

Abstract
The Histone Deacetylase 9 (HDAC9) polymorphism rs2107595 is associated with an increased risk for large vessel atherosclerotic stroke (LVAS). In humans, there remains a need to better understand this HDAC9 polymorphism’s contribution to large vessel stroke. In this pilot study, we evaluated whether the HDAC9 polymorphism rs2107595 is associated with differences in leukocyte gene expression in patients with LVAS. HDAC9 SNP rs2107595 was genotyped in 155 patients (43 LVAS and 112 vascular risk factor controls). RNA isolated from blood was processed on whole genome microarrays. Gene expression was compared between HDAC9 risk allele positive and risk allele negative LVAS patients and controls. Functional analysis identified canonical pathways and molecular functions associated with rs2107595 in LVAS. In HDAC9 SNP rs2107595 risk allele positive LVAS patients there were 155 genes differentially expressed compared to risk allele negative patients (fold change >|1.2|, p<0.05). The 155 genes separated the risk allele positive and negative LVAS patients on a Principal Components Analysis. Pathways associated with HDAC9 risk allele positive status involved IL6 signaling, cholesterol efflux, and platelet aggregation. These preliminary data suggest an association with the HDAC9 rs2107595 risk allele and peripheral immune, lipid, and clotting systems in LVAS. Further study is required to evaluate whether these differences are related to large vessel atherosclerosis and stroke risk.
Keywords: SNP, polymorphism, gene expression, large vessel stroke, ischemic stroke, atherosclerosis
Introduction
Large vessel atherosclerosis is an important cause of ischemic stroke. Understanding genomic differences that place individuals at risk of large vessel stroke could aid in the development of novel stroke prevention strategies. Histone Deacetylase 9 (HDAC9) is a class IIa histone deacetylase located at 7p211, 2. A GWAS study conducted by the International Stroke Genetics Consortium and the Wellcome Trust Case Control Consortium 2 demonstrated an association between HDAC9 and large vessel atherosclerotic stroke3. The A risk allele at HDAC9 single nucleotide polymorphism (SNP) rs2107595, which displays incomplete penetrance, increases odds for large vessel stroke4–6. However, the underlying functional relationship of rs2107595 to LVAS remains unclear.
HDAC9 regulates gene expression by removing acetyl groups from proteins that regulate chromatin state7. Hypo-acetylation at gene promoters typically leads to transcriptional repression by restricting accessibility of transcriptional machinery to chromatin8, 9. Balancing acetylation/deacetylation plays a key role in disease states such as atherosclerosis1, 8, 9.
HDAC9 is upregulated in human carotid and aortic atherosclerotic plaques4. Cells that express HDAC9 include monocytes, macrophages, T lymphocytes, vascular endothelial cells, and smooth muscle cells1. Coronary artery disease patients exhibit significantly elevated expression of HDAC9 in plasma6.
HDAC9 deletion disrupts lipid homeostasis and genes involved in inflammation10. Loss of HDAC9 increases Histone-3 acetylation at macrophage ABCA1, ABCG1, and PPAR-γ promoters, resulting in increased HDL-mediated cholesterol efflux and macrophage polarization toward the anti-inflammatory M2 phenotype10. HDAC9 deficiency also reduces atherosclerotic lesion size in ApoE−/− mice1.
In humans, there remains a need to better understand how HDAC9 SNP rs2107595 contributes to LVAS. In this novel pilot study, we evaluated how HDAC9 SNP rs2107595 relates to blood cell gene expression in patients with LVAS and in vascular risk factor controls (VRFC).
Methods
Study Subjects
LVAS patients and VRFC were prospectively recruited from the University of California, Davis from 2012-2014. The institutional review board approved the study protocol, and written informed consent was obtained from each patient. Stroke diagnosis criteria are available in the Supplementary Methods. VRFC had no clinical history of stroke or cardiovascular disease, and no recent infection. Controls were of similar age, sex, and vascular risk factor profile as stroke patients.
SNP Genotyping
Blood collection and RNA/DNA isolation methods are available in Supplementary Methods. The SNP rs2107595 was genotyped using the Single Tube TaqMan SNP Genotyping Assay per manufacturer protocol (Life Technologies Corporation, NY). PCR amplification was performed using 50ng of gDNA with the Applied Biosystems-7900HT Fast Real-Time PCR System. TaqMan Genotyper Software was used to plot Rn values based on fluorescence signals from each sample and used to determine allele representation. Assays were performed in triplicate and included positive controls of two homozygotes and one heterozygote on each replicate. Positive control DNA was obtained from the Coriell Biorepository (Coriell Institute for Medical Research, Camden, NJ). Genotype distribution of rs2107595 did not significantly deviate from Hardy-Weinberg equilibrium in both LVAS and control groups (p>0.05). Subjects exhibiting one or both of risk alleles were deemed rs2107595 risk allele positive. Subjects exhibiting two non-risk alleles were deemed rs2107595 risk allele negative.
Gene Expression Array Hybridization and Statistical Analysis
RNA samples underwent two-cycle target labeling, and cDNA was hybridized to Affymetrix Human Transcriptome 2.0 Arrays per the manufacturer’s GeneChip protocol (Affymetrix, Santa Clara, CA). These arrays cover all known protein coding genes (Affymetrix GeneChip HTA Data Sheet). To minimize batch effect and technical variation, each batch contained equal numbers of randomly allocated stroke and control patients.
Raw gene expression data was normalized using Robust Multichip Averaging (RMA) and log2 transformed as previously described11. Statistical analysis and principal components analysis were performed using Partek Genomics Suite 6.6. After removing unannotated transcripts, 33,007 probe sets were used for analysis. Gene expression of risk allele positive LVAS patients was compared to risk allele negative LVAS patients. Similarly, gene expression of risk allele positive VRFC patients was compared to risk allele negative VRFC patients. Univariate analysis was performed to identify potential confounding variables including age, sex, race, and vascular risk factors. Variables with p<0.05 were then included in the multivariate model. Probesets with a p<0.05 and fold change ≥|1.2| were considered significant, as previously described12–15. Functional pathways represented greater than expected by chance (Fisher’s exact test) based on the differentially expressed genes were identified using Ingenuity Pathway Analysis (IPA, QIAGEN, Redwood City, CA, www.ingenuity.com) and literature review.
Results
Patient Characteristics
A total of 43 LVAS and 112 VRFC patients were included in this study. There were no statistically significant differences in patient demographics including age, sex, vascular risk factors, or smoking status between LVAS and VRFC groups (Table 1). Of 43 LVAS patients, 24 were rs2107595 risk allele positive and 19 risk allele negative. Of the 112 VRFC patients, 31 were rs2107595 risk allele positive and 81 risk allele negative. There were significantly more rs2107595 risk allele positive LVAS patients (56%) than rs2107595 risk allele positive VRFC patients (28%) (p<0.001, chi square). There were no significant differences between rs2107595 risk allele positive and risk allele negative LVAS groups (Supplementary Table 1), nor were there significant differences between VRFC risk allele positive and negative groups (Supplementary Table 2).
Table 1.
Demographics for patients with large vessel atherosclerotic stroke (LVAS) and vascular risk factor controls (VRFC).
| Variables | LVAS(n=43) | VRFC(n=112) | p-value |
|---|---|---|---|
| Mean Age(years) | 65.49±1.59 | 63.83±1.15 | 0.43 |
| Sex, male(%) | 30 (69.8%) | 59 (52.7%) | 0.07 |
| Hypertension(%) | 35 (81.4%) | 76 (67.8%) | 0.38 |
| Diabetes(%) | 18 (41.2%) | 26 (23.2%) | 0.07 |
| Hyperlipidemia(%) | 28 (65.1%) | 62 (55.3%) | 0.58 |
| Smoking Status (%) | 13 (30.2%) | 18 (16.1%) | 0.10 |
Differentially Expressed Genes (DEG)
In LVAS there were 155 DEG in rs2107595 risk allele positive compared to risk allele negative patients (Supplementary Table 3). Of these, 139(89.1%) were upregulated and 16(10.9%) were downregulated in risk allele positive patients. A principal components analysis plot of these 155 genes demonstrates separation of rs2107595 risk allele positive and risk allele negative LVAS by gene expression (Figure 1). In VRFC there were 8 DEG in rs2107595 risk allele positive compared to risk allele negative patients (Supplementary Table 4). The CHML (Choroideremia-Like Protein) gene was shared by comparisons of risk allele+/− LVAS to risk allele+/− VRFC.
Figure 1.
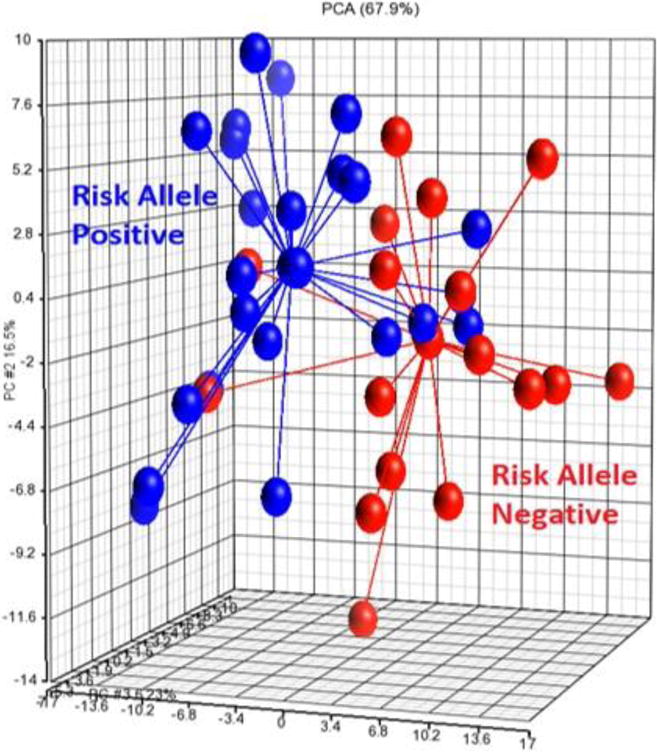
Principal components analysis showing separation of rs2107595 risk allele positive and risk allele negative LVAS patients based on the expression of 155 DEG.
Functional Analysis
Pathway analysis for the 155 genes associated with rs2107595 identified canonical pathways involving IL6 signaling, ERK/MAPK signaling, LXR/RXR activation, and the role of macrophages, fibroblasts, and endothelial cells in rheumatoid arthritis (Table 2). SNP rs2107595 risk allele positive LVAS was associated with leukocyte recruitment, chronic inflammation, immune response of macrophages, cholesterol efflux, and platelet aggregation (Table 2). GMFG (Glial growth factor γ, FC: 1.24, p=0.02) and LRP1 (Low-density lipoprotein receptor related protein 1, FC: −1.23, p=0.01) are involved in endothelial cell function and angiogenesis16, 17 and were differentially expressed in LVAS risk allele positive patients.
Table 2.
Functional analysis of the 155 genes differentially expressed in HDAC9 SNP rs2107595 risk allele positive compared to risk allele negative LVAS patients.
| rs2107595 RAP vs RAN | Genes | p-value | |
|---|---|---|---|
| Canonical Pathways | IL-6 Signaling | CEBPB, CXCL8, IL1R2, TNFAIP6 | 9.25E-03 |
| Role of Macrophages, Fibroblasts, and Endothelial Cells in RA | CEBPB, CXCL8, IL1R2, LRP1, LTB, PLCG2 | 1.51E-02 | |
| ERK/MAPK Signaling | DUSP1, PLCG2, PRKAR1A, TLN1 | 4.00E-02 | |
| LXR/RXR Activation | IL1R2, LY96, S100A8 | 4.35E-02 | |
| Molecular Functions | Leukocytes Recruitment | CLEC4E, CX3CR1, CXCL8, HCAR2, HMGB1, LRP1, LY96, S100A8, THBS1, TPT1 | 1.77E-04 |
| Chronic Inflammation | CCDC59, CEBPB, CLEC4E, CX3CR1, CXCL8, CYP4F3, DUSP1, HMGB1, IGF1R, IL1R2, LTB, PANK2, PDE3B, PTGS1, RPS13, S100A8, THBS1, TNFAIP6, TP53INP1, USP15 | 2.38E-04 | |
| Macrophage Immune Response | ABCA7, CEBPB, HMGB1, LY96, mir-142, THBS1 | 4.44E-04 | |
| Cholesterol Efflux | ABCA7, HCAR2, LRP1, S100A8 | 5.98E-03 | |
| Platelet Aggregation | DAB2, PLCG2, RAB27B, THBS1, TLN1 | 1.26E-02 |
Literature review indicates the following genes have previously been associated with atherosclerosis and were differentially expressed in a pro-atherogenic manner between risk allele positive and negative patients: CX3CR1, HCAR2, HMGB1, JUNB, LRP1, PDE3B, PTGS1, S100A8, THBS1, TUBB2A, and VCAN. Of note were HMGB1 (High mobility group box 1, FC: 1.20, p=0.03) and THBS1 (Thrombospondin 1, FC: −1.25, p=0.04).
Discussion
The HDAC9 risk allele at rs2107595 was associated with differences in blood cell gene expression in patients with LVAS. Risk allele positive LVAS patients had increased gene expression involved in inflammation, lipid metabolism and platelet aggregation. Mechanisms by which these changes occur require further study, but may include HDAC9 risk allele regulation of gene expression in leukocytes, or indirect regulation through leukocyte interactions with endothelial cells, or smooth muscle cells affected by the HDAC9 risk allele.
In this study the risk allele does not seem to alter HDAC9 expression levels. However, a previous study showed a significant increase of HDAC9 expression in both heterozygous and homozygous patients with the risk allele1. Explanations for why the two studies differ include: the 2015 study was performed on peripheral blood mononuclear cells (monocytes, lymphocytes), whereas the current study was performed on whole blood; and the current study utilized controls matched for vascular risk factors (hypertension, hyperlipidemia, diabetes, and statin therapy) compared to the LVAS group, as opposed to healthy patients. The different findings do raise the possibility that HDAC9 expression is regulated differently in different leukocyte subsets. Thus, the risk allele, whether through changes in HDAC9 expression or not, may affect deacetylase activity differently than the non-risk allele, leading to changes in gene expression. Indeed, both HDAC5 and HDAC7 alter gene expression through deacetylase actions on gene promoters18, 19. The risk allele at rs2107595 may affect HDAC9’s deacetylase activity and alter gene expression in leukocytes. Alternatively, HDAC9 could affect endothelial cells20, 21 and/or vascular smooth muscle cells, which could alter leukocyte interactions with these cells and produce the differential blood cell gene expression found in this study.
The risk allele at rs210759 is not causative for LVAS. Many people that exhibit the risk allele do not have stroke. It is possible that other factors interact with the HDAC9 polymorphism to contribute to stroke risk. The HDAC9 polymorphism displays incomplete penetrance which may be influenced by polymorphic alleles at other loci, epigenetic regulation, or environmental modifiers. This has been described in other diseases associated with a genetic risk such as BRCA1 and BRCA2 in breast cancer22, 23. This may explain why in our study risk allele positive LVAS patients had differential gene expression involved in lipid metabolism and inflammation while controls did not. Risk allele positive VRFC may have compensatory mechanisms or a different genetic and environmental background. Indeed, environmental factors such as diabetes and body mass index are known to influence risk of coronary artery disease in patients with the rs2107595 risk allele6, and microbial environment can influence HDAC9 deacetylase activity24. Specifically, statins have been reported to affect histone acetylation25, though there were no differences of statin usage in the risk allele positive and negative LVAS patients (Supplementary Table 1), and no differences in statin usage in the risk allele positive and negative VRFC patients (Supplementary Table 2). In addition, it is known that valproate is an HDAC inhibitor26, 27. In our study, no patients were treated with valproate.
Inflammation
HDAC9 SNP rs2107595 may increase risk of LVAS via its effect on leukocyte Treg function. HDAC9 deacetylates and downregulates FOXP3 (Forkhead Box P3). This impairs T-regulatory cell immunosuppression, resulting in increased inflammation which has been implicated in atherosclerosis28. Downregulation of FOXP3 is associated with increased HMGB1 (High Mobility Group Box 1) and IL6 (Interleukin 6) expression, which we observed in rs2107595 risk allele positive LVAS patients (Table 1). This immune activation may contribute to atherosclerosis and stroke.
FOXP3 also decreases thrombospondin (THBS1) which was downregulated in rs2107595 risk allele positive patients. THBS1 plays a protective role in atherosclerosis, impeding plaque maturation and rupture and suppressing the inflammatory response29, 30. THBS1 is downregulated in patients with atherosclerosis, promoting plaque inflammation and maturation29, 31. Thus the rs2107595 risk allele may contribute to LVAS via its effect on Treg function and inflammation.
Lipid Metabolism
ABCA7 and HCAR2 were both upregulated in LVAS HDAC9 risk allele positive patients. ABCA7 is implicated in macrophage lipid efflux and clearance of apoptotic cells. It also plays a role in T cell proliferation32 and is an Alzheimer’s disease risk gene33. HCAR2 (hydroxyl-carboxylic receptor 2) is a niacin receptor gene that mediates niacin decrease of Lipoprotein(a)34. Nicotinic acid action on HCAR2 causes atherosclerosis to regress in humans and mice.
LRP1 (low-density lipoprotein receptor-related protein-1), is a signaling receptor that was downregulated in LVAS risk allele positive patients. When expressed on adipocytes, it protects against diet-induced atherosclerosis and modulates atherosclerosis by regulating inflammation35. LRP1 regulates cholesterol accumulation in macrophages. This is important as excessive accumulation leads to foam cell formation which are precursors of atherosclerosis36. LRP1 also regulates plasma levels of blood coagulation factor VIII and other factors affecting coagulation37 which could predispose to stroke.
Calgranulin S100A8 is a proinflammatory mediator of atherosclerosis and was upregulated in LVAS risk allele positive patients. It is a major component of neutrophils, is upregulated in macrophages and foam cells, and influences leukocyte recruitment and inflammation by binding TLR4 and/or the receptor for advanced glycation end products (RAGE)38. Increased plasma levels of S100A8 predict cardiovascular events in humans, and deletion of this gene partly protects Apoe−/− mice from atherosclerosis39. Stroke associated carotid plaques have high levels of S100A8.
Platelet Aggregation
Disabled-2 (DAB2) is an adapter protein that is up-regulated during megakaryocytic differentiation of hematopoietic cells. It is abundantly expressed in platelets where it is a key regulator of platelet signaling40. Dab2 is released from platelet granules and controls the extent of clotting reaction, platelet-fibrinogen interactions and outside-in signaling40. DAB2 is required for platelet aggregation, fibrinogen uptake, and integrin αIIbβ3 activation stimulated by low concentrations of thrombin. As a result, bleeding time is prolonged and thrombus formation is impaired Dab2 deficient mice40. Moreover, the combination of Arh and Dab2 is responsible for the majority of adaptor function in LDLR endocytosis and LDLR-mediated cholesterol homeostasis41. Thus, Dab2 could play a role in clotting and atherosclerosis associated with large vessel stroke.
PLCG2 (PLCgamma2) is tightly regulated to ensure efficient but limited platelet activation at sites of vascular injury42. Gain of function mutations in PLCG2 cause platelet hyperactivity and a prothrombotic state42. Platelet activation and thrombin generation are crucial steps in primary and secondary hemostasis. Thrombospondin-1 (TSP1) levels in human blood correlated with monocyte-platelet aggregates and thrombin generation indicating a pivotal role in regulating thrombosis43. Indeed, platelet-derived TSP1 modulates arterial thrombosis in vivo. Young patients with high cardiovascular risk and atherosclerosis have high levels of thrombospondin-1 platelet microparticles. Thrombospondin-1 levels correlate with carotid atherosclerotic plaque size and irregularity44; and thrombospondin-1 levels immediately after stroke predict six-month mortality and morbidity. Finally, TLN1 (talin 1) plays a role in αIIbβ3 inactivation in procoagulant platelets45 and thus also plays a role in coagulation that could be crucial for outcomes in ischemic large vessel stroke.
Limitations
This is a novel pilot study of human stroke showing a relationship between an HDAC9 polymorphism and whole genome gene expression differences. Sample size was small, thus evaluation in larger cohorts is warranted to confirm findings. There was no significant difference in phenotype between the few LVAS patients who were homozygous in comparison to LVAS patients who were heterozygous for the risk allele. Similarly, there was no significant difference in phenotype between the few VRFC who were homozygous in comparison to VRFC who were heterozygous for the risk allele. Due to the limited number of homozygous subjects (n=4), we were unable to analyze gene expression differences between homozygous and heterozygous subjects in the LVAS and VRFC groups. Future studies are needed to assess the possibility of an allelic dosage effect on gene expression. Previous studies have shown that the risk allele at rs2107595 associates only with LVAS, and not with any other stroke subtypes. However, future studies could utilize stroke patients without LVAS as controls in order to specifically assess the risk allele’s pro-atherogenic effects in comparison to the risk allele’s pro-stroke effects.
Additional studies are needed to assess the relationship between leukocytes, endothelial cells, and smooth muscle cells with the risk allele. We were unable to determine whether observed changes in leukocyte gene expression relate to HDAC9 risk allele regulation in leukocytes, or an indirect effect of leukocytes interaction with endothelium or vascular smooth muscles cells affected by HDAC9 risk allele. We unfortunately do not have data sufficiently detailed to evaluate whether infarction of the insula or other particular brain location may affect gene expression. This will be important to consider in future studies given potential sympathetic effects on the spleen and peripheral cells.
While HDAC9 risk allele positivity is associated with LVAS, additional genetic or environmental factors may be required to result in stroke. While not every gene has the same pattern of expression across LVAS patients, certain pathways are consistently over represented in these patients. Thus, RAP patients consistently over express genes involved in IL6 signaling, leukocyte recruitment, chronic inflammation, cholesterol efflux, and platelet aggregation. However, whether the identified gene genes and pathways directly increase stroke risk remains unclear. Experimental rodent stroke data support the importance of HDACs in stroke46–48 and the need for further studies to evaluate the role of HDAC9 in stroke.
Supplementary Material
Acknowledgments
This study was completed with funds from the American Heart Association (GCJ), and the National Institutes of Health (FRS, BS, GCJ: NS075035, NS079153, NS097000).
Footnotes
Compliance with ethical standards
Conflict of interest: all authors declare they have no conflicts of interest.
Ethical approval: all procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards. All patients or their surrogates provided Informed Consent for this study.
References
- 1.Azghandi S, Prell C, van der Laan SW, et al. Deficiency of the stroke relevant HDAC9 gene attenuates atherosclerosis in accord with allele-specific effects at 7p21.1. Stroke. 2015;46:197–202. doi: 10.1161/STROKEAHA.114.007213. [DOI] [PubMed] [Google Scholar]
- 2.Ferronato S, Gelati M, Scuro A, et al. HDAC9, TWIST1 and FERD3L gene expression in asymptomatic stable and unstable carotid plaques. Inflamm Res. 2016;65:261–263. doi: 10.1007/s00011-015-0904-z. [DOI] [PubMed] [Google Scholar]
- 3.Bellenguez C, Bevan S, Gschwendtner A, et al. Genome-wide association study identifies a variant in HDAC9 associated with large vessel ischemic stroke. Nat Genet. 2012;44:328–333. doi: 10.1038/ng.1081. 2012/02/07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Markus HS, Makela KM, Bevan S, et al. Evidence HDAC9 genetic variant associated with ischemic stroke increases risk via promoting carotid atherosclerosis. Stroke. 2013;44:1220–1225. doi: 10.1161/strokeaha.111.000217. 2013/03/02. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Traylor M, Farrall M, Holliday EG, et al. Genetic risk factors for ischaemic stroke and its subtypes (the METASTROKE Collaboration): a meta-analysis of genome-wide association studies. The Lancet Neurology. 2012;11:951–962. doi: 10.1016/S1474-4422(12)70234-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang XB, Han YD, Sabina S, et al. HDAC9 Variant Rs2107595 Modifies Susceptibility to Coronary Artery Disease and the Severity of Coronary Atherosclerosis in a Chinese Han Population. PloS one. 2016;11:e0160449. doi: 10.1371/journal.pone.0160449. 2016/08/06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhou X, Marks PA, Rifkind RA, et al. Cloning and characterization of a histone deacetylase, HDAC9. Proc Natl Acad Sci U S A. 2001;98:10572–10577. doi: 10.1073/pnas.191375098. 2001/09/06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wierda RJ, Geutskens SB, Jukema JW, et al. Epigenetics in atherosclerosis and inflammation. Journal of Cellular and Molecular Medicine. 2010;14:1225–1240. doi: 10.1111/j.1582-4934.2010.01022.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang BK, Lai X, Jia SJ. Epigenetics in atherosclerosis: a clinical perspective. Discovery medicine. 2015;19:73–80. 2015/03/01. [PubMed] [Google Scholar]
- 10.Cao Q, Rong S, Repa JJ, et al. Histone deacetylase 9 represses cholesterol efflux and alternatively activated macrophages in atherosclerosis development. Arteriosclerosis, thrombosis, and vascular biology. 2014;34:1871–1879. doi: 10.1161/atvbaha.114.303393. 2014/07/19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Irizarry RA, Hobbs B, Collin F, et al. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003;4:249–264. doi: 10.1093/biostatistics/4.2.249. 2003/08/20. [DOI] [PubMed] [Google Scholar]
- 12.Dykstra-Aiello C, Jickling GC, Ander BP, et al. Altered Expression of Long Noncoding RNAs in Blood After Ischemic Stroke and Proximity to Putative Stroke Risk Loci. Stroke. 2016;47:2896–2903. doi: 10.1161/strokeaha.116.013869. 2016/11/12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stamova B, Jickling GC, Ander BP, et al. Gene expression in peripheral immune cells following cardioembolic stroke is sexually dimorphic. PloS one. 2014;9:e102550. doi: 10.1371/journal.pone.0102550. 2014/07/19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bai Z, Stamova B, Xu H, et al. Distinctive RNA Expression Profiles in Blood Associated with Alzheimer’s Disease after Accounting for White Matter Hyperintensities. Alzheimer disease and associated disorders. 2014;28:226–233. doi: 10.1097/WAD.0000000000000022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schumann CM, Sharp FR, Ander BP, et al. Possible sexually dimorphic role of miRNA and other sncRNA in ASD brain. Molecular Autism. 2017;8:4. doi: 10.1186/s13229-017-0117-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ikeda K, Kundu RK, Ikeda S, et al. Glia maturation factor-gamma is preferentially expressed in microvascular endothelial and inflammatory cells and modulates actin cytoskeleton reorganization. Circulation research. 2006;99:424–433. doi: 10.1161/01.RES.0000237662.23539.0b. 2006/07/29. [DOI] [PubMed] [Google Scholar]
- 17.Strickland DK, Muratoglu SC. LRP in Endothelial Cells. A Little Goes a Long Way. 2016;36:213–216. doi: 10.1161/atvbaha.115.306895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mottet D, Bellahcene A, Pirotte S, et al. Histone deacetylase 7 silencing alters endothelial cell migration, a key step in angiogenesis. Circulation research. 2007;101:1237–1246. doi: 10.1161/circresaha.107.149377. 2007/10/20. [DOI] [PubMed] [Google Scholar]
- 19.Urbich C, Rossig L, Kaluza D, et al. HDAC5 is a repressor of angiogenesis and determines the angiogenic gene expression pattern of endothelial cells. Blood. 2009;113:5669–5679. doi: 10.1182/blood-2009-01-196485. 2009/04/09. [DOI] [PubMed] [Google Scholar]
- 20.Han X, Han X, Wang Z, et al. HDAC9 regulates ox-LDL-induced endothelial cell apoptosis by participating in inflammatory reactions. Frontiers in bioscience (Landmark edition) 2016;21:907–917. doi: 10.2741/4428. 2016/04/23. [DOI] [PubMed] [Google Scholar]
- 21.Shi W, Wei X, Wang Z, et al. HDAC9 exacerbates endothelial injury in cerebral ischaemia/reperfusion injury. J Cell Mol Med. 2016;20:1139–1149. doi: 10.1111/jcmm.12803. 2016/02/13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Narod SA. Modifiers of risk of hereditary breast cancer. Oncogene. 2006;25:5832–5836. doi: 10.1038/sj.onc.1209870. [DOI] [PubMed] [Google Scholar]
- 23.Shawky RM. Reduced penetrance in human inherited disease. Egyptian Journal of Medical Human Genetics. 2014;15:103–111. doi: 10.1016/j.ejmhg.2014.01.003. [DOI] [Google Scholar]
- 24.Sanford JA, Zhang L-J, Williams MR, et al. Inhibition of HDAC8 and HDAC9 by microbial short-chain fatty acids breaks immune tolerance of the epidermis to TLR ligands. Science Immunology. 2016;1 doi: 10.1126/sciimmunol.aah4609. [DOI] [PubMed] [Google Scholar]
- 25.Dje N’Guessan P, Riediger F, Vardarova K, et al. Statins control oxidized LDL-mediated histone modifications and gene expression in cultured human endothelial cells. Arteriosclerosis, thrombosis, and vascular biology. 2009;29:380–386. doi: 10.1161/atvbaha.108.178319. 2009/01/06. [DOI] [PubMed] [Google Scholar]
- 26.Göttlicher M, Minucci S, Zhu P, et al. Valproic acid defines a novel class of HDAC inhibitors inducing differentiation of transformed cells. The EMBO Journal. 2001;20:6969–6978. doi: 10.1093/emboj/20.24.6969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Phiel CJ, Zhang F, Huang EY, et al. Histone deacetylase is a direct target of valproic acid, a potent anticonvulsant, mood stabilizer, and teratogen. The Journal of biological chemistry. 2001;276:36734–36741. doi: 10.1074/jbc.M101287200. 2001/07/27. [DOI] [PubMed] [Google Scholar]
- 28.Foks AC, Lichtman AH, Kuiper J. Treating Atherosclerosis With Regulatory T Cells. Arteriosclerosis, Thrombosis, and Vascular Biology. 2014 doi: 10.1161/atvbaha.114.303568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stenina OI, Plow EF. Counterbalancing forces: What is thrombospondin-1 doing in atherosclerotic lesions? Circulation research. 2008;103:1053–1055. doi: 10.1161/CIRCRESAHA.108.188870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lopez-Dee Z, Pidcock K, Gutierrez LS. Thrombospondin-1: Multiple Paths to Inflammation. Mediators of Inflammation. 2011;2011:10. doi: 10.1155/2011/296069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Moura R, Tjwa M, Vandervoort P, et al. Thrombospondin-1 Deficiency Accelerates Atherosclerotic Plaque Maturation in ApoE−/− Mice. Circulation Research. 2008;103:1181–1189. doi: 10.1161/circresaha.108.185645. [DOI] [PubMed] [Google Scholar]
- 32.Meurs I, Calpe-Berdiel L, Habets KL, et al. Effects of deletion of macrophage ABCA7 on lipid metabolism and the development of atherosclerosis in the presence and absence of ABCA1. PloS one. 2012;7:e30984. doi: 10.1371/journal.pone.0030984. 2012/03/10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Karch CM, Goate AM. Alzheimer’s disease risk genes and mechanisms of disease pathogenesis. Biol Psychiatry. 2015;77:43–51. doi: 10.1016/j.biopsych.2014.05.006. 2014/06/22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tuteja S, Wang L, Dunbar RL, et al. Genetic coding variants in the niacin receptor, hydroxyl-carboxylic acid receptor 2, and response to niacin therapy. Pharmacogenetics and genomics. 2017 doi: 10.1097/fpc.0000000000000289. 2017/06/20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gonias SL, Campana WM. LDL receptor-related protein-1: a regulator of inflammation in atherosclerosis, cancer, and injury to the nervous system. The American journal of pathology. 2014;184:18–27. doi: 10.1016/j.ajpath.2013.08.029. 2013/10/17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lillis AP, Muratoglu SC, Au DT, et al. LDL Receptor-Related Protein-1 (LRP1) Regulates Cholesterol Accumulation in Macrophages. PloS one. 2015;10:e0128903. doi: 10.1371/journal.pone.0128903. 2015/06/11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Strickland DK, Au DT, Cunfer P, et al. Low-density lipoprotein receptor-related protein-1: role in the regulation of vascular integrity. Arteriosclerosis, thrombosis, and vascular biology. 2014;34:487–498. doi: 10.1161/atvbaha.113.301924. 2014/02/08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Geczy CL, Chung YM, Hiroshima Y. Calgranulins may contribute vascular protection in atherogenesis. Circulation journal: official journal of the Japanese Circulation Society. 2014;78:271–280. doi: 10.1253/circj.cj-13-1505. 2014/01/07. [DOI] [PubMed] [Google Scholar]
- 39.Averill MM, Kerkhoff C, Bornfeldt KE. S100A8 and S100A9 in cardiovascular biology and disease. Arteriosclerosis, thrombosis, and vascular biology. 2012;32:223–229. doi: 10.1161/atvbaha.111.236927. 2011/11/19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tsai HJ, Tseng CP. The adaptor protein Disabled-2: new insights into platelet biology and integrin signaling. Thrombosis journal. 2016;14:28. doi: 10.1186/s12959-016-0101-5. 2016/10/22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tao W, Moore R, Meng Y, et al. Endocytic adaptors Arh and Dab2 control homeostasis of circulatory cholesterol. Journal of lipid research. 2016;57:809–817. doi: 10.1194/jlr.M063065. 2016/03/24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Elvers M, Pozgaj R, Pleines I, et al. Platelet hyperreactivity and a prothrombotic phenotype in mice with a gain-of-function mutation in phospholipase Cgamma2. Journal of thrombosis and haemostasis: JTH. 2010;8:1353–1363. doi: 10.1111/j.1538-7836.2010.03838.x. 2010/03/17. [DOI] [PubMed] [Google Scholar]
- 43.Gremmel T, Ay C, Riedl J, et al. Platelet-specific markers are associated with monocyte-platelet aggregate formation and thrombin generation potential in advanced atherosclerosis. Thrombosis and haemostasis. 2016;115:615–621. doi: 10.1160/th15-07-0598. 2015/10/16. [DOI] [PubMed] [Google Scholar]
- 44.Della-Morte D, Beecham A, Dong C, et al. Association between variations in coagulation system genes and carotid plaque. Journal of the neurological sciences. 2012;323:93–98. doi: 10.1016/j.jns.2012.08.020. 2012/09/18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mattheij NJ, Gilio K, van Kruchten R, et al. Dual mechanism of integrin alphaIIbbeta3 closure in procoagulant platelets. The Journal of biological chemistry. 2013;288:13325–13336. doi: 10.1074/jbc.M112.428359. 2013/03/23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Park MJ, Sohrabji F. The histone deacetylase inhibitor, sodium butyrate, exhibits neuroprotective effects for ischemic stroke in middle-aged female rats. Journal of neuroinflammation. 2016;13:300. doi: 10.1186/s12974-016-0765-6. 2016/12/03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kassis H, Shehadah A, Li C, et al. Class IIa histone deacetylases affect neuronal remodeling and functional outcome after stroke. Neurochemistry international. 2016;96:24–31. doi: 10.1016/j.neuint.2016.04.006. 2016/04/23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yildirim F, Ji S, Kronenberg G, et al. Histone acetylation and CREB binding protein are required for neuronal resistance against ischemic injury. PloS one. 2014;9:e95465. doi: 10.1371/journal.pone.0095465. 2014/04/22. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.