

Abstract
Background
Identification of pathogenic germline alterations (PGAs) has important clinical and therapeutic implications in pancreas cancer. We performed comprehensive germline testing (GT) in an unselected prospective cohort of patients with exocrine pancreatic neoplasms with genotype and phenotype association to facilitate identification of prognostic and/or predictive biomarkers and examine potential therapeutic implications.
Methods
Six hundred fifteen unselected patients with exocrine pancreatic neoplasms were prospectively consented for somatic tumor and matched sample profiling for 410–468 genes. GT for PGAs in 76 genes associated with cancer susceptibility was performed in an “identified” manner in 356 (57.9%) patients and in an “anonymized” manner in 259 (42.1%) patients, using an institutional review board–approved protocol. Detailed clinical and pathological features, response to platinum, and overall survival (OS) were collected for the identified cohort. OS was analyzed with Kaplan-Meier curves.
Results
PGAs were present in 122 (19.8%) of 615 patients involving 24 different genes, including BRCA1/2, ATM, PALB2, and multiple additional genes associated with the DNA damage response pathway. Of 122 patients with germline alterations, 41.8% did not meet current guidelines for GT. The difference in median OS was not statistically significant between patients with and without PGA (50.8 months, 95% confidence interval = 34.5 to not reached, two-sided P = .94). Loss of heterozygosity was found in 60.0% of BRCA1/2.
Conclusions
PGAs frequently occur in pancreas exocrine neoplasms and involve multiple genes beyond those previously associated with hereditary pancreatic cancer. These PGAs are therapeutically actionable in about 5% to 10% of patients. These data support routinely offering GT in all pancreatic ductal adenocarcimona patients with a broad panel of known hereditary cancer predisposition genes.
Pancreatic ductal adenocarcinoma (PDAC) has an overall five-year survival rate of about 8% and an annual disease-specific mortality that approaches the rising incidence of the disease (1). Currently, standard therapies for patients with metastatic PDAC and a good performance status consist of cytotoxic regimens of FOLFIRINOX or gemcitabine and nab-paclitaxel; median overall survival remains less than a year (2,3). Germline and somatic genetic testing or profiling (GT and ST, respectively) of PDAC is an emerging focus of current research and is paving the way for optimizing and refining currently available treatment options and the development of novel therapeutics (4–11).
The clinical and therapeutic implications of pathogenic germline alterations (PGAs) in PDAC have been most studied in relation to BRCA1/2 due to their high prevalence among PDAC (4–11). Specifically, the activity of PARP inhibitors in tumors defective in BRCA1/2 through a synthetically lethal interaction has spurred the development of drugs designed to exploit an acquired or inherited deficiency in double-stranded DNA break repair for therapeutic benefit (12–16). Several studies have reported encouraging results utilizing platinum agents with or without PARP inhibition in the setting of PDAC patients with BRCA1/2 mutations, and the spectrum of germline and somatic alterations previously identified in genes associated with the DNA damage response pathway suggests that there is a wider patient population with BRCA-like tumor characteristics who may benefit from a similar approach (13–17).
Genes implicated in predisposition to PDAC include BRCA1/2, ATM, PALB2, STK11, MLH1, MSH2, MSH6, PMS2, and CDKN2A, with other genes such as BARD1, ATR, RAD51, and CHEK2 having been suggested as potential candidate risk genes (4–11). Routine screening for PGAs in unselected patients with PDAC is currently not standard of care; rather, screening for PGAs is usually performed on a limited and targeted basis. The National Comprehensive Cancer Network (NCCN) and American College of Medical Genetics and Genomics (ACMG) recommend screening for PGAs in PDAC patients who have a strong family history of pancreatic, breast, and ovarian cancer; patients who meet criteria for familial pancreatic cancer (FPC)/Lynch syndrome (LS)/BRCA/familial atypical multiple mole (FAMM) syndrome; and those of Ashkenazi Jewish ancestry (18,19).
Notably, however, recent studies have identified multiple cohorts of unselected patients with PDAC who harbor PGA without having a family history or being of Ashkenazi Jewish descent (20–23). Furthermore, these patients did not meet current guidelines for screening, making it increasingly apparent that current screening guidelines do not adequately capture all patients with PGAs. Given the profound clinical implications that identifying PGAs may have, it is of critical relevance to reevaluate current screening guidelines for PGAs in PDAC. To further explore these observations, we sought to evaluate the spectrum of PGAs in patients with exocrine pancreatic neoplasms, evaluate therapeutic opportunities based on GT, assess the opportunity to identify at-risk family members, and assess and compare clinical outcomes among patients with and without PGAs.
We undertook a prospective study of PGAs among patients with pancreatic exocrine cancers receiving care at Memorial Sloan Kettering Cancer Center (MSKCC), unselected by personal and family history of cancer, ethnicity or age, using the Memorial Sloan Kettering–Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT) next-generation sequencing (NGS) platform.
Methods
Patients
Between January 2014 and July 2017, informed consent for ST under an institutional review board–approved protocol (NCT01775072) was prospectively obtained from 615 unselected patients with a diagnosis of exocrine pancreatic neoplasm. From November 2015 onward, informed consent was also prospectively obtained from patients for GT under the same protocol. Three hundred fifty-six (57.9%) patients consented to ST and GT, and 259 (42.1%) patients consented to only ST. Of note, all 336 patients in Lowery et al. (2017) were included in this cohort; however, these patients were not analyzed for PGAs in that report (24). Also, 162 (of 615) patients in the current cohort, including 39 who had PGAs, overlapped with Mandelker et al.’s (2017) cohort (22). The treating oncologist assessed clinical data and therapeutic responses. All patients who consented to ST were included in this study. ST across the full panel of 410–468 genes was performed on tumor samples obtained from resection or biopsy. GT performed on germline DNA obtained from normal peripheral blood was limited to 76 genes (Supplementary Table 1, available online). Clinical, demographic, and pathologic information was collected on all patients in the study. However, information was not available for analysis in 259 patients who consented to only ST as their data were anonymized prior to linkage with genetic testing results, which is described below.
Sequencing, Variant Calling, and Results Reporting
DNA was isolated from tumor tissue and matched normal peripheral blood and was analyzed through a hybridization capture-based NGS assay to identify nucleotide variants, small insertions and deletions, copy number variations, and structural rearrangements, as previously described (25,26). For 356 patients who consented to both ST and GT, variants were called using a combination of MuTect and Genomic Analysis Toolkit (GATK) HaplotypeCaller, with mapping and base quality scores of 20 or higher, 25% variant frequency, and 20× coverage thresholds (27,28). Copy number variants were identified using an in-house developed pipeline and were confirmed by additional laboratory tests (29). For these patients, somatic and germline variants were reported to treating physician and patients.
For 259 patients who consented only to ST, only somatic variants were reported to the treating physician and patients. To evaluate and associate these patients’ PGAs with their respective clinical data, the clinical and genomic data were anonymized and linked through an institutional review board–approved protocol. Clinical data were “binned” to remove any possible unique identifying values, and then linked to binary alignment map (.bam) files of the nontumor DNA with a unique study identifier prior to variant calling. Identifying protected health information was deleted before generation of variant call format files from the corresponding .bam files. A custom pipeline then analyzed the germline data for this anonymized cohort subset in order to call variants (details provided in the Supplementary Methods, available online).
All variants identified were classified based on the ACMG criteria. Only pathogenic and likely pathogenic variants are reported in the analysis herein.
Loss of Heterozygosity Analysis
Loss of heterozygosity (LOH) was analyzed on tumor samples from the initial GT and matched normal blood sample. Seventy-five patients with PGAs were analyzed. Using the FACETS algorithm, we determined total and allele-specific copy number from the sequence coverage and genotypes of polymorphic single nucleotide polymorphisms across the genome, for which we can identify regions of LOH (30). Zygosity of each pathogenic PGA was inferred in the tumor according to its variant allele fraction and the FACETS segment on which it occurred.
Statistical Analysis
Kaplan-Meier curves (KM) were generated, and median overall survival (OS) was compared with log-rank testing. Categorical variables were compared using the Fisher exact test. P values of less than .05 were considered statistically significant. All statistical tests were two-sided.
Results
PGAs Identified
The median time for somatic data return was 45 days from time of consent and 20 days from time of DNA extraction from tumor tissue (24). Germline data results were typically available within seven days of somatic results. The demographics of the entire patient cohort (n = 615) are summarized in Table 1. Five hundred fifty (89.4%) patients self-identified as white, and patients of known Ashkenazi Jewish heritage comprised 20.2% of this group. At least one PGA was identified in 122 (19.8%) patients, and of that, 108 (88.5%) patients had PDAC. Ashkenazi Jewish patients had the highest frequency of PGAs, at 36.0%. The frequencies of PGAs in Caucasian patients (including Ashkenazi Jewish), black/Hispanic/Latino patients, and Asian patients were 19.5%, 24.3%, and 21.4%, respectively. Black and Hispanic/Latino patients were analyzed together given low numbers.
Table 1.
Demographics and clinical characteristics of entire cohort of patients with exocrine pancreatic cancer
| Clinical characteristics | No. of patients (%) (n = 615) | PGA No. of patients (%) |
|
|---|---|---|---|
| Yes | No | ||
| Median age at diagnosis, y | |||
| <40 | 28 (4.6) | 9 (32.1) | 19 (67.9) |
| 40–50 | 54 (8.8) | 14 (25.9) | 40 (74.1) |
| 50–60 | 141 (22.9) | 37 (26.2) | 104 (73.8) |
| 60–70 | 218 (35.4) | 39 (17.9) | 179 (82.1) |
| 70–80 | 148 (24.1) | 20 (13.5) | 128 (86.5) |
| >80 | 26 (4.2) | 3 (11.5) | 23 (88.5) |
| Median age at diagnosis, y | 65 | 60 | 66 |
| PGA present | 122 (19.8) | ||
| PGA negative | 493 (80.2) | ||
| Sex | |||
| Male | 325 (52.8) | 71 (21.8) | 254 (78.2) |
| Female | 290 (47.2) | 51 (17.6) | 239 (82.4) |
| Race | |||
| White | 550 (89.4) | 107 (19.5) | 443 (80.5) |
| Non-Ashkenazi Jewish | 439 (79.8) | 67 (15.3) | 372 (84.7) |
| Ashkenazi Jewish | 111 (20.2) | 40 (36.0) | 71 (64.0) |
| Black/Hispanic/Latino | 37 (6.0) | 9 (24.3) | 28 (75.7) |
| Asian | 28 (4.6) | 6 (21.4) | 22 (78.6) |
| Smoking status | |||
| Yes | 307 (49.9) | 57 (18.6) | 250 (81.4) |
| No | 308 (50.1) | 65 (21.1) | 243 (78.9) |
| Diabetes | |||
| Yes | 139 (22.6) | 25 (18.0) | 114 (82.0) |
| No | 476 (77.4) | 97 (20.4) | 379 (79.6) |
| Stage at diagnosis | |||
| IPMN | 6 (0.9) | 2 (33.3) | 4 (66.7) |
| I/II | 271 (44.1) | 44 (16.2) | 227 (83.8) |
| III | 89 (14.5) | 18 (20.2) | 71 (79.8) |
| IV | 249 (40.5) | 58 (23.3) | 191 (76.7) |
| Pathology | |||
| Adenocarcinoma | 554 (90.1) | 108 (19.5) | 446 (80.5) |
| Adenosquamous carcinoma | 17 (2.8) | 4 (23.5) | 13 (76.5) |
| Acinar cell carcinoma of the pancreas | 17 (2.8) | 3 (17.6) | 14 (82.4) |
| Undifferentiated | 7 (1.1) | 1 (14.3) | 6 (85.7) |
| MCN/ITPN/MPAC/IPMN/COLLOID/SPNP* | 20 (3.3) | 6 (30.0) | 14 (70.0) |
COLLOID = colloid intraductal papillary mucinous neoplasm; IPMN = intrapapillary mucinous neoplasm; ITPN = intraductal tubullopapillary neoplasm; MCN = mucinous cystic neoplasm; MPAC = mixed pancreatic and acinar cell carcinoma of the pancreas; PGA = pathogenic germline alteration; SPNP = solid pseudopapillary neoplasm of the pancreas.
Fifty-one (41.8%) patients with PGAs did not meet current guidelines for GT. Table 2 shows the number of guidelines that patients who were found to have PGAs met for screening under NCCN hereditary breast and ovarian cancer (HBOC) and/or ACMG HBOC/FPC/LS/FAMM guidelines for screening (17,18).
Table 2.
Screening guidelines met by patients with and without PGA*
| Guidelines met (NCCN HBOC, ACMG: HBOC, FPC, LS, FAMM) | PGA, No. of patients (%) |
Total No. of patients (%) | |
|---|---|---|---|
| Yes | No | ||
| 0 | 51 (41.8) | 362 (73.4) | 413 (67.2) |
| 1 | 12 (9.8) | 32 (6.5) | 44 (7.2) |
| 2 | 32 (26.2) | 55 (11.1) | 87 (14.1) |
| 3 | 23 (18.9) | 43 (8.7) | 66 (10.7) |
| 4 | 3 (2.5) | 1 (0.2) | 4 (0.7) |
| 5 | 1 (0.8) | — | 1 (0.2) |
| Total | 122 (19.8) | 493 (80.2) | 615 |
— denotes 0. ACMG = American College of Medical Genetics and Genomics; FAMM = familial atypical multiple mole melanoma syndrome; FPC = familial pancreatic cancer; HBOC = hereditary breast ovary cancer; LS = Lynch syndrome; NCCN = The National Comprehensive Cancer Network; PGA = pathogenic germline alteration.
Fifty (8.1%) patients had a BRCA1 or BRCA2 mutation; 35 (5.7%) had a BRCA2 mutation, and 15 (2.4%) had a BRCA1 mutation. Among the 111 Ashkenazi Jewish patients, 18% harbored one of the three common BRCA Ashkenazi Jewish founder’s mutations (Tables 3 and 4). Twenty-six (52.0%) of the 50 patients with BRCA1/2 had one of the three common BRCA Ashkenazi Jewish founder’s mutations. PGAs were found in an additional 22 genes, including high-penetrance genes such as ATM (n = 11, 1.8%) and CDKN2A (n = 6, 1.0%). See Table 3 and Supplementary Figure 1 (available online) for further details of other PGAs found. Two PGAs occurring in separate genes were found in nine patients (Table 4).
Table 3.
PGA details: Founder status, number who met greater than 1 criteria, and penetrance
| Gene (transcript) | No. | Founder | Met ≥1 guideline | Penetrance |
|---|---|---|---|---|
| APC (NM_000038) | 13 | Yes | 7 | Low |
| c.3920T>A (p.Ile1307Lys) | ||||
| ATM (NM_000051) | 11 | 4 | Moderate | |
| c.5908C>T (p.Gln1970*) | 1 | Yes | ||
| c.7630-2A>C | 1 | No | ||
| Intragenic del ex62-63 | 1 | No | ||
| c.103C>T (p.Arg35*) | 1 | Yes | ||
| c.1027_1030del GAAA (p.Glu343Ilefs*2) | 1 | No | ||
| c.1057_1058delTG (p.Cys353Serfs*5) | 1 | No | ||
| c.2413C>T (p.Arg805*) | 1 | No | ||
| c.5712dupA (p.Ser1905Ilefs*25) | 1 | No | ||
| c.5712dupA (p. Ser1905llefs*25) | 1 | No | ||
| c.1285_1288delAACT (p.Asn429Valfs*7) | 1 | No | ||
| c.3340A>T (p.Lys1114*) | 1 | No | ||
| BARD1 (NM_000465) | 1 | 1 | Uncertain | |
| c.1690C>T (p.Gln564*) | 1 | Unknown | ||
| BLM (NM_000057) | 3 | 2 | Recessive | |
| c.2207_2212delATCTGAinsTAGATTC (p.Tyr736Leufs*5) | 1 | Yes | ||
| c.1642 C>T (p.Gln548*) | 2 | Unknown | ||
| BRCA1 (NM_007294) | 13 | 13 | High | |
| c.427G>T (p.Glu143*) | 1 | No | ||
| c.4986 + 5G>A | 1 | No | ||
| c.5266dupC (p.Gln1756Profs*74)† | 2 | Yes | ||
| c.65T>C (p.Leu22Ser) | 1 | No | ||
| c.4524G>A (p.Trp1508*) | 1 | No | ||
| c.68_69delAG (p.Glu23Valfs*17)† | 5 | Yes | ||
| c.181T>G (p.Cys61Gly) | 1 | Yes | ||
| c.798_799delTT (p.Ser267Lysfs*19) | 1 | No | ||
| BRCA2 (NM_000059) | 31 | 20 | High | |
| c.5946delT (p.Ser1982Argfs*22)† | 15 | Yes | ||
| c.4829_4830delTG (p.Val1610Glyfs*4) | 1 | No | ||
| c.5569delG (p.Glu1857Lysfs*6) | 1 | No | ||
| c.4638delT (p.Phe1546Leufs*22) | 1 | No | ||
| c.793 + 1G>A | 1 | No | ||
| c.9371A>T (p.Asn3124Ile) | 1 | Yes | ||
| c.5614A>T (p.Lys1872*) | 1 | No | ||
| c.6259delA (p.Arg2087Glufs*32) | 1 | No | ||
| c.3847_3848del (p.Val1283Lysfs*2) | 2 | No | ||
| c.3264dupT (p.Gln1089Serfs*10) | 1 | No | ||
| c.9070_9073delAACA (p.Asn3024Tyrfs*3) | 1 | No | ||
| c.2830A>T (p.Lys944*) | 1 | No | ||
| c.8537_8538delAG (p.Glu2846Glyfs*22) | 1 | No | ||
| c.2330dupA (p.Asp777Glufs*11) | 1 | No | ||
| c.9810dupC (p.Asp3272Glyfs*5) | 1 | No | ||
| c.1405_1406delGA (p.Asp469*) | 1 | No | ||
| CDKN2A (NM_000077) | 6 | 3 | High | |
| c.340_355delCCCGTGGACCTGGCTG (p.Pro114Argfs*27) | 1 | No | ||
| c.457G>C (p.Asp153His) | 1 | No | ||
| c.143C>G (p.Pro48Arg) | 1 | No | ||
| c.71G>C (p.Arg24Pro) | 2 | No | ||
| c.176T>G (p.Val59Gly) | 1 | No | ||
| CHEK2 (NM_007194) | 12 | 7 | Moderate | |
| c.1111C>T (p.His371Tyr) | 1 | No | ||
| c.470T>C (p.Ile157Thr) | 4 | No | ||
| c.1283C>T (p.Ser428Phe) | 4 | Yes | ||
| c.1100delC (p.Thr367Metfs*15) | 1 | Yes | ||
| c.470T>C (p.Ile157Thr) | 1 | No | ||
| c.1283C>T (p.Ser428Phe) | 1 | Yes | ||
| FH (NM_000143) | 1 | 1 | Recessive | |
| FH c.1431_1433dupAAA (p.Lys477dup) | 1 | Unknown | ||
| MITF (NM_000248) | 2 | 1 | Moderate | |
| MITF c.952G>A (p.Glu318Lys) | 2 | |||
| MLH1 (NM_000249) | 1 | No | 1 | High |
| c.1731G>A (p.Ser577=) | 1 | |||
| MSH2 (NM_000251) | 1 | Unknown | 1 | High |
| c.1906G>C (p.Ala636Pro) | 1 | |||
| MSH6 (NM_000179) | 1 | Unknown | High | |
| c.3268G>T (p.Glu1090*) | 1 | |||
| MUTYH (NM_001128425) | 6 | 1 | Low | |
| c.1356delA (p.Tyr453Ilefs*14) | 1 | |||
| c.1187G>A (p.Gly396Asp) | 4 | YES | ||
| c.536A>G (p.Tyr179Cys) | 1 | YES | ||
| NBN (NM_002485) | 1 | 1 | Moderate | |
| c.1903A>T (p.Lys635*) | 1 | No | ||
| NF1 (NM_000267) | 1 | 1 | High | |
| c.4600C>T (p.Arg1534*) | 1 | |||
| PALB2 (NM_024675) | 1 | High | ||
| c.2727_2728delTT (p.Thr911Leufs*16) | 1 | |||
| PMS2 (NM_000535) | 2 | High | ||
| deletions exons11-14 | 1 | |||
| c.1076dupT (p.Leu359Phefs) | 1 | No | ||
| RAD50 (NM_005732) | 2 | Uncertain | ||
| c.832C>T (p.Arg278*) | 1 | |||
| c.3655G>T (p.Glu1219*) | 1 | No | ||
| RAD51D (NM_002878) | 1 | No | Moderate | |
| c.263 + 1G>A | 1 | |||
| RECQL4 (NM_004260) | 1 | No | Recessive | |
| c.2077C>T (p.Gln693*) | 1 | |||
| STK11 (NM_000455) | 1 | No | High | |
| c.630C>A (p.Cys210*) | 1 | |||
| TP53 (NM_000546) | 1 | No | High | |
| c.844C>T (p.Arg282Trp) | 1 |
These three variants represent known Ashkenazi founder mutations: NM_007294 (BRCA1): c·5266dupC (p·Gln1756Profs*74), also known as c·5382insC and c·5385insC, NM_007294 (BRCA1): c·68_69delAG (p·Glu23Valfs*17), also known as BRCA1 c·185delAG or c·187delAG, NM_000059 (BRCA2): c·5946delT (p·Ser1982Argfs*22), also known as c·6174delT. PGA = pathogenic germline alteration.
Table 4.
Clinical characteristics and screening guidelines met in patients with two PGAs
| Gene | Nucleotide | Age, y | Sex | Stage | Family history | Guidelines met | Ethnicity |
|---|---|---|---|---|---|---|---|
| APC + CHEK2 | c.3920T>A (p.Ile1307Lys) + c.1283C>T (p.Ser428Phe) | 60–70 | Female | III | Colon, breast | NCCN HBOC, ACMG (HBOC) | Ashkenazi Jewish |
| BRCA2 + CHEK2 | c.5946delT (p.Ser1982Argfs*22) + c.1283C>T (p.Ser428Phe) | 45–60 | Male | IV | Prostate, breast | NCCN HBOC, ACMG (HBOC) | Ashkenazi Jewish |
| BRCA2 + CHEK2 | c.9148C>T (p.Gln3050*) + c.1283C>T (p.Ser428Phe) | 60–70 | Female | I/II | Breast | NCCN HBOC, ACMG (HBOC) | White |
| BRCA2 + APC | c.5946delT (p.Ser1982Argfs*22) + c.3920T>A (p.Ile1307Lys) | 60–70 | Male | IV | Breast, Pancreas, Ovary | NCCN HBOC, ACMG (HBOC, FPC, LS) | Ashkenazi Jewish |
| BRCA2 + PMS2 | c.6468_6469del (p.Gln2157Ilefs*18) + c.1831delinsTT (p.Ile611Phefs*2) | 70–80 | Male | I/II | Bladder | N | White |
| FAM175 A+ MUTYH† | c.442C>T (p.Arg148*) + -c.1187G>A (p.Gly396Asp) | 56–60 | Male | III | Pancreas, Breast, Ovary | NCCN HBOC, ACMG (HBOC, FPC, LS) | White |
| BRCA1 + CHEK2 | c.68_69delAG (p.Glu23Valfs*17) + c.1283C>T (p.Ser428Phe) | >80 | Male | IV | Y | ACMG (FPC, HBOC), NCCN HBOC | Ashkenazi Jewish |
| BRCA1 + BLM | c.68_69delAG (p.Glu23Valfs*17)+ c.2207_2212delATCTGAinsTAGATTC (p.Tyr736Leufs*5) | 51–55 | Female | I/II | Y | ACMG (FPC, HBOC), NCCN HBOC | Ashkenazi Jewish |
| PMS2+ CHEK2 | c.1076dupT (p.Leu359Phefs*6)+ c.1283C>T (p.Ser428Phe) | 60–70 | Female | IV | Y | N | White |
FAM175A Transcript (NM_139076). ACMG = American College of Medical Genetics and Genomics; FPC = familial pancreatic cancer; HBOC = hereditary breast ovary cancer; LS = Lynch syndrome; NCCN = The National Comprehensive Cancer Network; PGA = pathogenic germline alteration.
Supplementary Table 2 details the demographic data for identified patients with BRCA1/2 mutations (n = 32). Of the 32 patients, 16 had Ashkenazi Jewish founder’s mutations. Two (12.5%) of these 16 patients identified as non-Ashkenazi Jewish. Supplementary Table 3 details the specific mutation, founder mutation status, and guidelines met for screening. Five (22.7%) patients with BRCA2 mutations did not fall under any guidelines for screening (Supplementary Table 4, available online).
Response to Platinum in Patients With Advanced PDAC by PGA Presence and Type
Of 493 patients (80.2%) without PGA, 287 (58.2%) received platinum for advanced disease and 102 (35.5%) had a therapeutic response. Among 122 patients with PGAs, 84 (68.9%) received platinum for advanced disease, with 48 (57.1%) patients having treatment response. Twenty-nine (76.3%) of 38 BRCA1/2 patients who received platinum had treatment response. Table 5 and Supplementary Table 5 (available online) delineate response rates to platinum for advanced disease among all patients with PGAs.
Table 5.
Platinum response for patients with pathogenic germline alterations vs without PGAs*
| Gene | No. (%) | Received platinum, No. (%) | Response, No. (%) |
|---|---|---|---|
| No pathogenic germline alteration | 493 | 287 (58.2) | 102 (35.5) |
| Pathogenic germline alteration identified | |||
| BRCA2 | 31 (25.4) | 28 (90.3) | 21 (75.0) |
| CHEK2 | 14 (11.5) | 8 (57.1) | 2 (25.0) |
| APC | 13 (10.7) | 5 (38.5) | 1 (20.0) |
| BRCA1 | 12 (9.8) | 8 (66.7) | 7 (87.5) |
| ATM | 11 (9.0) | 9 (81.8) | 5 (55.6) |
| CDKN2A | 6 (4.9) | 5 (83.3) | 2 (40.0) |
| MUTYH | 6 (4.9) | 4 (66.7) | 3 (75.0) |
| BLM | 3 (2.5) | 1 (33.3) | — |
| RAD50 | 2 (1.6) | 2 (100.0) | — |
| MITF | 2 (1.6) | 1 (50.0) | — |
| BRCA2 + CHEK2 | 2 (1.6) | — | — |
| APC + CHEK2 | 1 (0.8) | 1 (100.0) | 1 (100.0) |
| BRCA2 + APC | 1 (0.8) | 1 (100.0) | 1 (100.0) |
| FH | 1 (0.8) | 1 (100.0) | 1 (100.0) |
| NF1 | 1 (0.8) | 1 (100.0) | 1 (100.0) |
| PMS2 | 1 (0.8) | 1 (100.0) | 1 (100.0) |
| STK11 | 1 (0.8) | 1 (100.0) | 1 (100.0) |
| TP53 | 1 (0.8) | 1 (100.0) | 1 (100.0) |
| RECQL4 | 1 (0.8) | 1 (100.0) | — |
| PMS2 + CHEK2 | 1 (0.8) | 1 (100.0) | — |
| NBN | 1 (0.8) | 1 (100.0) | — |
| BRCA1 + CHEK2 | 1 (0.8) | 1 (100.0) | — |
| MLH1 | 1 (0.8) | 1 (100.0) | — |
| MSH6 | 1 (0.8) | 1 (100.0) | — |
| MSH2 | 1 (0.8) | — | — |
| BRCA1 + BLM | 1 (0.8) | — | — |
| RAD51D | 1 (0.8) | — | — |
| BARD1 | 1 (0.8) | — | — |
| PALB2 | 1 (0.8) | — | — |
| BRCA2 + PMS2 | 1 (0.8) | — | — |
| FAM175A+MUTYH | 1 (0.8) | — | — |
| Total | 122 | 84 (68.9) | 48 (57.1) |
— denotes 0. PGA = pathogenic germline alteration.
Clinical Outcomes
For 356 patients who consented to GT, there was no statistically significant difference in median OS between patients with PGAs, who had a median OS of 48.4 months (95% CI = 26.3 to not reached), and patients without PGAs, who had a median OS of 50.8 months (95% CI = 34.5 to not reached, P = .94) (Figure 1). When restricted to patients with metastatic/recurrent disease, no statistically significant difference was found between patients with PGAs, who had a median OS of 33.5 months (95% CI = 16.7 to 53.3), and patients without PGAs, who had a median OS of 23.1 months (95% CI = 17.5 to 29.2, P = .42) (Figure 2). No statistically significant difference was found between patients with BRCA mutations, who had a median OS of 53.3 months (95% CI = 23.9 to infinity), and patients without BRCA mutations, who had a median OS of 48.4 months (95% CI = 34.5 to infinity, P = .47) (Supplementary Figure 2, available online). No statistically significant difference was found between BRCA patients with LOH, who had a median OS of 53.3 months (95% CI = 16.4 to 53.3), and patients without LOH, who had a median OS that was not reached (P = .48) (Supplementary Figure 3, available online).
Figure 1.
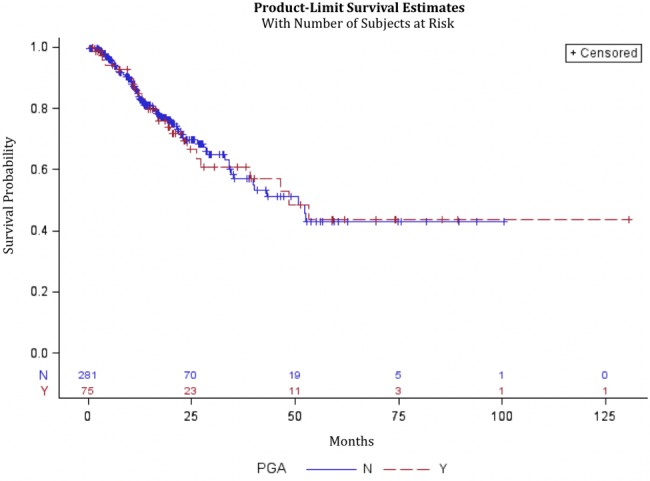
Kaplan-Meier curve for patients who consented to germline testing with and without pathogenic germline mutations from diagnosis date. Data from 356 patients were analyzed, 281 without pathogenic germline alterations (PGAs) and 75 with PGAs. PGA = pathogenic germline alteration.
Figure 2.
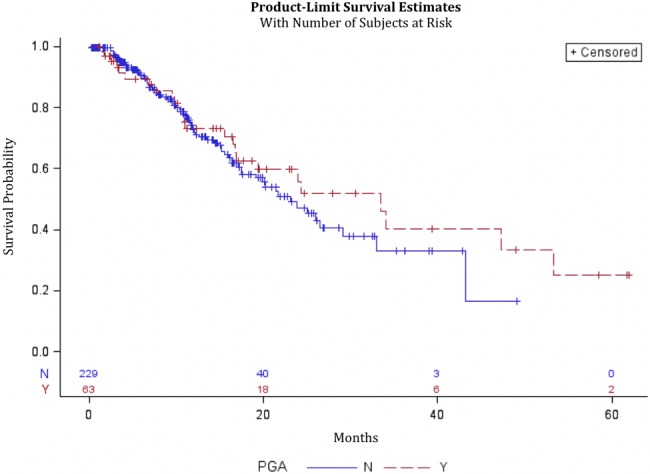
Kaplan-Meier curve for patients with and without pathogenic germline mutations, restricted to those with metastatic or recurrent disease who consented to germline testing. Data from 292 patients were analyzed, 229 without pathogenic germline alterations (PGAs) and 63 without PGAs. PGA = pathogenic germline alteration.
LOH Analysis
Table 6 details the number of wild-type (WT), LOH, and homozygous deletions in 75 identified patients with adequate tissue for analysis. Most notably, LOH was found in 12 (60.0%) BRCA2 mutations and six (60.0%) BRCA1 mutations. Six (30.0%) BRCA2 mutations and three (30.0%) BRCA1 mutations were WT. ATM also had substantial LOH, with five (62.5%) with LOH and two (25.0%) WT.
Table 6.
Loss of heterozygosity in PGAs*
| Gene | Wild-type, No. (%) | LOH, No. (%) | Copy neutral LOH, No. (%) | Homozygous deletion, No. (%) | Indeterminate, No. (%) | Total, No. |
|---|---|---|---|---|---|---|
| PMS2 | 1 (100.0) | — | — | — | — | 1 |
| APC | 8 (100.0) | — | — | — | — | 8 |
| ATM | 2 (25.0) | 3 (37.5) | 2 (25.0) | — | 1 (12.5) | 8 |
| BARD1 | 1 (100.0) | — | — | — | — | 1 |
| BLM | 2 (100.0) | — | — | — | — | 2 |
| BRCA1 | 3 (30.0) | 3 (30.0) | 3 (30.0) | 1 (10.0) | — | 10 |
| BRCA2 | 6 (30.0) | 9 (45.0) | 3 (15.0) | 1 (5.0) | 1 (5.0) | 20 |
| CDKN2A | 4 (66.7) | 1 (16.7) | — | — | 1 (16.7) | 6 |
| CHEK2 | 7 (58.3) | 2 (16.7) | 2 (16.7) | — | 1 (8.3) | 12 |
| FH | — | 1 (100.0) | — | — | — | 1 |
| MITF | 1 (100.0) | — | — | — | — | 1 |
| MUTYH | — | — | — | — | 2 (100.0) | 2 |
| NF1 | — | 1 (100.0) | — | — | — | 1 |
| PALB2 | 1 (100.0) | — | — | — | — | 1 |
| RAD50 | 1 (100.0) | — | — | — | — | 1 |
— denotes 0. LOH = loss of heterozygosity; PGA = pathogenic germline alteration.
Discussion
Previous observations have established that a small but clinically significant number of patients with PDAC harbor deleterious PGAs in BRCA1/2 and several other rare but high-penetrance genes, predominantly through study of patients with a personal or family history of cancer or of an ethnic background associated with increased carrier frequency of hereditary cancer predisposition syndromes (7–9,20–23).
Several publications have indicated a higher incidence of PGAs in a number of high- and moderate-penetrance genes that were not previously associated with an increased lifetime risk of pancreatic cancer incidence (21–23). We report results of GT using a 76-gene panel in an unselected population of 615 patients with exocrine pancreas neoplasms, primarily PDAC. We found at least one PGA in 122 (19.8%) of patients, the majority of whom were between age 50 and 60 years and were of Caucasian ethnicity without known Ashkenazi Jewish heritage. Notably, 41.8% of patients who had a confirmed PGA did not meet any screening criteria for GT in PDAC. While PGAs were most frequently identified in BRCA1/2, 22 additional genes were found to have deleterious alterations, of which the majority were genes associated with the DNA damage response pathway, including BLM, CHEK2, BARD1, ATM, RAD51D, and RAD50. Nine (1.5%) patients had PGAs in two different genes, typically including known founder mutations in the Ashkenazi Jewish population. These results lend strong support to the proposal that GT for a broad panel of known hereditary cancer predisposition genes should be offered routinely to patients with PDAC, regardless of age, ethnicity, and family/personal history of cancer.
The potential benefits of identifying PGAs in patients with PDAC are, first, the ability to test at-risk relatives and enroll carriers in prospective screening programs to decrease morbidity and mortality associated with increased cancer incidence both related to other cancers and potentially PDAC, and second, the possibility to exploit a known PGA for therapeutic benefit. While the development of effective tools for screening and early detection of PDAC remains a considerable challenge, the ability to screen for or undertake preventative surgery to reduce the risk of breast, colon, and ovarian cancer is likely to result in overall clinical benefit for patient relatives in whom a PGA is identified. With increased screening, an ongoing challenge will be how to advise patients in whom a PGA in a low/moderate-penetrance gene is identified whereby the clinical implications of such an alternation are not well understood and the overall increased lifetime risk of cancer is modest. Careful prospective study of cancer incidence and mortality, impact of lifestyle/environmental factors, and analysis of risk reduction interventions will be crucial to developing appropriate clinical guidelines for management of these patients.
The potential to utilize PGAs for therapeutic benefit relates predominantly to the identification of tumors with defective DNA-damage response repair genes that have increased sensitivity to DNA-damaging agents or targeted therapies that are synthetically lethal with defective double-strand break repair, or, alternatively, the use of checkpoint inhibitors in patients with hypermutated tumors associated with a PGA in one of the mismatch repair genes. Based on this supposition, we postulate that about 5% to 10% of PDAC patients will have therapeutically actionable PGAs. In patients for whom we had complete data regarding clinical outcomes, we did not identify a statistically significant difference in median OS in patients with vs without PGA. However, there was longer median OS, though statistically nonsignificant, when analysis was restricted to metastatic/recurrent disease and in patients with BRCA mutations vs patients without. These observations may have been limited by several factors. Only 356 (57.9%) patients had clinical outcomes data included in analysis. Additionally, not all patients with PGAs may have had a therapeutically actionable PGA, nor did every patient with a therapeutically actionable PGA receive corresponding therapy. The lack of a statistically significantly longer median OS does, however, suggest that prognostically patients with PGAs may not have a different outcome compared with patients without PGAs.
The benefits described do depend on timely results of GT. Our study had a median reporting time of 45 days from consent. Furthermore, effective reporting of results to the treating physician and deceased patient’s relatives should be in place; should the results become available posthumously, reporting to patients’ relatives can inform them of the need for testing and screening.
We found that 48 (57.1%) patients with a PGA who received platinum had a therapeutic response, while 102 (35.5%) patients without a PGA responded to platinum in the advanced disease setting. Our data further demonstrate that patients with BRCA mutations have the best response to platinum therapy while patients with non-BRCA PGAs respond more than patients without PGAs but not as well as BRCA patients. Our data support the hypothesis that there is increased tumor response to platinum in patients who carry a PGA in genes associated with DNA-damage response—a concept that is currently being tested in prospective studies of platinum therapy in patients with BRCA1/2 mutation–associated PDAC (13,14,16). Early results indicate that we may consider prospective evaluation of agents targeting defective DNA damage repair pathways in a broader patient population than previously considered, including those with pathogenic PGA in BLM, CHEK2, ATM, and other less frequently altered genes. This response to platinum is further supported by our LOH analysis, where we found 18 (60%) BRCA1/2 mutations with LOH. The prevalence of LOH in BRCA patients likely confers sensitivity to platinum and allows for therapies that are synthetically lethal. We also found PGAs such as CHEK2, ATM, NF1, and FH to have LOH, which further raises the possible use of synthetically lethal therapies in the same manner that is currently being studied in BRCA LOH (13,14,16). Interestingly, however, not all BRCA1/2 mutations had LOH, a finding that will need further study to evaluate the clinical significance. To date, the value of ST in patients with PDAC is limited from a therapeutic standpoint; in contrast, our findings reported in this manuscript indicate that GT is potentially of more clinically meaningful value in the identification of targetable genetic alterations in this population (24,31).
Our study has several limitations, including the low proportion of non-Caucasian patients, related to referral patterns to MSKCC. Furthermore, our patient population has a high frequency (18%) of Ashkenazi Jewish heritage. Additionally, it is likely that some patients who identified only as Caucasian were of Ashkenazi Jewish heritage. This explains, in part, the increased incidence of PGA that we identified in our study in comparison with recently published series, as some findings reflect carrier status of low-penetrance genes (21,23). However, we did find PGA in patients of African American, Asian, and Hispanic ethnicity at similar frequency to the Caucasian population, acknowledging relatively small numbers. Additional studies in these non-Caucasian patient populations are needed to characterize the frequency in patients of varied ethnic backgrounds.
Our study demonstrates the acceptability, feasibility, and clinical value of broad GT in patients with PDAC, independent of ethnicity, age, and family cancer history. Future development of precision medicine in patients with PDAC is likely to be guided by the identification of both germline and somatic genetic alterations, offering opportunity for improvement in both preventative and therapeutic treatment approaches. Looking forward, we propose the incorporation of GT as standard of care for patients with PDAC and the consideration of stratification by PGA in prospective studies evaluating the use of rational combinations of targeted and cytotoxic therapies.
Funding
This work was supported by the David M. Rubenstein Pancreatic Cancer Research Center, Robert and Kate Niehaus Center for Inherited Cancer Genomics, Bonnie Reiss Pancreatic Cancer Family Foundation, Suzanne Cohn Simon Pancreatic Cancer Research Fund, Cancer Center Support Grant P30-CA008748-50 from the National Cancer Institute, Marie-Josée and Henry R. Kravis Center for Molecular Oncology, and the National Cancer Institute of the National Institutes of Health (R25CA020449) to JWL.
Notes
Affiliations of authors: Department of Medicine (MAL, WW, EJJ, JWL, KHY, AV, DPK, RB, EK, DMH, GKA, CT, CC, HM, MER, KO ZKS, EOR), Robert and Kate Niehaus Center for Inherited Genomics (YK, JV, AZ, ES, AGA, VR, SM, MER, KO, ZKS), Department of Epidemiology and Biostatistics (MC), David M. Rubenstein Center for Pancreatic Cancer Research (MAL, KHY, AV, DPK, RB, EK, PA, GA, SDL, CID, EOR), Center for Molecular Oncology (JV, AZ, VR, MFB, DMH), Department of Pathology (DM, MB, DK, GA), Department of Surgery (PJA, SDL), Department of Medicine, Weill Cornell (WW, KHY, AV, DPK, DMH, GKA, ZKS, EOR).
The funder had no role in the design of the study; the collection, analysis, or interpretation of the data; the writing of the manuscript; or the decision to submit the manuscript for publication.
Supplementary Material
References
- 1. Rahib L, Smith B, Aizenberg R, et al. Projecting cancer incidence and deaths to 2030: The unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014;7411:2913–2921. [DOI] [PubMed] [Google Scholar]
- 2. Conroy T, Desseigne F, Ychou M, et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med. 2011;3658:768–769. [DOI] [PubMed] [Google Scholar]
- 3. Von Hoff D, Ervin T, Arena F, et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N Engl J Med. 2013;36918:1691–1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Knudsen E, O’Reilly EM, Brody J, Witkiewicz A.. Genetic diversity of pancreatic ductal adenocarcinoma and opportunities for precision medicine. Gastroenterology. 2016;1501:48–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Waddell N, Pajic M, Patch A, et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature. 2015;5187540:495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Goggins M, Schutte M, Lu J. et al. Germline BRCA2 gene mutations in patients with apparently sporadic pancreatic carcinomas. Cancer Res. 1996;56:5360–5364. [PubMed] [Google Scholar]
- 7. Grant R, Selander I, Connor A, et al. Prevalence of germline mutations in cancer predisposition genes in patients with pancreatic cancer. Gastroenterology. 2015;1483:556–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Roberts NJ, Norris AL, Petersen GM, et al. Whole genome sequencing defines the genetic heterogeneity of familial pancreatic cancer. Cancer Discov. 2016;62:166–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lee K, Yoo C, Kim K, et al. Germline BRCA mutations in Asian patients with pancreatic adenocarcinoma: A prospective study evaluating risk category for genetic testing. Invest New Drugs. 2018;361:163–169. [DOI] [PubMed] [Google Scholar]
- 10. Krantz BA, Yu KH, O'Reilly EM.. Pancreas adenocarcinoma: Novel therapeutics. Chin Clin Oncol. 2017;63:30. [DOI] [PubMed] [Google Scholar]
- 11. Lucas A, Shakya R, Lipsyc M, et al. High prevalence of BRCA1 and BRCA2 germline mutations with loss of heterozygosity in a series of resected pancreatic adenocarcinoma and other neoplastic lesions. Clin Cancer Res. 2013;1913:3396–3403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Moynahan ME, Pierce AJ, Jasin M.. BRCA2 is required for homology-directed repair of chromosomal breaks. Mol Cell. 2001;72:263–272. [DOI] [PubMed] [Google Scholar]
- 13. Lowery MA, Kelsen DP, Capanu M, et al. Phase II trial of veliparib in patients with previously-treated BRCA-mutated pancreas ductal adenocarcinoma. Eur J Cancer. 2018;89:19–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. O’Reilly EM, Lee JW, Lowery MA. et al. Phase I trial evaluating cisplatin, gemcitabine and veliparib in two patient cohorts: Germline-BRCA mutation carriers and BRCA wild-type pancreas ductal adenocarcinoma. Cancer. 2018; doi: 10.1002/cncr.31218. [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kaufman B, Shapira-Frommer R, Schmutzler R, et al. Olaparib monotherapy in patients with advanced cancer and a germline BRCA1/2 mutation. J Clin Oncol. 2015;333:244–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kindler HL, Locker G, Mann H, et al. POLO: A randomized phase III trial of olaparib tablets in patients with metastatic pancreatic cancer (mPC) and a germline BRCA1/2 mutation (gBRCAm) who have not progressed following first-line platinum based chemotherapy. J Clin Oncol. 2015;33(suppl);abstr TPS4149. [Google Scholar]
- 17. McCabe N., Turner N C, Lord C, et al. Deficiency in the repair of DNA damage by homologous recombination and sensitivity to poly(ADP-ribose) polymerase inhibition. Cancer Res. 2006:6616;8109–8115. [DOI] [PubMed] [Google Scholar]
- 18. Hampel H, Bennett R, Buchanan A, Pearlman R, Wiesner G.. A practice guideline from the American College of Medical Genetics and Genomics and the National Society of Genetic Counselors: Referral indications for cancer predisposition assessment. Genet Med. 2014;171:70–87. [DOI] [PubMed] [Google Scholar]
- 19. National Comprehensive Cancer Network. NCCN Clinical Practice Guidelines in Oncology: Genetic/Familial High-Risk Assessment: Breast and Ovarian. Version 1. Fort Washington, PA: National Comprehensive Cancer Network; 2018. https://www.nccn.org/professionals/physician_gls/pdf/genetics_screening.pdf. Accessed October 10, 2017. [Google Scholar]
- 20. Salo-Mullen E, O'Reilly E, Kelsen D, et al. Identification of germline genetic mutations in patients with pancreatic cancer. Cancer. 2015;12124:4382–4388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Shindo K, Yu J, Suenaga M, et al. Deleterious germline mutations in patients with apparently sporadic pancreatic adenocarcinoma. J Clin Oncol. 2017;3530:3382–3390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mandelker D, Zhang L, Kemel Y, et al. Mutation detection in patients with advanced cancer by universal sequencing of cancer-related genes in tumor and normal DNA vs guideline-based germline testing. JAMA. 2017;3189:825–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chaffee KG, Oberg AL, Mcwilliams RR, et al. Prevalence of germ-line mutations in cancer genes among pancreatic cancer patients with a positive family history. Genet Med. 2018;201:119–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lowery MA, Jordan EJ, Basturk O, et al. Real-time genomic profiling of pancreatic ductal adenocarcinoma: Potential actionability and correlation with clinical phenotype. Clin Cancer Res. 2017;2320:6094–6100. [DOI] [PubMed] [Google Scholar]
- 25. McKenna A, Hanna M, Banks E, et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;209:1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zehir A, Benayed R, Shah RH, et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat Med. 2017;236:703–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Cheng DT, Mitchell TN, Zehir A, et al. Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT): A hybridization capture-based next-generation sequencing clinical assay for solid tumor molecular oncology. J Mol Diagn. 2015;17:251–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Cibulskis K, Lawrence MS, Carter SL, et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol. 2013;313:213–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Schouten JP, McElgunn CJ, Waaijer R, et al. Relative quantification of 40 nucleic acid sequences by multiplex ligation-dependent probe amplification. Nucleic Acids Res. 2002;3012:57e57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Shen R, Seshan V.. FACETS: Allele-specific copy number and clonal heterogeneity analysis tool for high-throughput DNA sequencing. Nucleic Acids Res. 2016;4416:e131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chantrill LA, Nagrial AM, Watson C, et al. Precision medicine for advanced pancreas cancer: The individualized molecular pancreatic cancer therapy (impact) trial. Clin Cancer Res. 2015;219:2029–2037. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.