

Abstract
Objective
Maternal immune activation (MIA) is associated with an increased risk of autism spectrum disorder (ASD) in offspring. Herein, we investigate the altered expression of microRNAs (miRNA), and that of their target genes, in the brains of MIA mouse offspring.
Methods
To generate MIA model mice, pregnant mice were injected with polyriboinosinic:polyribocytidylic acid on embryonic day 12.5. We performed miRNA microarray and mRNA sequencing in order to determine the differential expression of miRNA and mRNA between MIA mice and controls, at 3 weeks of age. We further identified predicted target genes of dysregulated miRNAs, and miRNA‐target interactions, based on the inverse correlation of their expression levels.
Results
Mice prenatally subjected to MIA exhibited behavioral abnormalities typical of ASD, such as a lack of preference for social novelty and reduced prepulse inhibition. We found 29 differentially expressed miRNAs (8 upregulated and 21 downregulated) and 758 differentially expressed mRNAs (542 upregulated and 216 downregulated) in MIA offspring compared to controls. Based on expression levels of the predicted target genes, 18 downregulated miRNAs (340 target genes) and three upregulated miRNAs (60 target genes) were found to be significantly enriched among the differentially expressed genes. miRNA and target gene interactions were most significant between mmu‐miR‐466i‐3p and Hfm1 (ATP‐dependent DNA helicase homolog), and between mmu‐miR‐877‐3p and Aqp6 (aquaporin 6).
Interpretation
Our results provide novel information regarding miRNA expression changes and their putative targets in the early postnatal period of brain development. Further studies will be needed to evaluate potential pathogenic roles of the dysregulated miRNAs.
Introduction
Autism spectrum disorder (ASD) is a neurodevelopmental disorder characterized by persistent deficits in social communication and social interaction, and restricted, repetitive patterns of behavior, interests, or activities, which occur in the early developmental period.1 It has been well established that ASD is a complex disorder caused by both genetic and environmental factors.2 Because no single genetic variation or mutation can account for a majority of ASD cases, the converging actions of ASD‐related genes on common pathways, as well as interaction effects with non‐genetic factors, are considered to be a likely explanation for ASD pathophysiology.2
Epigenetic modification is an important regulatory mechanism for controlling gene expression without the involvement of DNA mutations or polymorphisms.3 Epigenetic regulation, including DNA methylation and histone modification, is essential for normal brain development, and dysregulation of the epigenetic machinery has been implicated in various neurodevelopmental and neuropsychiatric disorders, including ASD.4 microRNA (miRNA) is a well‐known epigenetic component that post‐transcriptionally regulates gene expression by binding to complementary nucleotide sequences in the 3′ untranslated region of target mRNAs.5 Altered miRNA expression has been associated with neurological disorders, including Alzheimer's disease,6 schizophrenia,7 and ASD.8, 9 Therefore, miRNAs may act as epigenetic factors in complex ASD etiologies, however, there have been substantial differences reported in miRNA expression profiles among different studies.10 Furthermore, studies of human ASD cases cannot investigate miRNA changes during earlier developmental stages.
Epidemiological studies have shown that maternal infection is associated with a higher risk of neurodevelopmental disorders, particularly schizophrenia and ASD, in offspring.11 Moreover, animal model research suggests that maternal immune activation (MIA), even without exposure to infectious pathogens, can cause behavioral and histological phenotypes of ASD in offspring.12 The prenatal administration of polyriboinosinic:polyribocytidylic acid (poly[I:C]) is one of the most widely used methods for generating an MIA model in mice. Poly (I:C), a synthetic analog of double‐stranded RNA, activates the innate immune response by stimulating toll‐like receptor 3, mimicking a viral infection‐like acute phase response.13 Poly (I:C) administration strongly induces pro‐inflammatory cytokines, and among these, interleukin (IL)‐6 and IL‐17a are key mediators for abnormal cortical development and behavioral phenotypes in the MIA model.14, 15 Although numerous preclinical studies have been conducted using the MIA animal model, little information exists about the effect of MIA on miRNA expression associated with mRNA expression in the brain. In particular, it remains unknown whether prenatal immune activation affects miRNA and gene regulation during postnatal development of the brain. Considering that brain development, such as myelination, synaptogenesis, and synaptic pruning, continues after birth, miRNA dysregulation in the early postnatal period might play a role in the pathogenesis of ASD and other neurodevelopmental disorders.
In this study, we aimed to investigate miRNA and mRNA expression profiles in the brains of juvenile MIA offspring. Gene annotation analysis was conducted to identify functional annotations enriched in differentially expressed genes in ASD. In addition, we performed in silico analysis to identify target genes of differentially expressed miRNAs, and to understand the interactions of the miRNAs with the target genes.
Materials and Methods
MIA mouse model
To generate the MIA model, pregnant C57BL6 mice were given intraperitoneal injections of 20 mg/kg of poly(I:C) (P9582, Sigma‐Aldrich, St. Louis, MO) on embryonic day 12.5 (E12.5), according to methods previously described.14, 16 Poly(I:C) lyophilized powder was reconstituted in nuclease‐free distilled water (10 mg/mL) to yield Poly(I:C) in an isotonic buffer solution, according to the manufacturer's instructions. Control pregnant mice were intraperitoneally injected with PBS on E12.5. Offspring were weaned at 3 weeks of age and housed in same‐sex groups of 2–5 mice. Thereafter, we only examined male mice, in order to exclude sex‐related differences of MIA. The mice were housed with ad libitum access to food and water under a 12:12 h light‐dark cycle. Mouse brains (n = 4 per group) were extracted from the skull under deep anesthesia at 3 weeks of age, as previously described.17 Olfactory bulbs and cerebellum were dissected immediately. The study protocol was reviewed and approved by the Institutional Animal Care and Use Committee (IACUC) of Seoul National University Hospital (IACUC No. 13‐0385‐S1A0).
Behavioral testing
Mouse behavioral tasks were performed at 7 weeks of age, and consisted of the three‐chamber test, prepulse inhibition (PPI), stereotyped behaviors, and nest‐building performance. All behavioral procedures were video recorded and the recorded data were analyzed by an experimenter unaware of the treatments.
A three‐chambered plastic box was used to test sociability and preference for social novelty, based on a previously described protocol.18 First, for habituation, mice (MIA and control) were placed in the middle chamber and allowed to explore for 10 min. During habituation, each of the two side chambers contained an empty cylinder cage. Following a 10‐min habituation, for the sociability test, an unfamiliar mouse (social object, age‐matched, same‐sex mouse), enclosed in a cylinder cage, was placed in one of the side chambers. An inanimate object, enclosed in a cylinder cage, was placed in the other side chamber, and then the subject mouse was allowed to explore for 10 min. In the subsequent social novelty preference test, another novel mouse (novel social object, age‐matched, same‐sex mouse) was replaced with the inanimate object, and the subject mouse was allowed to explore the two social objects, for 10 min. In the sociability and social novelty preference tests, the time spent exploring each object in the side chambers was analyzed. Exploration behavior was defined as sniffing and contacting a cylinder cage, or as when a mouse faces the cage with the distance between the nose and the cage less than 1 inch. In the sociability test, preference for a social object was calculated as (time spent exploring social object)/(time spent exploring social object + time spent exploring inanimate object). In the social novelty test, preference for a novel mouse was calculated as (time spent exploring novel mouse)/(time spent exploring novel mouse + time spent exploring familiar mouse).
For the PPI test, we used the SR‐LAB™ startle response system (San Diego Instruments, San Diego, CA, USA). Startle response of tested mice was recorded by a piezoelectric sensor within the chamber. This experiment was conducted as previously described.16 A five‐min habituation period, with background noise of 65 dB, was provided 1 day before, and immediately prior to, the PPI testing. After habituation, a test session began, with six consecutive startle (120‐dB burst for 40 ms) trials, which were followed by 50 prepulse trials in a randomized sequence. There were five types of stimulus in the prepulse trials: startle stimulus (120‐dB burst for 40 ms) only, startle stimulus preceded by 20‐ms prepulse stimulus at 68, 71, and 77‐dB intensities, and no stimulus. The onset‐to‐onset interval between prepulse and startle stimuli was 100 ms. Each test session ended with six consecutive startle trials. The inter‐trial interval was set at 15 s. PPI was calculated as (startle only stimulus–prepulse and startle stimulus)/startle only stimulus.
To investigate repetitive/stereotyped behaviors,19 we observed jumping and self‐grooming behaviors of tested mice in a clean, empty box for 10 min. The box was made of white plastic (40 × 40 × 40 cm). Before the test, a 10‐min habituation period was provided for each mouse. Behaviors were video‐recorded and analyzed for the number of jumping and self‐grooming events, and the total time spent self‐grooming.
Assessment for nest building was performed as previously described.20 Briefly, the tested mice were moved to a cage with wood‐chip bedding and one Nestlet (Ancare, Bellmore, NY, USA), ~1 h before the dark phase. The following morning, we assessed the nest‐building status on a rating scale from 1 to 5: 1, Nestlet largely untouched (>90% intact); 2, Nestlet partially torn up (50–90% remaining intact); 3, Nestlet mostly shredded (<50% remaining intact), but no identifiable nest site; 4, An identifiable, but flat nest (walls higher than mouse height <50% of the nest circumference); 5, A perfect nest with a crater (walls higher than mouse height ≥50% of the nest circumference).
As a result, the MIA mouse model at 7 weeks of age exhibited behavioral abnormalities typical of ASD, such as lack of preference for social novelty, reduced PPI, increased stereotyped behaviors, and impaired nest‐building behaviors (Data S1 and Figure S1).
MicroRNA microarray
Total RNA was isolated from individual brain samples using TRIzol (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's protocol. The eluted RNA was quantified using an ND‐1000 spectrophotometer (NanoDrop Technologies, Inc., Wilmington, DE, USA). The quality of the RNA was verified by 1% agarose denaturing gel and also with an Agilent 2100 bio‐analyzer (Agilent Technologies, Palo Alto, CA, USA). The synthesis of target miRNA probes and hybridization were performed using Agilent's miRNA Labeling Reagent and Hybridization kit (Agilent Technologies), according to the manufacturer's instructions. Briefly, 100 ng of total RNA was dephosphorylated with 15 Units of calf intestine alkaline phosphatase (CIP), followed by RNA denaturation with 40% DMSO and a 10‐min incubation at 100°C. Dephosphorylated RNA was ligated with pCp‐Cy3 mononucleotides and purified with MicroBioSpin 6 columns (Bio‐rad, Hercules, CA, USA). After purification, labeled samples were resuspended with Gene Expression blocking Reagent and Hi‐RPM Hybridization buffer (Agilent Technologies), followed by boiling for 5 min at 100°C, and then chilled on ice for 5 min. Finally, denatured labeled probes were pipetted onto assembled Agilent SurePrint G3 Mouse miRNA Microarrays (Release 19, 8 × 60K), and hybridized for 20 h at 55°C with 20 RPM rotating, in an Agilent Hybridization oven (Agilent Technologies). The hybridized microarrays were washed according to the manufacturer's washing protocol (Agilent Technologies). Thereafter, the hybridized images were scanned using Agilent's DNA microarray scanner and quantified with Feature Extraction Software (Agilent Technologies). All data normalization and selection of fold‐changed probes were performed using GeneSpringGX 7.3 (Agilent Technologies). We performed data transformation (set measurements less than 0.01–0.01) and per chip (normalize to 75th percentile) normalization. The microarray analysis was performed by eBiogen (Seoul, South Korea). Upregulation of miRNA was defined as a >twofold increase, while downregulation of miRNA was defined as a <0.5‐fold decrease, compared with controls. P‐values were corrected for multiple testing by the Benjamini‐Hochberg method.
mRNA sequencing
Total RNA was isolated using TRIzol (Invitrogen), as noted above. Construction of libraries was performed using QuantSeq 3′ mRNA‐Seq Library Prep Kit (Lexogen, Inc., Vienna, Austria), according to the manufacturer's instructions. In brief, 500 ng of total RNA was prepared and an oligo‐dT primer containing an Illumina‐compatible sequence at its 5′ end was hybridized to the RNA and reverse transcription was performed. After degradation of the RNA template, second strand synthesis was initiated by a random primer containing an Illumina‐compatible linker sequence at its 5′ end. The double‐stranded library was purified by using magnetic beads to remove all reaction components. The library was amplified to add the complete adapter sequences required for cluster generation. The finished library was purified from the polymerase chain reaction (PCR) components. High‐throughput sequencing was performed using NextSeq 500 (Illumina, Inc., San Diego, CA, USA), with single‐end 75‐bp reads.
For data analysis, QuantSeq 3′ mRNA‐Seq reads were aligned using the Bowtie2 alignment tool21. Bowtie2 indices were generated either from the genome assembly sequence or the representative transcript sequences, for alignment to the genome and transcriptome. The alignment file was used for assembling transcripts, estimating their abundance, and detecting differential expression of genes. Differentially expressed gene were determined based on counts from unique and multiple alignments, using coverage in BEDTools22. The read count data were processed based on Quantile‐Quantile normalization method, using EdgeR within R, using Bioconductor.23 Genes were determined to be differentially expressed when log2 transformed normalized read counts were greater than four and the fold change (FC) was >1.5 or <0.67, with a P < 0.05. Gene classification was based on the Database for Annotation, Visualization, and Integrated Discovery (DAVID, http://david.abcc.ncifcrf.gov/), and on Medline databases (http://www.ncbi.nlm.nih.gov/). The mRNA sequencing and analysis were performed by eBiogen (Seoul, South Korea).
Gene Ontology enrichment analysis
We performed Gene Ontology (GO) enrichment analysis for differentially expressed genes by using the DAVID Bioinformatics Resources.24 Briefly, the GO terms shared by differentially expressed genes in the MIA offspring were compared to the proportion of genes annotated to the GO term in the whole genome. A statistical significance for enrichment was estimated by the hypergeometric distribution, and the Benjamini‐Hochberg method was used for multiple testing correction; a corrected P < 0.05 indicated significant enrichment of the corresponding GO terms. Additionally, we investigated the fold enrichment of the significantly enriched GO terms.
microRNA‐target gene interactions
First, putative target genes of dysregulated miRNAs were predicted using the miRWalk 2.0 algorithm.25 Databases for predicted target genes consisted of miRanda,26 RNA22,27 and Targetscan,28 as well as miRWalk 2.0.25 Putative target genes were identified when the miRNA‐target interactions were verified by three or more databases. Thereafter, we performed a right‐sided hypergeometric test to identify significantly enriched miRNA in the genes differentially expressed in the opposite direction: upregulated miRNA‐downregulated mRNA and downregulated miRNA‐upregulated mRNA, respectively. We applied the Benjamini‐Hochberg method for multiple testing correction, and a corrected P < 0.05 was considered to indicate significant enrichment. We determined whether the target genes of the differentially expressed miRNA were overrepresented among the differentially expressed genes in the transcriptome. For measuring the negative correlation between expression levels of miRNAs and target genes, we measured an FC ratio; the FC of upregulated mRNA divided by the FC of downregulated miRNA, or the FC of upregulated miRNA divided by the FC of downregulated mRNA. Additionally, we used Cytoscape for visualizing significant miRNA‐target gene interaction networks.29
Quantitative RT‐PCR
For measuring the expression level of mRNAs, total RNA was reverse transcribed with the SuperScript® III First‐Strand Synthesis System (Invitrogen, Catalog no. 18080‐051). Using the product as a template, quantitative real time PCR was performed with primers specific for the target genes using an ABI PRISM 7500 sequence detection system (Applied Biosystems, Foster City, CA, USA), in triplicate. Relative expression levels of the target genes were calculated using the comparative threshold cycle (Ct) method (2−ΔCt) and were normalized to those of the housekeeping gene, glyceraldehyde 3‐phosphate dehydrogenase (Gapdh). Primer sequences used in this study were as follows: Hfm1 forward, CAAGTCTCGGCGGAAGTAAG; Hfm1 reverse, TCAGCGGTCTCCTCTCTTGT; Slc39a2 forward, CCTGCTTGCTCTTCTGGTTC; Slc39a2 reverse, CCTCCAGAGCTTCAGCAGTC; Fzd5 forward, ACATGGAACGATTCCGCTAC; Fzd5 reverse, GGCCATGCCAAAGAAATAGA; Tfap2b forward, CCAAGAAGTGGGCTCAGAAG; Tfap2b reverse, TGGCATCTTCAACTGACTGC; Gapdh forward, ACAATGAATACGGCTACAG; Gapdh reverse, GGTCCAGGGTTTCTTACT.
Statistical analysis
Data are presented as mean ± standard error of the mean, or number (%), unless otherwise noted. We used two‐way ANOVA with factors of object and group in the three‐chamber test and repeated‐measures ANOVA with factors of prepulse intensity and group in the PPI. In addition, we used Wilcoxon rank‐sum test for comparing stereotyped behaviors and nest‐building performances between the two groups. A two‐tailed P < 0.05 was considered statistically significant, and statistical analyses were conducted using SPSS version 18 (SPSS Inc., Chicago, IL, USA).
Results
Dysregulated miRNAs and mRNAs in the MIA model
We evaluated miRNA expression profiles from the brain of juvenile MIA offspring, compared to control mice, at 3 weeks of age. This is the typical age of weaning in mice, at which the brain reaches approximately 90% of its adult weight, corresponding to approximately 2–3 years of age for humans.30 Significant dysregulation of miRNA was found in the MIA mice (corrected P < 0.05, Table 1); eight miRNAs were upregulated (FC > 2) and 21 miRNAs were downregulated (FC < 0.5). A heatmap combined with hierarchical clustering of differentially expressed miRNAs demonstrated a clear separation between the MIA and control mice (Figure 1). Additionally, we analyzed mRNA sequencing data to identify differentially expressed genes in 3‐week‐old MIA offspring. We found 542 upregulated (FC > 1.5) mRNAs and 216 downregulated (FC < 0.67) mRNAs in the MIA group compared to the control group. A list of differentially expressed mRNAs is shown in Table S1.
Table 1.
Differentially expressed microRNAs in the maternal immune activation (MIA) offspring
| Upregulated microRNA | Fold change | P | Downregulated microRNA | Fold change | P |
|---|---|---|---|---|---|
| mmu‐miR‐877‐3p | 101.636 | 1.61E‐03 | mmu‐miR‐135a‐5p | 0.274 | 2.91E‐02 |
| mmu‐miR‐680 | 86.444 | 4.86E‐03 | mmu‐miR‐133a‐3p | 0.307 | 1.14E‐02 |
| mmu‐miR‐3474 | 69.974 | 1.10E‐02 | mmu‐miR‐133b‐3p | 0.308 | 4.63E‐02 |
| mmu‐miR‐5119 | 34.652 | 1.24E‐02 | mmu‐miR‐6402 | 0.33 | 5.56E‐03 |
| mmu‐miR‐1196‐5p | 3.505 | 4.94E‐03 | mmu‐miR‐466i‐3p | 0.335 | 1.14E‐02 |
| mmu‐miR‐3092‐3p | 2.36 | 2.01E‐02 | mmu‐miR‐669c‐3p | 0.367 | 9.77E‐03 |
| mmu‐miR‐705 | 2.25 | 2.91E‐02 | mmu‐miR‐1a‐3p | 0.375 | 6.10E‐03 |
| mmu‐miR‐702‐3p | 2.003 | 2.61E‐02 | mmu‐miR‐466f‐3p | 0.402 | 4.58E‐02 |
| mmu‐miR‐466q | 0.406 | 2.01E‐02 | |||
| mmu‐miR‐34a‐5p | 0.423 | 2.26E‐02 | |||
| mmu‐miR‐6412 | 0.435 | 9.09E‐03 | |||
| mmu‐miR‐126‐5p | 0.444 | 1.03E‐02 | |||
| mmu‐miR‐223‐3p | 0.446 | 4.60E‐02 | |||
| mmu‐miR‐29a‐3p | 0.458 | 1.24E‐02 | |||
| mmu‐miR‐29b‐3p | 0.459 | 1.31E‐02 | |||
| mmu‐miR‐669p‐3p | 0.461 | 2.69E‐02 | |||
| mmu‐miR‐338‐3p | 0.474 | 1.19E‐02 | |||
| mmu‐miR‐212‐5p | 0.475 | 2.54E‐02 | |||
| mmu‐miR‐328‐3p | 0.479 | 2.67E‐02 | |||
| mmu‐miR‐30c‐5p | 0.481 | 1.27E‐02 | |||
| mmu‐miR‐219‐5p | 0.496 | 1.98E‐02 |
The P‐values are corrected by the Benjamini–Hochberg method. The expression levels of the MIA offspring at 3 weeks of age were compared to those of the control.
Figure 1.

Hierarchical clustering heatmap of miRNA expression in the brain sample of the maternal immune activation mouse model. Eight miRNAs were upregulated and 21 miRNAs were downregulated (corrected P < 0.05; n = 4 per group). The color code indicates the normalized expression level of each miRNA. Red indicates upregulation and blow indicates downregulation. MIA, maternal immune activation.
Gene Ontology analysis of differentially expressed mRNAs
We used the DAVID Bioinformatics Resources to perform GO enrichment analysis for differentially expressed genes. Upregulated genes in the MIA offspring were significantly associated with five biological processes, 10 cellular components, and 10 molecular functions (corrected P < 0.05; Figure 2). Among them, photoreceptor cell maintenance, euchromatin, and methylcytosine dioxygenase activity showed the highest fold enrichment in biological processes, cellular components, and molecular functions, respectively. Conversely, only three GO terms in the cellular component category were significantly enriched in the downregulated genes of the MIA model: extracellular exosome, mitochondrion, and cytosol (corrected P < 0.05). There was no significantly over‐represented biological process or molecular function in the downregulated genes.
Figure 2.
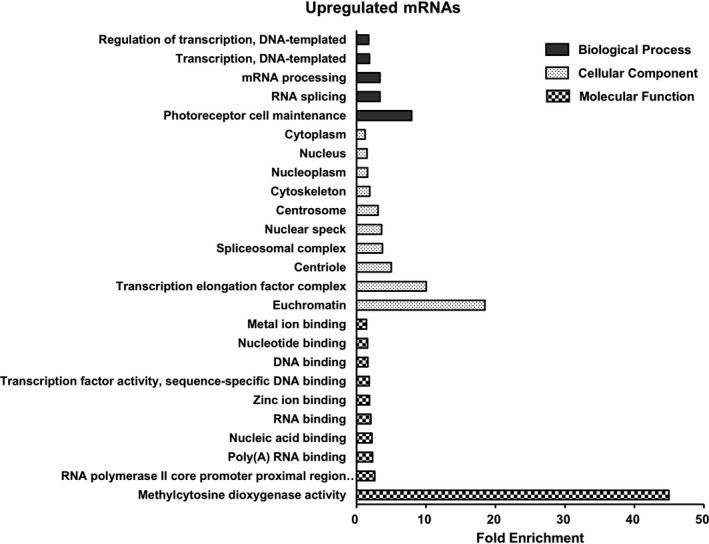
Gene Ontology terms significantly enriched in the upregulated mRNAs. There are five biological processes, 10 cellular components, and 10 molecular functions (corrected P < 0.05). Each bar indicates fold enrichment of the corresponding GO term.
MicroRNA‐target gene interactions
To investigate miRNA‐target gene interactions, we analyzed miRNA enrichment for their putative targets among differentially expressed genes in the MIA model. Among 21 downregulated miRNAs, 18 miRNAs were significantly enriched in the upregulated genes (corrected P < 0.05). We found a total of 340 target genes whose expression was negatively correlated with the pertinent miRNA expression (Figure 3A). Of eight upregulated miRNAs, three miRNAs showed a significant enrichment in the downregulated target genes (corrected P < 0.05): mmu‐miR‐3092‐3p, mmu‐miR‐680, and mmu‐miR‐877‐3p (Figure 3B). We identified 60 genes as predicted targets of the upregulated miRNAs. These miRNA‐target gene interactions in the MIA mouse brain are visualized as a network in Figure 4.
Figure 3.
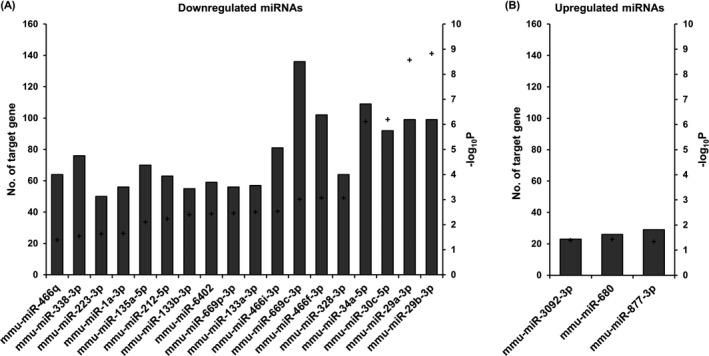
Significantly enriched miRNAs in differentially expressed genes. (A) Eighteen downregulated miRNAs. (B) Three upregulated miRNAs. Each bar indicates the number of target genes which had a negative correlation with the corresponding miRNAs. Plus sign indicates the P‐values (‐log10P) for enrichment of each miRNA. P values were corrected by the Benjamini‐Hochberg method.
Figure 4.
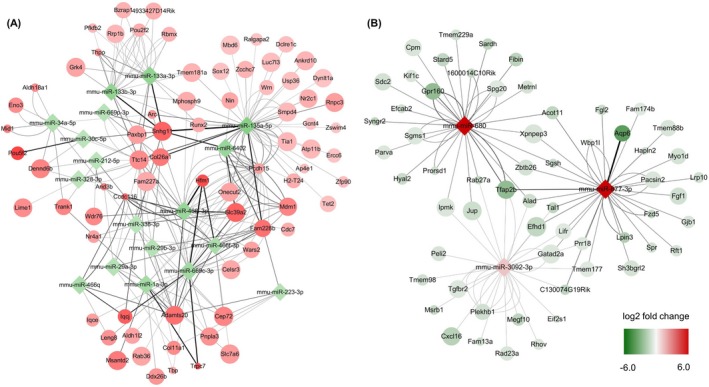
Network of significantly enriched miRNAs and their target genes. (A) Eighteen downregulated miRNAs and negatively correlated target genes. Only 74 upregulated target genes with the fold change ratio of >7 were included in this network. (B) Three upregulated miRNAs and 60 downregulated target genes. Node color indicates the log twofold change of the miRNA and target gene expression. Node size indicates the significance of the differential expression; larger sizes indicate smaller P‐values. Edge thickness corresponds to the fold change ratio of the negative correlation between miRNAs and target genes.
Next, we calculated the FC ratio as a measure of the interaction between miRNAs and target genes. A higher FC ratio indicates a higher degree of negative correlation between the expression levels of miRNAs and their target genes. The highest interaction among the downregulated miRNAs occurred between mmu‐miR‐466i‐3p and Hfm1 (ATP‐dependent DNA helicase homolog). Moreover, negative correlation between mmu‐miR‐877‐3p and Aqp6 (aquaporin 6) was the highest among the upregulated miRNAs. A list of the top 10 interactions in upregulated and downregulated miRNAs is shown in Table 2.
Table 2.
The top 10 miRNA–target gene interactions
| miRNA | Gene | Fold change ratio |
|---|---|---|
| Downregulated miRNA–upregulated gene | ||
| mmu‐miR‐466i‐3p | Hfm1 | 20.5 |
| mmu‐miR‐135a‐5p | Snhg11 | 19.8 |
| mmu‐miR‐669c‐3p | Hfm1 | 18.7 |
| mmu‐miR‐133a‐3p | Snhg11 | 17.6 |
| mmu‐miR‐133b‐3p | Snhg11 | 17.6 |
| mmu‐miR‐669c‐3p | Trpc7 | 17.5 |
| mmu‐miR‐135a‐5p | Fam228b | 17.4 |
| mmu‐miR‐466f‐3p | Hfm1 | 17.1 |
| mmu‐miR‐1a‐3p | Trpc7 | 17.1 |
| mmu‐miR‐6402 | Slc39a2 | 16.5 |
| Upregulated miRNA–downregulated gene | ||
| mmu‐miR‐877‐3p | Aqp6 | 1379.7 |
| mmu‐miR‐877‐3p | Tfap2b | 556.9 |
| mmu‐miR‐680 | Tfap2b | 473.7 |
| mmu‐miR‐680 | Gpr160 | 464.6 |
| mmu‐miR‐877‐3p | Lpin3 | 330.6 |
| mmu‐miR‐877‐3p | C130074G19Rik | 266.7 |
| mmu‐miR‐877‐3p | Acot11 | 261.3 |
| mmu‐miR‐877‐3p | Tal1 | 260.3 |
| mmu‐miR‐877‐3p | Efhd1 | 245.5 |
| mmu‐miR‐877‐3p | Fzd5 | 243.0 |
Thereafter, we performed quantitative RT‐PCR to measure the expression levels of selected target genes: Hfm1, Slc39a2, Fzd5, and Tfap2b. As shown in Figure 5, the relative expression levels of those target genes were largely consistent with those from the RNA sequencing data. These findings corroborate the altered gene expression profile of the MIA model shown in this study.
Figure 5.
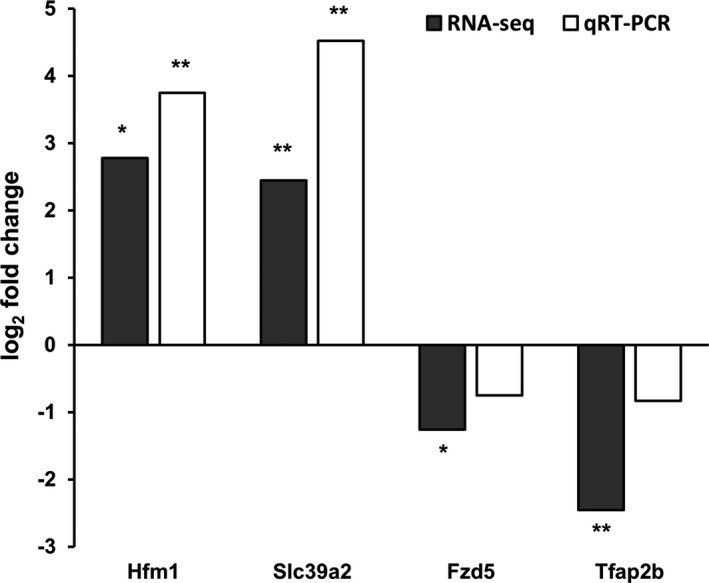
Relative expression level of target genes. Bars indicate fold change (log 2) of gene expression in the MIA offspring compared to the controls, which was measured by both RNA sequencing and quantitative RT‐PCR. *P < 0.05 and **P < 0.01 by Student's t‐test (RNA sequencing) and Wilcoxon rank‐sum test (quantitative RT‐PCR) for comparison of expression levels between the MIA and control groups; n = 4 per group.
Discussion
In this study, we evaluated miRNA and mRNA expression profiles in the whole brain of the MIA mouse model for ASD. Adult mice prenatally exposed to poly (I:C) exhibited typical behavioral phenotypes of ASD, including reduced preference for social novelty, impaired PPI of the startle reflex, and abnormal stereotyped behaviors. Moreover, altered expression of both miRNAs and mRNAs in the brain was found in early postnatal MIA offspring at 3 weeks of age. We identified target genes of dysregulated miRNAs through in silico analysis and negative regulatory interactions between differentially expressed miRNAs and putative targets in ASD. Although gene and miRNA expression profiles have been previously studied, they have typically involved non‐cerebral tissue, such as blood or lymphoblast cell lines, or postmortem brains in human cases.10 Considering that ASD is a developmental disorder of the brain, brain tissue from adult cases as well as non‐cerebral tissue may not be optimal targets for investigating disease processes during the early developmental stage. However, it is essentially impossible to obtain fetal or early postnatal brain tissue from human ASD cases. In this regard, an animal model can represent an appropriate alternative, although there have been few studies on mRNA and miRNA profiling in animal models of ASD.
The effect of maternal inflammation on gene regulation in the offspring brain has been reported previously.31 Although the precise mechanism still remains unclear, it is generally accepted that cytokines induced by maternal immune system affect transcriptional and developmental regulation of the offspring brains. Among pro‐inflammatory cytokines, IL‐6 acts as a key mediator for transcriptional changes in the MIA model, because anti‐IL‐6 treatment normalized altered gene expression evoked by MIA.14 Furthermore, IL‐17a, produced by T helper 17 cells, was found to act downstream of IL‐6, leading to abnormal cortical development and behavioral phenotypes in MIA offspring.15 Previous studies examined miRNA levels in the embryonic or adult brain of ASD mice.32, 33 It should be noted that this is the first study to analyze the whole‐brain expression of ASD‐associated mRNAs and miRNAs at the transcriptome level, particularly in the early postnatal mouse brain, which is the main strength of this study. MIA was previously reported to cause long‐lasting changes in cytokine expression in the postnatal mouse brain.34 This is in agreement with our results showing that an effect of MIA on miRNA and gene expression continues during postnatal development of the brain. Brain development at 3 weeks of age in rodents is characterized by a peak in synaptogenesis and myelination.30 Furthermore, as synaptic dysfunction is considered a major pathophysiology of ASD,35 the transcriptional changes observed in our study might be involved in synaptic structure and function. Future studies will be needed to understand the functional roles of dysregulated miRNAs and their targets in ASD.
Among the differentially expressed miRNAs in our study, miR‐212‐5p has been shown to be downregulated in patients with ASD and schizophrenia.9, 36 Reduced expression of miR‐212 and miR‐132 was also observed in human Alzheimer's disease brains.37 The miR‐132/212 family has been functionally implicated in synaptic transmission, dendritic development, regulation of brain‐derived neurotrophic factor (BDNF) expression, and neuronal survival.38 The miR‐132/212 knockout decreased basal synaptic transmission in the hippocampus and neocortex, while not affecting neuronal morphology.39 Moreover, the miR‐132 knockdown mouse demonstrated impairment in temporal memory acquisition, which provides a pathogenic link between miR‐132/212 downregulation and the temporal processing deficit in ASD.40 Hara et al. reported conflicting results; prenatal exposure to valproic acid upregulated miR‐132‐5p expression in the embryonic mouse brain.33 However, the effect of miR‐132‐5p upregulation was transient, lasting only 18–24 h after valproic acid exposure, which might explain the opposite direction of miR‐132‐5p dysregulation, compared to other studies in human cases.8, 9
Downregulation of miR‐219‐5p, miR‐133b, miR‐34a‐5p, and miR‐328‐3p in our MIA model is also consistent with previous reports. Sarachana et al. reported downregulation of miR‐219‐5p and miR‐133b in lymphoblast cell lines from ASD patients.8 In particular, they confirmed that miR‐219‐5p targeted Polo like kinase 2 (PLK2), whose activation in neurons is dependent on synaptic activity and is involved in homeostatic regulation of synaptic plasticity.41 Given that PLK2 induction resulted in dendritic spine dysmorphogenesis,42 downregulation of miR‐219‐5p might contributed to synaptic dysfunction in ASD pathophysiology. Although ASD is a neurodevelopmental disorder, there are several comorbidities, such as gastrointestinal distress and immune dysregulation, observed in patients with ASD.43 Moreover, a gut‐microbiome‐brain connection was found to contribute to ASD pathogenesis,16 further suggesting that ASD is a systemic disorder caused by both genetic and environmental factors. In this regard, peripheral blood samples have been used to identify pathophysiology and biomarker of ASD. In addition, miR‐219, which were downregulated in ASD derived lymphoid cell lines, is brain‐specific miRNA, supporting that miRNA dysregulation in peripheral blood might reflect system‐level transcriptional changes in ASD. In addition, reduced expression of miR‐34a‐5p is consistent with previous RNA sequencing data obtained from ASD brains.44 Brain‐enriched miR‐34a was also shown to modulate embryonic neural development, dendritic spine morphology, and synaptic transmission.45 Modi et al. reported that miR‐34a targeted cyclin D1 and promoted neuronal differentiation by inhibiting cell cycle reentry, while downregulation of miR‐34a caused cell cycle‐related neuronal apoptosis.46 Fear memory consolidation was also mediated by miR‐34, which inhibits the Notch signaling pathway in the amygdala.47 Taken together, these results supported a potential involvement of miRNA dysregulation in neurodevelopmental disease processes.
There was an over‐representation of genes for biological processes associated with RNA splicing, mRNA processing, and regulation of transcription, in MIA mice compared to controls. This is consistent with previous results showing that genes associated with transcriptional regulation were downregulated in ASD.48 It has been well established that epigenetic and post‐transcriptional regulation have pivotal roles in the pathogenesis of neurodevelopmental disorders, including ASD.49 In agreement with this finding, genome‐wide transcriptomic analysis identified differential splicing events in post‐mortem ASD brains, which were associated with neuronal activity‐dependent gene regulation.50 Differential alternative splicing was also found in blood samples from ASD children, and target genes were involved in oxidative stress, the mammalian target of rapamycin (mTOR) pathway, and nerve growth factor (NGF) signaling, as well as splicing regulation.51 In addition, atypical splicing of neuroligin, the mutation of which is associated with ASD phenotypes and synaptic dysfunction, was found in lymphoblastoid cell lines of ASD patients.52
We investigated a specific cell type of 29 differentially expressed miRNAs based on the previous report.53 Four miRNAs (mmu‐miR‐1a‐3p, mmu‐miR‐29a‐3p, mmu‐miR‐29b‐3p, and mmu‐miR‐212‐5p) are characterized by neocortex‐specific expression, while 4 miRNAs (mmu‐miR‐877‐3p, mmu‐miR‐133a‐3p, mmu‐miR‐133b‐3p, and mmu‐miR‐34a‐5p) were expressed higher in cerebellum (particularly, Purkinje cells) than in neocortex. In addition, the expression of mmu‐miR‐135a‐5p is specific to glutamatergic pyramidal neurons, while the expression of mmu‐miR‐669c‐3p and mmu‐miR‐669p‐3p is specific to cortical GABAergic interneurons. Further research about cell‐type specific miRNA dysregulation and their target genes will help us understand the patho‐mechanisms of ASD.
In this study, putative targets of miRNAs were identified by in silico analysis, however, molecular interactions were not verified by in vitro testing, such as luciferase assays. Furthermore, there is little information published on their functional implications with ASD or other neurodevelopmental disorders. Thus, although our data provide additional information on the transcriptional landscape and miRNA expression profiles in ASD, a pathophysiological association cannot be concluded from this study, and further research will be needed to address this concern. Additionally, because microarrays are based on hybridization with DNA probes included on a matrix, we might have missed altered expression of miRNAs not included in the microarray probes. Moreover, microarray data for miRNA expression was not validated by qRT‐PCR, which might have led to false positive findings.
Author Contributions
J.S.S, D.J, S.T.L, J.M, K.H.J, K.I.P, K.Y.J, M.K, S.K.L, and K.C contributed to conception and design of the study. J.S.S, D.J, J.S.Y, D.K.P, J.Y.B, D.Y.L, S.K, and K.C contributed to acquisition and analysis of data. J.S.S, D.J, and K.C contributed to drafting a significant portion of the manuscript.
Conflict of Interest
None declared.
Supporting information
Data S1. Behavioral phenotype of MIA offspring.
Figure S1. Behavior phenotypes of the offspring exposed to maternal immune activation.
Table S1. List of differentially expressed genes in the maternal immune activation mouse brain.
Acknowledgment
This work was supported by a grant from Myung In Pharmaceutical Company (1220140004) and Ildong Pharmaceutical Company (0620180350).
Funding Information
This work was supported by a grant from Myung In Pharmaceutical Company (1220140004) and Ildong Pharmaceutical Company (0620180350).
References
- 1. American Psychiatric Association . Diagnostic and statistical manual of mental disorders, 5th ed Washington, DC: American Psychiatric Association, 2013. [Google Scholar]
- 2. Kim YS, Leventhal BL. Genetic epidemiology and insights into interactive genetic and environmental effects in autism spectrum disorders. Biol Psychiatry 2015;77:66–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Jaenisch R, Bird A. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat Genet 2003;33(Suppl):245–254. [DOI] [PubMed] [Google Scholar]
- 4. Tsankova N, Renthal W, Kumar A, Nestler EJ. Epigenetic regulation in psychiatric disorders. Nat Rev Neurosci 2007;8:355–367. [DOI] [PubMed] [Google Scholar]
- 5. Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell 2009;136:215–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lee ST, Chu K, Jung KH, et al. miR‐206 regulates brain‐derived neurotrophic factor in Alzheimer disease model. Ann Neurol 2012;72:269–277. [DOI] [PubMed] [Google Scholar]
- 7. Lai CY, Lee SY, Scarr E, et al. Aberrant expression of microRNAs as biomarker for schizophrenia: from acute state to partial remission, and from peripheral blood to cortical tissue. Transl Psychiat 2016;6:e717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sarachana T, Zhou R, Chen G, et al. Investigation of post‐transcriptional gene regulatory networks associated with autism spectrum disorders by microRNA expression profiling of lymphoblastoid cell lines. Genome Med 2010;2:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Abu‐Elneel K, Liu T, Gazzaniga FS, et al. Heterogeneous dysregulation of microRNAs across the autism spectrum. Neurogenetics 2008;9:153–161. [DOI] [PubMed] [Google Scholar]
- 10. Hicks SD, Middleton FA. A comparative review of microRNA expression patterns in autism spectrum disorder. Front Psychiatry 2016;7:176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Brown AS. Epidemiologic studies of exposure to prenatal infection and risk of schizophrenia and autism. Dev Neurobiol 2012;72:1272–1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Estes ML, McAllister AK. Maternal immune activation: implications for neuropsychiatric disorders. Science 2016;353:772–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Meyer U. Prenatal poly(I:C) exposure and other developmental immune activation models in rodent systems. Biol Psychiatry 2014;75:307–315. [DOI] [PubMed] [Google Scholar]
- 14. Smith SE, Li J, Garbett K, et al. Maternal immune activation alters fetal brain development through interleukin‐6. J Neurosci 2007;27:10695–10702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Choi GB, Yim YS, Wong H, et al. The maternal interleukin‐17a pathway in mice promotes autism‐like phenotypes in offspring. Science 2016;351:933–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hsiao EY, McBride SW, Hsien S, et al. Microbiota modulate behavioral and physiological abnormalities associated with neurodevelopmental disorders. Cell 2013;155:1451–1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lee ST, Chu K, Jung KH, et al. Slowed progression in models of Huntington disease by adipose stem cell transplantation. Ann Neurol 2009;66:671–681. [DOI] [PubMed] [Google Scholar]
- 18. Moy SS, Nadler JJ, Perez A, et al. Sociability and preference for social novelty in five inbred strains: an approach to assess autistic‐like behavior in mice. Genes Brain Behav 2004;3:287–302. [DOI] [PubMed] [Google Scholar]
- 19. Malkova NV, Yu CZ, Hsiao EY, et al. Maternal immune activation yields offspring displaying mouse versions of the three core symptoms of autism. Brain Behav Immun 2012;26:607–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Deacon RM. Assessing nest building in mice. Nat Protoc 2006;1:1117–1119. [DOI] [PubMed] [Google Scholar]
- 21. Langmead B, Salzberg SL. Fast gapped‐read alignment with Bowtie 2. Nat Methods 2012;9:357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 2010;26:841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gentleman RC, Carey VJ, Bates DM, et al. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol 2004;5:R80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. da Huang W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 2009;4:44–57. [DOI] [PubMed] [Google Scholar]
- 25. Dweep H, Gretz N. miRWalk2.0: a comprehensive atlas of microRNA‐target interactions. Nat Methods 2015;12:697. [DOI] [PubMed] [Google Scholar]
- 26. Betel D, Wilson M, Gabow A, et al. The microRNA.org resource: targets and expression. Nucleic Acids Res 2008;36:D149–D153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Miranda KC, Huynh T, Tay Y, et al. A pattern‐based method for the identification of MicroRNA binding sites and their corresponding heteroduplexes. Cell 2006;126:1203–1217. [DOI] [PubMed] [Google Scholar]
- 28. Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 2005;120:15–20. [DOI] [PubMed] [Google Scholar]
- 29. Shannon P, Markiel A, Ozier O, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 2003;13:2498–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Semple BD, Blomgren K, Gimlin K, et al. Brain development in rodents and humans: identifying benchmarks of maturation and vulnerability to injury across species. Prog Neurobiol 2013;106–107:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Fatemi SH, Pearce DA, Brooks AI, Sidwell RW. Prenatal viral infection in mouse causes differential expression of genes in brains of mouse progeny: a potential animal model for schizophrenia and autism. Synapse 2005;57:91–99. [DOI] [PubMed] [Google Scholar]
- 32. Olde Loohuis NF, Kole K, Glennon JC, et al. Elevated microRNA‐181c and microRNA‐30d levels in the enlarged amygdala of the valproic acid rat model of autism. Neurobiol Dis 2015;80:42–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hara Y, Ago Y, Takano E, et al. Prenatal exposure to valproic acid increases miR‐132 levels in the mouse embryonic brain. Mol Autism 2017;8:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Garay PA, Hsiao EY, Patterson PH, McAllister AK. Maternal immune activation causes age‐ and region‐specific changes in brain cytokines in offspring throughout development. Brain Behav Immun 2013;31:54–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zoghbi HY, Bear MF. Synaptic dysfunction in neurodevelopmental disorders associated with autism and intellectual disabilities. Cold Spring Harb Perspect Biol 2012;4:a009886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Perkins DO, Jeffries CD, Jarskog LF, et al. microRNA expression in the prefrontal cortex of individuals with schizophrenia and schizoaffective disorder. Genome Biol 2007;8:R27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Cogswell JP, Ward J, Taylor IA, et al. Identification of miRNA changes in Alzheimer's disease brain and CSF yields putative biomarkers and insights into disease pathways. J Alzheimers Dis 2008;14:27–41. [DOI] [PubMed] [Google Scholar]
- 38. Wanet A, Tacheny A, Arnould T, Renard P. miR‐212/132 expression and functions: within and beyond the neuronal compartment. Nucleic Acids Res 2012;40:4742–4753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Remenyi J, van den Bosch MWM, Palygin O, et al. miR‐132/212 knockout mice reveal roles for these miRNAs in Regulating cortical synaptic transmission and plasticity. PLoS One 2013;8:e62509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wang RY, Phang RZ, Hsu PH, et al. In vivo knockdown of hippocampal miR‐132 expression impairs memory acquisition of trace fear conditioning. Hippocampus 2013;23:625–633. [DOI] [PubMed] [Google Scholar]
- 41. Seeburg DP, Pak D, Sheng M. Polo‐like kinases in the nervous system. Oncogene 2005;24:292–298. [DOI] [PubMed] [Google Scholar]
- 42. Pak DTS, Sheng M. Targeted protein degradation and synapse remodeling by an inducible protein kinase. Science 2003;302:1368. [DOI] [PubMed] [Google Scholar]
- 43. Gesundheit B, Rosenzweig JP, Naor D, et al. Immunological and autoimmune considerations of Autism Spectrum Disorders. J Autoimmun 2013;44:1–7. [DOI] [PubMed] [Google Scholar]
- 44. Mor M, Nardone S, Sams DS, Elliott E. Hypomethylation of miR‐142 promoter and upregulation of microRNAs that target the oxytocin receptor gene in the autism prefrontal cortex. Mol Autism 2015;6:46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Agostini M, Tucci P, Steinert JR, et al. microRNA‐34a regulates neurite outgrowth, spinal morphology, and function. Proc Natl Acad Sci USA 2011;108:21099–21104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Modi PK, Jaiswal S, Sharma P. Regulation of neuronal cell cycle and apoptosis by MicroRNA 34a. Mol Cell Biol 2016;36:84–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Dias BG, Goodman JV, Ahluwalia R, et al. Amygdala‐dependent fear memory consolidation via miR‐34a and Notch signaling. Neuron 2014;83:906–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Alter MD, Kharkar R, Ramsey KE, et al. Autism and increased paternal age related changes in global levels of gene expression regulation. PLoS One 2011;6:e16715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Smith RM, Sadee W. Synaptic signaling and aberrant RNA splicing in autism spectrum disorders. Front Synaptic Neurosci 2011;3:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Parikshak NN, Swarup V, Belgard TG, et al. Genome‐wide changes in lncRNA, splicing, and regional gene expression patterns in autism. Nature 2016;540:423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Stamova BS, Tian Y, Nordahl CW, et al. Evidence for differential alternative splicing in blood of young boys with autism spectrum disorders. Mol Autism 2013;4:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Talebizadeh Z, Lam DY, Theodoro MF, et al. Novel splice isoforms for NLGN3 and NLGN4 with possible implications in autism. J Med Genet 2006;43:e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. He M, Liu Y, Wang X, et al. Cell‐type based analysis of microRNA profiles in the mouse brain. Neuron 2012;73:35–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Behavioral phenotype of MIA offspring.
Figure S1. Behavior phenotypes of the offspring exposed to maternal immune activation.
Table S1. List of differentially expressed genes in the maternal immune activation mouse brain.