuate the imbalance of MMPs and TIMPS that occurs following cardiac injury.
Inflammation plays a critical role in adverse ventricular remodeling and decreased cardiac function following injury.42 In this context, TLRs are proposed to be one of the main targets for endogenous ligands released after cardiac insult to promote the recruitment of inflammatory cells into the injured myocardium.64 Interestingly, FN acts as a ligand for TLR-2 and TLR-4. We found TLR-2 expression to be significantly reduced in the LV of pUR4-treated animals 7 days post-I/R. Further, the in vitro adhesion studies indicated that pUR4 significantly reduced neutrophil adhesion to TNFα activated MHEC which may explain the decreased myocardial neutrophil recruitment observed in vivo after I/R cardiac injury. The observation that pUR4 selectively impacts neutrophil arrest but not neutrophil rolling suggests that pUR4 regulates the expression of heart endothelial cell integrin ligands, but not selectins, involved in the firm arrest and rolling steps of the leukocyte recruitment cascade, respectively. These data are consistent with published data showing that targeting FN expression or polymerization resulted in a suppressed inflammatory response.31, 32, 65 In fact, the data demonstrate that inhibiting FN polymerization does not have an impact on neutrophil integrins involved in cell recruitment suggesting that the observed effects of pUR4 in neutrophil recruitment in vivo and in neutrophil arrest in vitro, are mediated by pUR4 effects on heart endothelial cells integrin ligands (VCAM-1 and ICAM-1). Although flow cytometry data show that overall, pUR4 does not significantly decrease ICAM-1 and VCAM-1 surface expression, immunocytochemistry analysis revealed decreased total expression of both ligands. Several reports have demonstrated that activated neutrophils trigger an intense oxidative response66, 67 as well as produce proteases and collagenases68 to further contribute to the pathologic remodeling and the detrimental organ dysfunction exhibited following cardiac injury. Our observations that pUR4 specifically prevents neutrophil myocardial infiltration post-I/R in vivo suggest a significant role of FN in recruiting neutrophils to the injured myocardium, potentially through a regulation of TLRs and heart endothelial cell adhesion molecules, involved in neutrophil recruitment.
As outlined above, exaggerated ECM remodeling accelerates the pathological functional decline associated with HF progression, eliciting the formation of a mature scar that severely compromises organ compliance. Therefore, experiments were performed to test whether 7 days of daily pUR4 to inhibit FN immediately post-I/R would be salutary following myocardial injury up to 28 days post-I/R. Remarkably, 7 days of pUR4 treatment significantly reduced myocardial dysfunction up to 4 weeks post-I/R as measured by echocardiography. Additionally, pUR4 attenuated pathological changes in cardiac morphometry. Importantly, 7 days of pUR4 peptide injections dramatically reduced pathologic cardiac remodeling and fibrosis as observed up to 4 weeks post-I/R by a decrease in FN and collagen deposition in the LV. The decrease of FN in the heart after pUR4 treatment following injury reduced the fibrotic scar in the myocardium. Further, delayed pUR4 treatment 4 weeks post-I/R indicated a favorable downward trend in cardiac hypertrophy and fibrosis with no significant change in cardiac function compared to III-11C treatment. Although we could not detect a significant reduction in cardiac function at this stage despite a trend toward reduction of hypertrophy and fibrosis, this may be due to the short period of time of peptide administration initiated four weeks post-I/R as well as cessation of therapy for two weeks prior to sample analysis. Future experiments will further establish the optimal dosage and timing of treatment to potentially revert pathologic aspects of the pre-established scar concomitant with improvement in cardiac function.
Since CF are believed to be the most abundant contributors to FN production in the heart, we sought to validate the salutary effects observed with systemic pUR4 delivery in our novel, inducible, fibroblast-specific CF-FN-KO. To this end, FN expression was ablated in Tcf21+ expressing cells (quiescent CF before I/R and their activated MF progeny after cardiac injury).33, 34 FN ablation in Tcf21+-derived CF lineage attenuated cardiac hypertrophy and pathological fibrosis, including reduced CM cross sectional area and collagen content. Interestingly, addition of the FN inhibitor peptide pUR4 did not further preserve cardiac function in CF-FN-KO compared to III-11C. These findings suggest that the cardioprotective effects proffered by ablating FN expression or attenuating FN polymerization may be mediated through effects on FN from the CF lineage.
Although FN has been extensively studied, the role of blocking its polymerization or genetic ablation in CF populations in regulating cardiac cell behavior and the development of cardiac fibrosis has not yet been elucidated. Our data are the first to demonstrate that suppressing FN matrix deposition by utilizing a peptide to attenuate FN polymerization or ablating its gene expression specifically in CF populations preserves cardiac function, attenuates adverse left ventricular remodeling and limits cardiac fibrosis in a mouse model of HF. These findings reinforce the importance of understanding the role of FN and its polymerization in the heart, both during homeostasis and following cardiac injury, and suggest that targeting FN polymerization may be a new therapeutic strategy for treating cardiac fibrosis and HF.
Supplementary Material
Clinical perspective.
What is new?
- The FN polymerization inhibitor pUR4 attenuates the pathological phenotype exhibited by mouse and human myofibroblasts by decreasing fibronectin (FN) polymerization and collagen deposition into the ECM, as well as MF proliferation and migration.
- Inhibiting FN matrix deposition by pUR4 treatment or deleting FN gene expression in cardiac fibroblasts (Tcf21 lineage) confers cardioprotection against ischemia/reperfusion-induced injury by attenuating adverse left ventricular remodeling and cardiac fibrosis, thus preserving cardiac function.
What are the clinical implications?
- Targeting FN polymerization may be a new therapeutic strategy for treating cardiac fibrosis and HF and this principle may also extend to other fibrotic diseases.
Acknowledgments
We thank Evan Meyer and Matthew Kofron for microscopy assistance, Philip Khoury and Jareen Meinzen-Derr for statistical support and Michelle Tallquist for generously sharing the Tcf21 inducible-Cre mice.
Sources of Funding
This work was supported in part by NIH R01 HL132551, R01 HL133695, R01 HL134312 (BCB), P01 HL069779 (BCB and JDM), an American Heart Association–Great Rivers Affiliate Post-Doctoral Fellowship (No. 16POST30180015, IV-A), R35 GM119458 (TJDF) and NIH R01 HL123658 (PA).
Footnotes
Disclosures
None
Bibliography
- 1.Benjamin EJ, Blaha MJ, Chiuve SE, Cushman M, Das SR, Deo R, de Ferranti SD, Floyd J, Fornage M, Gillespie C, Isasi CR, Jimenez MC, Jordan LC, Judd SE, Lackland D, Lichtman JH, Lisabeth L, Liu S, Longenecker CT, Mackey RH, Matsushita K, Mozaffarian D, Mussolino ME, Nasir K, Neumar RW, Palaniappan L, Pandey DK, Thiagarajan RR, Reeves MJ, Ritchey M, Rodriguez CJ, Roth GA, Rosamond WD, Sasson C, Towfighi A, Tsao CW, Turner MB, Virani SS, Voeks JH, Willey JZ, Wilkins JT, Wu JH, Alger HM, Wong SS, Muntner P, American Heart Association Statistics C and Stroke Statistics S Heart Disease and Stroke Statistics-2017 Update: A Report From the American Heart Association. Circulation. 2017;135:e146–e603. doi: 10.1161/CIR.0000000000000485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Heidenreich PA, Albert NM, Allen LA, Bluemke DA, Butler J, Fonarow GC, Ikonomidis JS, Khavjou O, Konstam MA, Maddox TM, Nichol G, Pham M, Pina IL, Trogdon JG, American Heart Association Advocacy Coordinating C, Council on Arteriosclerosis T, Vascular B, Council on Cardiovascular R, Intervention, Council on Clinical C, Council on E, Prevention and Stroke C Forecasting the impact of heart failure in the United States: a policy statement from the American Heart Association. Circ Heart Fail. 2013;6:606–619. doi: 10.1161/HHF.0b013e318291329a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jarvelainen H, Sainio A, Koulu M, Wight TN, Penttinen R. Extracellular matrix molecules: potential targets in pharmacotherapy. Pharmacological reviews. 2009;61:198–223. doi: 10.1124/pr.109.001289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Valiente-Alandi I, Schafer AE, Blaxall BC. Extracellular matrix-mediated cellular communication in the heart. Journal of molecular and cellular cardiology. 2016;91:228–237. doi: 10.1016/j.yjmcc.2016.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Berk BC, Fujiwara K, Lehoux S. ECM remodeling in hypertensive heart disease. The Journal of clinical investigation. 2007;117:568–575. doi: 10.1172/JCI31044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Petrov VV, Fagard RH, Lijnen PJ. Stimulation of collagen production by transforming growth factor-beta1 during differentiation of cardiac fibroblasts to myofibroblasts. Hypertension. 2002;39:258–263. doi: 10.1161/hy0202.103268. [DOI] [PubMed] [Google Scholar]
- 7.Tomasek JJ, Gabbiani G, Hinz B, Chaponnier C, Brown RA. Myofibroblasts and mechano-regulation of connective tissue remodelling. Nature reviews Molecular cell biology. 2002;3:349–363. doi: 10.1038/nrm809. [DOI] [PubMed] [Google Scholar]
- 8.Johnson CM, Helgeson SC. Fibronectin biosynthesis and cell-surface expression by cardiac and non-cardiac endothelial cells. Am J Pathol. 1993;142:1401–1408. [PMC free article] [PubMed] [Google Scholar]
- 9.McKeown-Longo PJ, Mosher DF. Interaction of the 70,000-mol-wt amino-terminal fragment of fibronectin with the matrix-assembly receptor of fibroblasts. The Journal of cell biology. 1985;100:364–374. doi: 10.1083/jcb.100.2.364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dobaczewski M, Bujak M, Zymek P, Ren G, Entman ML, Frangogiannis NG. Extracellular matrix remodeling in canine and mouse myocardial infarcts. Cell and tissue research. 2006;324:475–488. doi: 10.1007/s00441-005-0144-6. [DOI] [PubMed] [Google Scholar]
- 11.Heling A, Zimmermann R, Kostin S, Maeno Y, Hein S, Devaux B, Bauer E, Klovekorn WP, Schlepper M, Schaper W, Schaper J. Increased expression of cytoskeletal, linkage, and extracellular proteins in failing human myocardium. Circulation research. 2000;86:846–853. doi: 10.1161/01.res.86.8.846. [DOI] [PubMed] [Google Scholar]
- 12.van Dijk A, Niessen HW, Ursem W, Twisk JW, Visser FC, van Milligen FJ. Accumulation of fibronectin in the heart after myocardial infarction: a putative stimulator of adhesion and proliferation of adipose-derived stem cells. Cell and tissue research. 2008;332:289–298. doi: 10.1007/s00441-008-0573-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.George EL, Georges-Labouesse EN, Patel-King RS, Rayburn H, Hynes RO. Defects in mesoderm, neural tube and vascular development in mouse embryos lacking fibronectin. Development. 1993;119:1079–1091. doi: 10.1242/dev.119.4.1079. [DOI] [PubMed] [Google Scholar]
- 14.Konstandin MH, Volkers M, Collins B, Quijada P, Quintana M, De La Torre A, Ormachea L, Din S, Gude N, Toko H, Sussman MA. Fibronectin contributes to pathological cardiac hypertrophy but not physiological growth. Basic Res Cardiol. 2013;108:375. doi: 10.1007/s00395-013-0375-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Konstandin MH, Toko H, Gastelum GM, Quijada P, De La Torre A, Quintana M, Collins B, Din S, Avitabile D, Volkers M, Gude N, Fassler R, Sussman MA. Fibronectin Is Essential for Reparative Cardiac Progenitor Cell Response After Myocardial Infarction. Circulation research. 2013;113:115–125. doi: 10.1161/CIRCRESAHA.113.301152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hernnas J, Nettelbladt O, Bjermer L, Sarnstrand B, Malmstrom A, Hallgren R. Alveolar accumulation of fibronectin and hyaluronan precedes bleomycin-induced pulmonary fibrosis in the rat. The European respiratory journal. 1992;5:404–410. [PubMed] [Google Scholar]
- 17.Muro AF, Moretti FA, Moore BB, Yan M, Atrasz RG, Wilke CA, Flaherty KR, Martinez FJ, Tsui JL, Sheppard D, Baralle FE, Toews GB, White ES. An essential role for fibronectin extra type III domain A in pulmonary fibrosis. American journal of respiratory and critical care medicine. 2008;177:638–645. doi: 10.1164/rccm.200708-1291OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jarnagin WR, Rockey DC, Koteliansky VE, Wang SS, Bissell DM. Expression of variant fibronectins in wound healing: cellular source and biological activity of the EIIIA segment in rat hepatic fibrogenesis. The Journal of cell biology. 1994;127:2037–2048. doi: 10.1083/jcb.127.6.2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sottile J, Shi F, Rublyevska I, Chiang HY, Lust J, Chandler J. Fibronectin-dependent collagen I deposition modulates the cell response to fibronectin. Am J Physiol Cell Physiol. 2007;293:C1934–1946. doi: 10.1152/ajpcell.00130.2007. [DOI] [PubMed] [Google Scholar]
- 20.Sottile J, Hocking DC. Fibronectin polymerization regulates the composition and stability of extracellular matrix fibrils and cell-matrix adhesions. Mol Biol Cell. 2002;13:3546–3559. doi: 10.1091/mbc.E02-01-0048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ruoslahti E. Fibronectin in cell adhesion and invasion. Cancer Metastasis Rev. 1984;3:43–51. doi: 10.1007/BF00047692. [DOI] [PubMed] [Google Scholar]
- 22.Cao Y, Liu X, Lu W, Chen Y, Wu X, Li M, Wang XA, Zhang F, Jiang L, Zhang Y, Hu Y, Xiang S, Shu Y, Bao R, Li H, Wu W, Weng H, Yen Y, Liu Y. Fibronectin promotes cell proliferation and invasion through mTOR signaling pathway activation in gallbladder cancer. Cancer Lett. 2015;360:141–150. doi: 10.1016/j.canlet.2015.01.041. [DOI] [PubMed] [Google Scholar]
- 23.Han SW, Roman J. Fibronectin induces cell proliferation and inhibits apoptosis in human bronchial epithelial cells: pro-oncogenic effects mediated by PI3-kinase and NF-kappa B. Oncogene. 2006;25:4341–4349. doi: 10.1038/sj.onc.1209460. [DOI] [PubMed] [Google Scholar]
- 24.Straus AH, Carter WG, Wayner EA, Hakomori S. Mechanism of fibronectin-mediated cell migration: dependence or independence of cell migration susceptibility on RGDS-directed receptor (integrin) Experimental cell research. 1989;183:126–139. doi: 10.1016/0014-4827(89)90423-0. [DOI] [PubMed] [Google Scholar]
- 25.Zou L, Cao S, Kang N, Huebert RC, Shah VH. Fibronectin induces endothelial cell migration through beta1 integrin and Src-dependent phosphorylation of fibroblast growth factor receptor-1 at tyrosines 653/654 and 766. The Journal of biological chemistry. 2012;287:7190–7202. doi: 10.1074/jbc.M111.304972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Farias E, Lu M, Li X, Schnapp LM. Integrin alpha8beta1-fibronectin interactions promote cell survival via PI3 kinase pathway. Biochem Biophys Res Commun. 2005;329:305–311. doi: 10.1016/j.bbrc.2005.01.125. [DOI] [PubMed] [Google Scholar]
- 27.Lin HY, Tsai CC, Chen LL, Chiou SH, Wang YJ, Hung SC. Fibronectin and laminin promote differentiation of human mesenchymal stem cells into insulin producing cells through activating Akt and ERK. J Biomed Sci. 2010;17:56. doi: 10.1186/1423-0127-17-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ensenberger MG, Annis DS, Mosher DF. Actions of the functional upstream domain of protein F1 of Streptococcus pyogenes on the conformation of fibronectin. Biophysical chemistry. 2004;112:201–207. doi: 10.1016/j.bpc.2004.07.020. [DOI] [PubMed] [Google Scholar]
- 29.Ensenberger MG, Tomasini-Johansson BR, Sottile J, Ozeri V, Hanski E, Mosher DF. Specific interactions between F1 adhesin of Streptococcus pyogenes and N-terminal modules of fibronectin. The Journal of biological chemistry. 2001;276:35606–35613. doi: 10.1074/jbc.M105417200. [DOI] [PubMed] [Google Scholar]
- 30.Tomasini-Johansson BR, Kaufman NR, Ensenberger MG, Ozeri V, Hanski E, Mosher DF. A 49-residue peptide from adhesin F1 of Streptococcus pyogenes inhibits fibronectin matrix assembly. J Biol Chem. 2001;276:23430–23439. doi: 10.1074/jbc.M103467200. [DOI] [PubMed] [Google Scholar]
- 31.Chiang HY, Korshunov VA, Serour A, Shi F, Sottile J. Fibronectin is an important regulator of flow-induced vascular remodeling. Arteriosclerosis, thrombosis, and vascular biology. 2009;29:1074–1079. doi: 10.1161/ATVBAHA.108.181081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Altrock E, Sens C, Wuerfel C, Vasel M, Kawelke N, Dooley S, Sottile J, Nakchbandi IA. Inhibition of fibronectin deposition improves experimental liver fibrosis. J Hepatol. 2015;62:625–633. doi: 10.1016/j.jhep.2014.06.010. [DOI] [PubMed] [Google Scholar]
- 33.Acharya A, Baek ST, Huang G, Eskiocak B, Goetsch S, Sung CY, Banfi S, Sauer MF, Olsen GS, Duffield JS, Olson EN, Tallquist MD. The bHLH transcription factor Tcf21 is required for lineage-specific EMT of cardiac fibroblast progenitors. Development. 2012;139:2139–2149. doi: 10.1242/dev.079970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kanisicak O, Khalil H, Ivey MJ, Karch J, Maliken BD, Correll RN, Brody MJ, SC JL, Aronow BJ, Tallquist MD, Molkentin JD. Genetic lineage tracing defines myofibroblast origin and function in the injured heart. Nat Commun. 2016;7:12260. doi: 10.1038/ncomms12260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chiang HY, Korshunov VA, Serour A, Shi F, Sottile J. Fibronectin Is an Important Regulator of Flow-Induced Vascular Remodeling. Arterioscl Throm Vas. 2009;29:1074–U157. doi: 10.1161/ATVBAHA.108.181081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gustafsson E, Fassler R. Insights into extracellular matrix functions from mutant mouse models. Exp Cell Res. 2000;261:52–68. doi: 10.1006/excr.2000.5042. [DOI] [PubMed] [Google Scholar]
- 37.Knowlton AA, Connelly CM, Romo GM, Mamuya W, Apstein CS, Brecher P. Rapid expression of fibronectin in the rabbit heart after myocardial infarction with and without reperfusion. The Journal of clinical investigation. 1992;89:1060–1068. doi: 10.1172/JCI115685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ulrich MM, Janssen AM, Daemen MJ, Rappaport L, Samuel JL, Contard F, Smits JF, Cleutjens JP. Increased expression of fibronectin isoforms after myocardial infarction in rats. Journal of molecular and cellular cardiology. 1997;29:2533–2543. doi: 10.1006/jmcc.1997.0486. [DOI] [PubMed] [Google Scholar]
- 39.Fan D, Takawale A, Lee J, Kassiri Z. Cardiac fibroblasts, fibrosis and extracellular matrix remodeling in heart disease. Fibrogenesis Tissue Repair. 2012;5:15. doi: 10.1186/1755-1536-5-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ohnishi T, Hiraga S, Izumoto S, Matsumura H, Kanemura Y, Arita N, Hayakawa T. Role of fibronectin-stimulated tumor cell migration in glioma invasion in vivo: clinical significance of fibronectin and fibronectin receptor expressed in human glioma tissues. Clin Exp Metastasis. 1998;16:729–741. doi: 10.1023/a:1006532812408. [DOI] [PubMed] [Google Scholar]
- 41.Coito AJ, de Sousa M, Kupiec-Weglinski JW. Fibronectin in immune responses in organ transplant recipients. Dev Immunol. 2000;7:239–248. doi: 10.1155/2000/98187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Frangogiannis NG. The inflammatory response in myocardial injury, repair, and remodelling. Nat Rev Cardiol. 2014;11:255–265. doi: 10.1038/nrcardio.2014.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Epelman S, Liu PP, Mann DL. Role of innate and adaptive immune mechanisms in cardiac injury and repair. Nat Rev Immunol. 2015;15:117–129. doi: 10.1038/nri3800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chigaev A, Sklar LA. Aspects of VLA-4 and LFA-1 regulation that may contribute to rolling and firm adhesion. Front Immunol. 2012;3:242. doi: 10.3389/fimmu.2012.00242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sans M, Panes J, Ardite E, Elizalde JI, Arce Y, Elena M, Palacin A, Fernandez-Checa JC, Anderson DC, Lobb R, Pique JM. VCAM-1 and ICAM-1 mediate leukocyte-endothelial cell adhesion in rat experimental colitis. Gastroenterology. 1999;116:874–883. doi: 10.1016/s0016-5085(99)70070-3. [DOI] [PubMed] [Google Scholar]
- 46.Chen X, Nadiarynkh O, Plotnikov S, Campagnola PJ. Second harmonic generation microscopy for quantitative analysis of collagen fibrillar structure. Nat Protoc. 2012;7:654–669. doi: 10.1038/nprot.2012.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cox G, Kable E, Jones A, Fraser I, Manconi F, Gorrell MD. 3-dimensional imaging of collagen using second harmonic generation. J Struct Biol. 2003;141:53–62. doi: 10.1016/s1047-8477(02)00576-2. [DOI] [PubMed] [Google Scholar]
- 48.Hinz B. Formation and function of the myofibroblast during tissue repair. J Invest Dermatol. 2007;127:526–537. doi: 10.1038/sj.jid.5700613. [DOI] [PubMed] [Google Scholar]
- 49.Baum J, Duffy HS. Fibroblasts and myofibroblasts: what are we talking about? J Cardiovasc Pharmacol. 2011;57:376–379. doi: 10.1097/FJC.0b013e3182116e39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gustafsson E, Fassler R. Insights into extracellular matrix functions from mutant mouse models. Experimental cell research. 2000;261:52–68. doi: 10.1006/excr.2000.5042. [DOI] [PubMed] [Google Scholar]
- 51.Velling T, Risteli J, Wennerberg K, Mosher DF, Johansson S. Polymerization of type I and III collagens is dependent on fibronectin and enhanced by integrins alpha 11beta 1 and alpha 2beta 1. The Journal of biological chemistry. 2002;277:37377–37381. doi: 10.1074/jbc.M206286200. [DOI] [PubMed] [Google Scholar]
- 52.Amati B, Alevizopoulos K, Vlach J. Myc and the cell cycle. Front Biosci. 1998;3:d250–268. doi: 10.2741/a239. [DOI] [PubMed] [Google Scholar]
- 53.Rajabi HN, Baluchamy S, Kolli S, Nag A, Srinivas R, Raychaudhuri P, Thimmapaya B. Effects of depletion of CREB-binding protein on c-Myc regulation and cell cycle G1-S transition. The Journal of biological chemistry. 2005;280:361–374. doi: 10.1074/jbc.M408633200. [DOI] [PubMed] [Google Scholar]
- 54.Danen EH, Sonneveld P, Brakebusch C, Fassler R, Sonnenberg A. The fibronectin-binding integrins alpha5beta1 and alphavbeta3 differentially modulate RhoA-GTP loading, organization of cell matrix adhesions, and fibronectin fibrillogenesis. The Journal of cell biology. 2002;159:1071–1086. doi: 10.1083/jcb.200205014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shi F, Sottile J. Caveolin-1-dependent beta1 integrin endocytosis is a critical regulator of fibronectin turnover. J Cell Sci. 2008;121:2360–2371. doi: 10.1242/jcs.014977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhao X, Guan JL. Focal adhesion kinase and its signaling pathways in cell migration and angiogenesis. Adv Drug Deliv Rev. 2011;63:610–615. doi: 10.1016/j.addr.2010.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hanks SK, Calalb MB, Harper MC, Patel SK. Focal adhesion protein-tyrosine kinase phosphorylated in response to cell attachment to fibronectin. Proc Natl Acad Sci U S A. 1992;89:8487–8491. doi: 10.1073/pnas.89.18.8487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fornaro M, Plescia J, Chheang S, Tallini G, Zhu YM, King M, Altieri DC, Languino LR. Fibronectin protects prostate cancer cells from tumor necrosis factor-alpha-induced apoptosis via the AKT/survivin pathway. The Journal of biological chemistry. 2003;278:50402–50411. doi: 10.1074/jbc.M307627200. [DOI] [PubMed] [Google Scholar]
- 59.Heng BC, Haider H, Sim EK, Cao T, Ng SC. Strategies for directing the differentiation of stem cells into the cardiomyogenic lineage in vitro. Cardiovasc Res. 2004;62:34–42. doi: 10.1016/j.cardiores.2003.12.022. [DOI] [PubMed] [Google Scholar]
- 60.Willems IE, Arends JW, Daemen MJ. Tenascin and fibronectin expression in healing human myocardial scars. J Pathol. 1996;179:321–325. doi: 10.1002/(SICI)1096-9896(199607)179:3<321::AID-PATH555>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 61.Froen JF, Larsen TH. Fibronectin penetration into heart myocytes subjected to experimental ischemia by coronary artery ligation. Acta Anat (Basel) 1995;152:119–126. doi: 10.1159/000147690. [DOI] [PubMed] [Google Scholar]
- 62.Ahmed SH, Clark LL, Pennington WR, Webb CS, Bonnema DD, Leonardi AH, McClure CD, Spinale FG, Zile MR. Matrix metalloproteinases/tissue inhibitors of metalloproteinases: relationship between changes in proteolytic determinants of matrix composition and structural, functional, and clinical manifestations of hypertensive heart disease. Circulation. 2006;113:2089–2096. doi: 10.1161/CIRCULATIONAHA.105.573865. [DOI] [PubMed] [Google Scholar]
- 63.Phatharajaree W, Phrommintikul A, Chattipakorn N. Matrix metalloproteinases and myocardial infarction. Can J Cardiol. 2007;23:727–733. doi: 10.1016/s0828-282x(07)70818-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Miyake K. Innate immune sensing of pathogens and danger signals by cell surface Toll-like receptors. Semin Immunol. 2007;19:3–10. doi: 10.1016/j.smim.2006.12.002. [DOI] [PubMed] [Google Scholar]
- 65.Arslan F, Smeets MB, Riem Vis PW, Karper JC, Quax PH, Bongartz LG, Peters JH, Hoefer IE, Doevendans PA, Pasterkamp G, de Kleijn DP. Lack of fibronectin-EDA promotes survival and prevents adverse remodeling and heart function deterioration after myocardial infarction. Circulation research. 2011;108:582–592. doi: 10.1161/CIRCRESAHA.110.224428. [DOI] [PubMed] [Google Scholar]
- 66.Entman ML, Youker K, Shoji T, Kukielka G, Shappell SB, Taylor AA, Smith CW. Neutrophil induced oxidative injury of cardiac myocytes. A compartmented system requiring CD11b/CD18-ICAM-1 adherence. The Journal of clinical investigation. 1992;90:1335–1345. doi: 10.1172/JCI115999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tyagi S, Klickstein LB, Nicholson-Weller A. C5a-stimulated human neutrophils use a subset of beta2 integrins to support the adhesion-dependent phase of superoxide production. J Leukoc Biol. 2000;68:679–786. [PubMed] [Google Scholar]
- 68.Kawakami R, Saito Y, Kishimoto I, Harada M, Kuwahara K, Takahashi N, Nakagawa Y, Nakanishi M, Tanimoto K, Usami S, Yasuno S, Kinoshita H, Chusho H, Tamura N, Ogawa Y, Nakao K. Overexpression of brain natriuretic peptide facilitates neutrophil infiltration and cardiac matrix metalloproteinase-9 expression after acute myocardial infarction. Circulation. 2004;110:3306–3312. doi: 10.1161/01.CIR.0000147829.78357.C5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.