

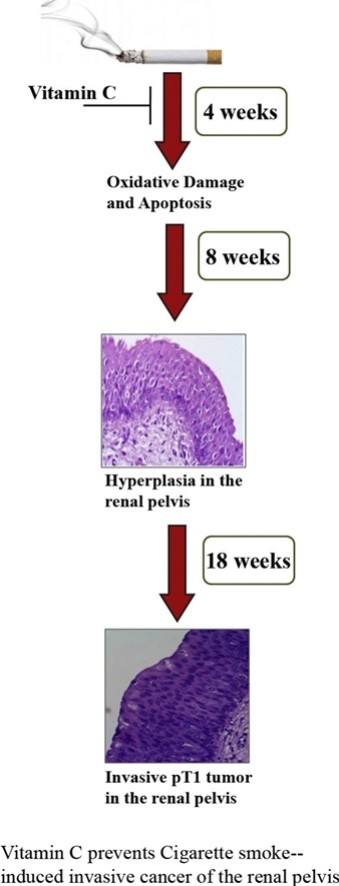
Graphical abstract
Abbreviations: CRP, cancer of the renal pelvis; UC, urothelial carcinoma; p-BSQ, parabenzosemiquinone; p-BQ, parabenzoquinone; CIS, carcinoma in situ; MAPK, Mitogen activated protein kinase; CS, cigarette smoke
Keywords: Invasive cancer of the renal pelvis, Cigarette smoke, Vitamin C, Oxidative damage, Persistent EGFR signaling, Cell cycle deregulation
Highlights
-
•
Cigarette smoke causes invasive urothelial cancer (pT1) in the renal pelvis.
-
•
The initial pathogeneses are oxidative damage followed by aberrant EGFR activation.
-
•
Oral vitamin C supplementation holistically prevents the tumor formation.
Abstract
Urothelial cancer of the renal pelvis (CRP) is predominantly associated with cigarette smoking. However, the molecular pathogenesis of initiation and progression of cigarette smoke (CS)-induced CRP is unknown. Majority of CRP is high grade and high stage at presentation and has a high recurrence rate even after surgery. Earlier we reported that prolonged treatment (24 weeks) of a guinea pig model with p-benzoquinone (p-BQ), a product of CS in vivo, produced carcinoma in situ in the renal pelvis, a noninvasive cancer. Since CS is known to induce invasive cancer, we investigated the effect of CS exposure to the guinea pigs. We observed that CS exposure for a short period (18 weeks) produced invasive tumor (pT1). pT1 was confirmed by immunohistochemistry showing increased immunoexpression of nuclear p53 indicating p53 mutation, aberrant CK20, increased Ki-67 and uniformly negative labeling of CD44. As observed earlier with p-BQ treatment, the initial events of CS exposure were oxidative damage and apoptosis that was followed by persistent signaling through EGFR and MAP kinase pathway. CS exposure also caused hyperphosphorylation of pRb, activation of cyclin E and cell cycle deregulation leading to infiltration of epithelial cells in lamina propria of the renal pelvis resulting in pT1 tumor. Oral supplementation of vitamin C (30 mg/kg guinea pig/day) inhibited oxidative damage and apoptosis and holistically prevented the tumor formation. We consider that our preclinical findings on the intake of adequate vitamin C, along with intense advice for cessation of smoking, will be helpful for the prevention of CS-induced CRP in smokers.
1. Introduction
Urothelial cancer of the renal pelvis (CRP) is a principal component of the upper tract urothelial carcinomas (UC) with an estimated incidence of 1-4 cases per 100,000 individuals per year [1]. CRP is usually more invasive than bladder cancer at diagnosis and is associated with worse prognosis. Almost 70% of CRP is high grade and high stage UC at presentation [2]. Till date, the gold standard treatment of CRP is radical nephroureterectomy. However, CRP is known to have a high recurrence rate even after surgery.
The predominant risk factor for CRP is cigarette smoking [3]. Cigarette smoke (CS) increases the risk of CRP by three to seven folds, compared to the general population [3]. Moreover, smoking-related CRP is associated with disease recurrence and cancer-specific mortality [3]. However, the molecular pathogenesis of initiation and progression of CS-related CRP is unknown.
Using a vitamin C-restricted guinea pig model, earlier we had shown that prolonged treatment (intramuscular injection) for 24 weeks with p-benzoquinone (p-BQ) produced carcinoma in situ (CIS) in the renal pelvis, a noninvasive cancer [4]. Although p-benzoquinone is not present in CS, it is produced from benzosemiquinone (p-BSQ) of CS by oxidation and disproportionation [4]. p-BSQ is present in substantial amounts (100–200 μg) in CS [4]. Since, besides p-BSQ, CS is a complex mixture of more than 4000 compounds, including a number of oxidants, toxic chemicals and carcinogens [5,6]. We therefore investigated the effect of CS exposure to the guinea pigs. We used the guinea pig model because of its similarity with CS-induced pathophysiology of humans, the details of which are given elsewhere [4]. Here, we showed that CS exposure produced invasive tumor (pT1) that has penetrated the basement membrane to invade the lamina propria in the renal pelvis. pT1 usually progresses to more invasive muscularis propria (pT2) and renal parenchyma (pT3) [7].
Most case-control studies have found an inverse association between dietary vitamin C intake and cancers of the lung, breast, colon or rectum, stomach, oral cavity, larynx or pharynx, and esophagus [[8], [9], [10], [11]]. Here, we also showed that the development of CS-induced pT1 tumor in the renal pelvis was prevented by prior oral supplementation of vitamin C (30 mg/kg guinea pig/day).
2. Materials and methods
2.1. Exposure of Guinea pigs to CS
All animal experiments were approved by the Institutional Animal Ethics Committee, University of Calcutta and were in strict conformity with NIH guidelines. Dunkin Hartley albino purebred male guinea pigs weighing 450–550 g were divided into the following weight-matched experimental groups (n = 18 for each group). (i) CS-exposed vitamin C-restricted group, given oral supplementation of 1 mg vitamin C/ animal /day; (ii) CS-exposed vitamin C-sufficient group, given oral supplementation of 30 mg vitamin C/kg body weight of animal/day; (iii) sham control exposed to air instead of CS and fed 1 mg vitamin C/animal/day. We did not include another group of sham control air-exposed animals fed 30 mg vitamin C in our study, because previous reports had shown that there was practically no difference between the air exposed and 30 mg vitamin C-fed animals [12]. Earlier we had shown that 30 mg vitamin C/kg body weight of animal/day prevented cigarette smoke-induced protein damage; 10 mg or 20 mg vitamin C/kg body weight of animal/day was insufficient to prevent cigarette smoke-induced damage [[13], [14], [15]]. The dosage of 30 mg vitamin C/kg body weight of animal/day vitamin C is ≈ 2 g vitamin C/adult human/day [12]. The guinea pigs were subjected to cigarette smoke exposure from 5 Kentucky Research cigarettes 3R4F (2 puffs per cigarette)/animal/day (6 days/week) in a smoke chamber, as described before, The details of smoke chamber and the duration of cigarette smoke exposure were described previously in details [14,16]. In brief, the smoke chamber was similar to that of a 2.5 L vacuum desiccator with an open tube at the top and a side tube fitted with a stopcock. The side tube was fitted with a pump through a cotton filter. The cigarette placed at the top was lit and CS was sucked into the chamber containing the guinea pig by applying a mild suction of 4 cm water through the side tube for 5 s. Thereafter, the vacuum was turned off and the guinea pig was subjected to whole body exposure to the accumulated smoke for 40 s. Two puffs were given per cigarette, allowing the animal to breathe air between each puff. The gap between one cigarette and the next was 1 h. Sham controls were subjected to air exposure instead of CS (Fig. 1).After treatment, the guinea pigs were euthanized under deep anesthesia using i.p. injection of ketamine hydrochloride (100 mg/kg body weight), tissues were collected and some portions were fixed in formalin and the other portions frozen at -80 °C for further experiments.
Fig. 1.

The smoker chamber used to expose guinea pigs to cigarette smoke. 1. The vacuum pump, 2. Pressure gauge, 3. Filter (to trap tar), 4. The vacuum desiccator that acts as the smoke chamber, 5. Cigarette holder containing the cigarette.
2.2. Tissue preparation and histological analysis
Both the left and right kidneys were surgically excised, fixed in formalin and stained with Hematoxylin and Eosin (H&E) (Sigma-Aldrich) by standard protocol. At least three sections per animal were analyzed for histopathology by a pathologist. The details of animals analyzed at each time point is given in Table 1.
Table 1.
Table indicating number of animals analysed at each experimental time points.
| Group | 2 weeks | 4 weeks | 6 weeks | 8 weeks | 18 weeks |
|---|---|---|---|---|---|
| Sham control (air-exposed fed 1 mg vitamin C/animal/day) | 3 | 3 | 3 | 3 | 6 |
| Vitamin C restricted (CS exposed fed 1 mg vitamin C/animal/day) | 3 | 3 | 3 | 3 | 6 |
| Vitamin C sufficient (CS exposed fed30 mg vitamin C/animal/day) | 3 | 3 | 3 | 3 | 6 |
2.3. Tissue lysate preparation
Thin layers of renal pelvis were cut out using a dissection microscope and the dissected tissue samples were lysed in buffer as described before [12].
2.4. Protein oxidation
Protein oxidation was measured as described earlier [12].
2.5. Immunoblotting
Renal pelvis tissue lysate (30 μg protein) was separated by SDS–PAGE and transferred to Immun-Blott PVDF membrane (GE Healthcare). The membrane was blocked in 5% skim milkin TBS for 1 h and then incubated with primary antibody (1:1000) dilutions of phospho p53(Cell Signalling Technologies; CST#2527 overnight at 4 °C), Bax(CST#2774 overnight at 4 °C), Bcl-2(CST#2872 overnight at 4 °C), caspase 3(CST#9662 overnight at 4 °C), cleaved caspase 3(CST#9661 overnight at 4 °C), erk1/2(CST#4695 overnight at 4 °C), phospho-erk1/2(CST#4370 overnight at 4 °C), c-Myc (CST#13987 overnight at 4 °C), p21 waf1/cip1(CST#2947 for 5 h at 37 °C), cyclin D1(CST#2987 for 5 h at 37 °C), phospho pRb (807/811)(CST#8516 overnight at 4 °C), anti-GTPase Hras(1:3000 dilution) (Abcam#ab201054 overnight at 4 °C). cyclin E2 (1:2000 dilution) (Abcam #ab32103 overnight at 4 °C). GAPDH (1:3000)(Biobharati, India overnight at 4 °C) was used as a loading control. After washing three times with TBS-T (20 mM Tris, 500 mM NaCl, 0.1% Tween-20, pH 7.5), the membrane was incubated with the goat anti-rabbit/mouse HRP-conjugated secondary antibody(Genei, Bangalore (1:3000 dilution) (for 1 h at room temperature. The membrane was further washed with TBS-T and developed with Lumiglo reagent (CST).
2.6. Immunoprecipitation
Two mg of protein A sepharose beads (GE Healthcare) were coated with anti-EGFR antibody (Abcam#ab131498) (1:100) in phosphate buffered saline (PBS) at room temperature for 4 h. Renal pelvis lysate (300 μg protein) was added to the beads and incubated overnight at 4 °C. Immunoprecipitates were washed with PBS containing 0.2% Triton X-100 and subjected to immunoblotting using 1:1000 dilution of primary anti-PY20(Abcam# ab10321 for 4 h at 37 °C), anti-phospho-Tyr845(CST#6963overnight at 4 °C), anti-phospho-Tyr 1045(CST#2237 overnight at 4 °C), anti-phospho-Tyr1068(CST#3777 overnight at 4 °C), anti-phospho-Tyr 1173(CST#440 overnight at 4 °C 7), anti-c-Cbl(CST#2747 overnight at 4 °C) and anti-ubiquitin antibody(CST#3933 overnight at 4 °C) After washing three times with TBS-T (20 mM Tris, 500 mM NaCl, 0.1% Tween-20, pH 7.5), the membrane was incubated with the goat anti-rabbit/mouse HRP-conjugated secondary antibody (Genei, Bangalore (1:3000 dilution) (for 1 h at room temperature). The membrane was further washed with TBS-T and developed with Lumiglo reagent (CST).
2.7. Immunohistochemistry
Five μm thick tissue sections were cut, deparaffinized in xylene and hydrated with graded alcohol following standard protocol. The sections were blocked with 3% hydrogen peroxide to quench endogenous peroxidase and subjected to heat-induced epitope retrieval in 10 mmol/L citrate buffer (pH 6.0) for 15 min in a pressure cooker. Sections were blocked using 10% rabbit serum followed by incubation with monoclonal or polyclonal primary antibody. Negative control sections were treated with just rabbit serum. Primary antibody dilutions (1: 100) and treatments were as follows: Ki-67(Abcam# ab15580), overnight at 4 °C; p53 (CST#2527), overnight at 4 °C, Cytokeratin 20 (Abcam# ab76126), 1 h at room temperature; CD44 (Abcam# ab157107), 1 h at room temperature, Cyclin E2 (Abcam# ab32103), overnight at 4 °C, followed by treatment with HRP conjugated secondary antibody for 1 h at room temperature.Images were acquired using digital Digieye 330/210 camera. Ki-67-positive and p53-positive cell counts were measured from ten images per section per animal using Dewinter Biowizard 4.1 imaging software and represented as %±SD. The sections were blinded to the histopathologist in order to reduce study bias.
2.8. Quantitation of immunofluorescence and immunoblots
Quantitation of immunofluorescence and immunoblots were done using NIH Image J software.
2.9. Statistical analysis
Statistical analysis was performed using one-way ANOVA and Fisher’s t-tests. P values were calculated using appropriate F statistics for ANOVAs and t statistics for t-tests. P < 0.05 was considered to be significant. The statistical analyses were performed using MINITAB software.
3. Results and discussion
3.1. Suppression of CS-induced oxidative protein damage and apoptosis is followed by hyperplasia: prevention by vitamin C
Like that reported before with p-BQ treatment [4], we observed that CS exposure caused significant (p < 0.001) protein oxidation and apoptosis (p < 0.001) in the renal pelvis up to 4 weeks of exposure (Fig. 2). Protein oxidation was evidenced by an increase in protein carbonyl formation after 4 weeks compared to sham control and vitamin C-sufficient animals (p < 0.005)(Fig. 1A, B). Apoptosis was evidenced by significant up-regulation of pro-apoptotic markers such as phospho-p53 (p < 0.005), Bax/Bcl-2 ratio (p < 0.005) and cleaved caspase 3 (p < 0.005)(Fig. 1C, D). However, on prolonging CS-exposure up to 8 weeks, both protein oxidation and apoptosis subsided. This was accompanied by hyperplasia (Fig. 2E) as revealed by markedly thickened mucosa with an increase in the number of cell layers without apparent change in the cytology of cells (Fig. 2E). No such changes were observed in either the sham control or the vitamin C-supplemented guinea pig.
Fig. 2.

Cigarette smoke (CS) induces oxidative protein damage and apoptosis that is followed by hyperplasia in the renal pelvis of guinea pigs. (A) Oxyblot depicting time-dependant increase in protein oxidation by CS exposure to guinea pigs. Bottom panel, cropped Ponceau stained blot used as loading control. (B) Quantitation of the protein carbonyl formation after time-dependant CS exposure (n = 3). (C) Immunoblots showing activation of p53 (formation of phospho–p53), expression of B-cell Lymphoma 2 (Bcl-2), overexpression of Bcl2 associated X protein (Bax), expression of caspase 3(c3) and cleaved caspase 3(cc3) after time-dependant CS exposure. (D) Quantitation of the apoptotic protein expression level after 2, 4, 6 and 8 weeks of CS exposure (n = 3). (E) Hematoxylin and Eosin stained sections of the renal pelvis depicting CS-induced hyperplasia. Upper and lower panel show image of 10X and 40X magnification respectively, of the same field (upper panel scale bar = 100 μm and lower panel scale bar = 25 μm). CT, white bars: sham control; CS-vitC, black bars: CS-exposed vitamin C-restricted animals; CS + vit C, gray bars: CS-exposed vitamin C-sufficient animals. Data were statistically analyzed by one-way ANOVA. Significant differences (*P ≤ 0.05, **P ≤ 0.005) were observed in comparison to sham control and vitamin C-sufficient animals. Data are represented as means ± SD (n = 3) of three independent experiments done under similar conditions.
It is well known that CS-induced oxidative damage leads to a plethora of diseases [[17], [18], [19]]. We have observed that the primary trigger for CS-induced pathophysiology is oxidative damage [[13], [14], [15]]. It is also known that oxidative damage plays a pivotal role in apoptosis. Cells that evade apoptosis emit persistent signaling resulting in overgrowth that is likely to contribute to the formation of cancer [20,21]. Suppression of apoptosis and persistent proliferation are hallmarks of most types of cancer [22]. Moreover, deregulated cell proliferation and suppressed cell death together provide the underlying platform for neoplastic progression [23].
3.2. CS-induced pT1 formation as revealed by histology and immunohistochemistry of p53, CD44, CK20, and Ki6
3.2.1. Histology
After continuation of CS exposure for 18 weeks, the animals showed signs of cachexia. At that time point we observed cancerous lesions in the renal pelvis in four out of six vitamin C-restricted animals (Fig. 3). H&E staining showed neoplastic cells as indicated by altered nucleus: cytoplasmic ratio, loss of polarity of cells, and lack of cytoplasmic clearing after 18 weeks of CS exposure. These cells had infiltrated the lamina propria, the subepithelial connective tissue, indicating pathological staging pT1 (Fig. 3A). On the other hand, animals showing flat urothelial hyperplasia after 8 weeks of CS exposure did not exhibit any abnormal cytology and showed sequential maturation from base to surface (Fig. 3A). The sham control and vitamin C-sufficient animals did not show any cancerous lesion.
Fig. 3.

Prolonged CS exposure led to cancer of the renal pelvis. (A) Panel showing H&E stained representative section of CS exposure after 8 and 18 weeks that caused hyperplasia and pT1 tumor in the renal urothelium respectively of vitamin C-restricted animals. (Bi) Panel showing p53 stained representative section of CS exposure after 8 and 18 weeks. (Bii) Panel showing CD44 stained representative section of CS exposure after 8 and 18 weeks. (Biii) Panel showing CK20 stained representative section of CS exposure after 8 and 18 weeks. (Biv) Panel showing Ki67 stained representative section of CS exposure after 8 and 18 weeks. CT, sham control; CS-vitC, CS-exposed vitamin C-restricted animals; CS + vit C, CS-exposed vitamin C-sufficient animals (10 fields, n = 3 for 8 weeks of CS-exposure, n = 4 for 18 weeks of CS exposure. 40X magnification (scale bar = 25 μm).
3.2.2. Immunohistochemistry of p53, CD44, CK20, and Ki67
Immunohistochemical studies have been useful in the staging of neoplastic lesions. p53, CD44, CK20, and Ki67 are four protein markers whose immunoreactivity plays potential role in distinguishing neoplasia from non-neoplastic urothelium [[24], [25], [26], [27]]. p53 has a short half-life and usually, nuclear staining of p53 is practically absent in normal urothelium but present in many urothelial cancer [28,29].
We observed that positive nuclear staining for p53 was absent in sham control, vitamin C-sufficient animals, and hyperplastic urothelium after 8 weeks of CS exposure in vitamin C-restricted animals (Fig. 3Bi). However, there was marked immunoexpression of p53 in 43% ± 5.28 SD cells at the pT1 stage after 18 weeks of CS exposure (Fig. 3Bi). Increased nuclear accumulation of p53 protein indicates mutations of the TP53 gene, which is a common event in the development of urothelial invasive neoplasms and is associated with higher tumor grade, more advanced stage, frequent tumor recurrences and worse prognosis [26,30]. Although we detected p53 protein accumulation in pT1 tumor, there was no accumulation of p53 up to the stage of hyperplasia after 8 weeks of CS exposure. This would indicate that p53 gene mutation was not the initial event in CS-induced CRP. CD44, a transmembrane glycoprotein, plays a key role in cell-cell and cell matrix adhesion. It is usually limited to the basal cells in normal urothelium, but absent in tumor [24,27]. We observed that while CD44 immunopositivity was restricted to the basal cells in sham control, vitamin C-sufficient, and hyperplastic urothelium after 8 weeks of CS-exposure (Fig. 3Bii), it was absent in pT1 urothelium (Fig. 3Bii).
Similarly, CK20 is reported to be present only in the umbrella/superficial cells of the normal epithelium [27]. We observed that while CK20 was present in umbrella cells of sham control, vitamin C-sufficient animals, and hyperplastic urothelium after 8 weeks CS exposure (Fig. 3Biii), it was present in full thickness of the neoplastic urothelium after 18 weeks of CS exposure (Fig. 3Biii). Immunopositivity of both CD44 and CK20 helps in predicting patient outcome in urothelial pT1 neoplasms [24,27].
Additionally, the proliferation marker Ki67 has been reported to be overexpressed in pT1 tumors [25]. Here, we showed that compared to negative staining for Ki67 in sham control and vitamin C-sufficient animals, the nuclear positivity of Ki67 is 54% ± 7.72 SD in pT1 urothelium (Fig. 3Biv). Hyperplastic urothelium of 8 weeks CS-exposed animals showed < 3% nuclear positivity for Ki67 (Fig. 3Biv).
3.3. CS-induced persistent proliferation through aberrant activation/phosphorylation of EGFR: prevention by vitamin C
EGFR activation/phosphorylation and its downstream signaling are involved in the development of cancer [[31], [32], [33]]. Previously we and others had shown that CS causes aberrant EGFR activation in cultured human lung cells [34]. Immunoblot of EGFR immunoprecipitate with anti PY20 antibody measures the total phosphorylation of the tyrosine residues. Here, we showed increased overall phosphorylation of PY20 after 18 weeks, compared to 8 weeks of CS exposure (p = 0.007) (Fig. 4A, B). We also showed that compared to both sham control and vitamin C-sufficient animals there was a significant increase in phosphorylation of Y845 after 8weeks (p < 0.005) and 18 weeks (p < 0.005) of CS exposure (Fig. 4A, B). Phosphorylation of Y1068 acts as the docking site for adaptor protein Grb2 that helps in downstream activation of Ras and Ras/Raf/MAPK (Erk1/2) pathway. We observed that after CS-exposure for 8 weeks, there was no significant increase in the phosphorylation of Y1068 (p > 0.1). On the other hand, after 18 weeks of CS exposure Y1068 increased significantly (p = 0.014) (Fig. 4A, B). There was no significant change in the phosphorylation of Y1173 (p > 0.1) after CS exposure for 18 weeks that produced stage pT1 of CRP (Fig. 4A, B). Phosphorylation of Y1045 is the docking site for c-cbl which in return recruits ubiquitin for subsequent ubiquitination leading to EGFR degradation that is essential for signal regulation [32]. Here, we showed that although there was no significant change in the phosphorylation of Y1045 (p > 0.1) after 8 weeks of CS exposure, phosphorylation of Y1045 markedly decreased on prolonging the CS exposure up to 18 weeks (p = 0.004) (Fig. 4A, B).
Fig. 4.

CS causes aberrant EGFR phosphorylation leading to persistent signaling. (A) Immunoblot of EGFR immunoprecipitate showing the different phosphorylation status of Tyrosine (Y) residues in the cytoplasmic domain of EGFR. (B) Quantitation of phosphorylation of Tyr residues after 8 and 18 weeks of CS exposure. (C) Immunoblot showing binding of c-cbl and ubiquitin to EGFR. (D) Quantitation of c-Cbl and ubiquitin binding to EGFR after 8 and 18 weeks of exposure. CT, white bars: sham control; CS-vitC, black bars: CS-exposed vitamin C-restricted animals; CS + vit C, gray bars: CS-exposed vitamin C-sufficient animals where n = 3, for 8 weeks and n = 4 for 18 weeks of CS exposure. Data represented as mean ± SD of experiments done in triplicate under similar conditions. Data were statistically analyzed by one-way ANOVA and Fischer’s t-test. Significant differences (*P ≤ 0.05, **P ≤ 0.005) were observed in comparison to sham control and vitamin C-sufficient animals.
Phosphorylation of Tyr 1045 acts as the docking site for E3 ubiquitin ligase c-Cbl. This ligase recruits ubiquitin, which results in EGFR ubiquitination and proteasomal degradation [32]. Unphosphorylated Tyr 1045 is unable to dock c-Cbl and thereby prevents ubiquitination and degradation of EGFR causing recycling of the receptor leading to persistent MAP kinase signaling and continued proliferation that disrupts homeostasis of the cell leading to cancer [33].Immunoblotting of EGFR immunoprecipitates revealed that after 18 weeks of CS exposure, the level of E3 ubiquitin-protein ligase c-Cbl (p < 0.01) and ubiquitin binding to EGFR (p < 0.005) were practically absent compared to sham control and vitamin C-sufficient animals (Fig. 4C, D). Thus the activated EGFR was neither ubiquitinated nor subsequently degraded due to its inability to bind the E3-ligase, c-Cbl. This allowed EGFR to remain active for a longer period, thereby causing prolonged survival signals
3.4. CS-induced activation of MAPK (erk1/2) and c-Myc: prevention by vitamin C
The Ras/Raf/MAPK (Erk1/2) cascade is one of the major and best studied EGFR downstream pathways, which links extracellular signals to important cellular processes including cell growth, proliferation and cancer [33,35]. Here we show that compared to sham control and vitamin C-sufficient animals, CS exposure for 8 weeks resulted in significant increase in the GTPase Hras protein level (p < 0.05) (Fig. 5A, B). On prolonging the exposure for 18 weeks, the expression level of GTPase Hras increased further (p < 0.05) (Fig. 5A, B). The active Hras ultimately activates erk1/2 (p44/p42 MAPK) by phosphorylation [35]. We also showed that the protein expression level of phosphorylated erk1/2 (p44/p42 MAPK), which increased after 8 weeks CS exposure (p = 0.001), increased further after prolonging the exposure for 18 weeks (p < 0.05) (Fig. 5A, B). Activated MAPK in return is reported to activate various transcription factors including c-Myc that is reported to be overexpressed in a number of tumors [36,37]. Our results also showed that c-Myc protein expression level increased significantly after 18 weeks of CS exposure (p < 0.05) (Fig. 5A, B).
Fig. 5.

CS-induced HRas, and erk1/2 activation.(A) Immunoblots showing expression of GTPase Hras, erk1/2, p-erk1/2 (phospho-erk1/2) and c-myc. (B) Quantitation of expression of GTPase Hras, erk1/2, p-erk1/2 and c-myc after 8 and 18 weeks of CS exposure. CT, white bars: sham control; CS-vitC, black bars: CS-exposed vitamin C-restricted animals; CS + vit C, gray bars: CS-exposed vitamin C-sufficient animals where n = 3, for 8 weeks and n = 4 for 18 weeks of CS exposure. Data represented as mean ± SD of experiments done in triplicate under similar conditions. Data were statistically analyzed by one-way ANOVA and Fischer’s t-test. Significant differences (*P ≤ 0.05, **P ≤ 0.005) were observed in comparison to sham control and vitamin C-sufficient animals.
3.5. Cell cycle deregulation: prevention by vitamin C
Cyclin D1 expression has been reported to be decreased in several types of invasive urothelial carcinoma [30,38]. We also observed that cyclin D1 protein expression decreased markedly after CS exposure for 18 weeks (p < 0.001) (Fig. 6A, B), when pT1 developed. Overexpression of cyclin E protein plays important roles in cell cycle deregulation and CRP [[39], [40], [41]]. Using both immunoblot (Fig. 6A) and immunohistochemistry (Fig. 6E) we observed that compared to sham controls and vitamin C-sufficient guinea pigs, the expression cyclin E2 protein increased significantly in CRP after 18 weeks of CS exposure (p < 0.001). Cyclin E protein overexpression in association with p53 alteration is an important prognostic indicator of tumor aggressiveness [28]. We further observed that the expression of p21waf1/cip1, the cyclin-dependent kinase inhibitor and a key mediator of the p53-dependent cell cycle, decreased significantly (p < 0.05) (Fig. 6C, D). Downregulation of p21waf1/cip1 causes inactivation of retinoblastoma (pRb) protein through hyperphosphorylation at serine 807/811 resulting in cell cycle progression [29]. We also observed marked increase in hyperphosphorylation of pRb at serine 807/811 after 18 weeks of CS exposure (p < 0.05) (Fig. 6C, D). The pRb protein has a central role in restricting the cell cycle progression at the G1/S transition [42]. In the early G1 and G0, pRb remains hypophosphorylated and bound with the E2F family of transcription factors and prevents excessive cell growth. As the cells progress into late G1 and early S phase, pRb becomes hyperphosphorylated and dysfunctional. This disrupts its association with E2F family resulting in excessive proliferation [30]. The results indicate that exposure to CS causes cell cycle deregulation at the G1/S checkpoint, the most important checkpoint in the mammalian cell cycle.
Fig. 6.

CS-induced cell cycle deregulation (A) Immunoblots showing protein expression of Cyclin D1, Cyclin E2, p21 waf1/Cip1 and hyperphosphorylated Retinoblastoma (pRb) at Ser 807/811. (B) Quantitation of protein expression of cell cycle regulatory proteins after 8 and 18 weeks of CS exposure. (C) Immunohistochemistry of Cyclin E2 protein after 18 weeks of CS exposure at 40X magnification (scale bar = 25 μm). CT, white bars: sham control; CS-vitC, black bars: CS-exposed vitamin C-restricted animals; CS + vit C, gray bars: CS-exposed vitamin C-sufficient animals where n = 3, for 8 weeks and n = 4 for 18 weeks of CS exposure. Data represented as mean ± SD of experiments done in triplicate under similar conditions. Data were statistically analyzed by one-way ANOVA and Fischer’s t-test. Significant differences (*P ≤ 0.05, **P ≤ 0.005) were observed in comparison to sham control and vitamin C-sufficient animals.
Usually CRP is diagnosed at advanced stages when treatment option is radical nephroureterectomy that has high risk of recurrence. According to WHO, chemoprevention of cancer is a basic tenet and the goal of therapeutic intervention. In the case of CS-induced CRP, oxidative damage appears to be the initial event that triggers apoptosis and persistent signaling ultimately leading to advanced cancer. Here, we showed that oral supplementation of vitamin C at a dose of 30 mg/kg body weight/day prevents CS-induced CRP. Possibly vitamin C, a strong antioxidant, holistically prevents CS-induced CRP by attenuating oxidative damage and subsequent pathogeneses. It is reported that vitamin C can prevent cancer through its antioxidant function, possibly by inhibiting oxidative damage [43]. Epidemiological and preclinical studies of large doses of vitamin C have been reported to show significant anticancer effects in animal models, tissue culture investigations and clinical trials [[8], [9], [10], [11],[44], [45], [46]]. Also, clinical trial indicates that high doses of vitamin C are well tolerated and safe [45].
3.6. Comparisons of tumor observed following CS exposure and p-BQ treatment (invasive vs. noninvasive cancer)
Earlier we had shown that p-BQ in amounts available from CS exposure caused carcinoma in situ (CIS) in the renal pelvis of guinea pigs that was prevented by oral administration of vitamin C (30 mg/kg body weight/day). p-BQ closely mimicked CS for producing oxidative damage, apoptosis, persistent proliferation and cell cycle degradation in the renal pelvis leading to development of CIS [4]. However, in contrast to CS-induced CRP (stage pT1), which is an invasive cancer, p-BQ-induced CIS is a noninvasive cancer. A major difference between CS-induced CRP and p-BQ-induced CIS was that cyclin E was overexpressed in CS-induced CRP, There was no such overexpression of cyclin E in p-BQ-induced CIS. Overexpression of cyclin E is known to be a frequent event in a variety of human malignancies, [40,41,47,48]. Moreover, it took 24 weeks to produce p-BQ-induced CIS, but CS-induced CRP developed in 18 weeks. CS is a complex mixture of many thousand chemicals [5]. It would thus appear that although CS-derived p-BQ is a causative factor for the initiation and progression of CRP, some other chemical(s) present in CS triggers the advancement of smoke-induced CIS to pT1. Further research will be needed to explore the nature of the chemical(s).
4. Conclusions
Cigarette smoke (CS) exposure produced invasive tumor (pT1) in the renal pelvis of guinea pigs that was prevented by vitamin C (30 mg/guinea pig/day). pT1 usually progresses to more invasive muscularis propria (pT2) and renal parenchyma (pT3). The gold standard treatment of cancer of the renal pelvis (CRP) is radical nephroureterectomy. However, there is a chance of high recurrence rate even after surgery. Obviously, early intervention would constitute a basic tenet of prevention of CRP. Although the best method of prevention of CS-induced CRP is cessation of smoking, it has proven difficult to achieve and yet to be accomplished, particularly in the developing countries. In many respects, CS-exposed guinea pigs have similarities with human smokers [4]. Therefore, the results may be applicable to humans. We consider that our preclinical findings, along with intense advice for cessation of smoking, will have mechanistic insights into the use of vitamin C for the prevention of CS-induced CRP in smokers. Our observations might also help scientists and clinicians to devise other effective strategies for early intervention of the disease.
Authors contribution
Conceived and designed the experiments: Indu B Chatterjee, Shinjini Ganguly.
Performed most of the experiments: Shinjini Ganguly. Analyzed most of the data: Indu B Chatterjee, Shinjini Ganguly
Statistical analysis done: Ayan Chandra.
Wrote the paper: Indu B Chatterjee, Shinjini Ganguly.
Financial support
The study was supported by CSIR; Juthika Research Foundation and Krishna & Sukhamoy Lahiri Cancer Research Foundation of Calcutta University. IBC is INSA Honorary Scientist; SG is CSIR (NET) Research Fellow.
Transparency document
Acknowledgments
We thank Dr. Sayeed M. Nadeem, MD, N. G. Medicare, Kolkata 700029, India, for carrying out histopathological analyses.
References
- 1.Ehsani L., Osunkoya A.O. Human epidermal growth factor receptor 2 expressions in urothelial carcinoma of the renal pelvis: correlation with clinicopathologic parameters. Int. J. Clin. Exp. Pathol. 2014;7:2544–2550. [PMC free article] [PubMed] [Google Scholar]
- 2.Jemal A., Siegel R., Ward E., Murray T., Xu J., Smigal C. Cancer statistics. CA Can. J. Clin. 2006;56:106–130. doi: 10.3322/canjclin.56.2.106. [DOI] [PubMed] [Google Scholar]
- 3.Mc Laughlin J.K., Silverman D.T., Hsing A.W., Ross R.K., Schoenberg J.B., Yu M.C. Cigarette smoking and cancers of the renal pelvis and ureter. Cancer Res. 1992;52:254–257. [PubMed] [Google Scholar]
- 4.Ganguly S., Chandra A., Chattopadhyay D.J., Chatterjee I.B. P-benzoquinone initiates non-invasive urothelial cancer through aberrant tyrosine phosphorylation of EGFR, MAP kinase activation and cell cycle deregulation: Prevention by vitamin C. Toxicol. Rep. 2017;4:296–305. doi: 10.1016/j.toxrep.2017.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stewart B.W., Kleihues P. International Agency 23 for Research on Cancer. 2003. World cancer report; pp. 21–22. Lyon, France. [Google Scholar]
- 6.U.S. Department of Health and Human Services . U.S. Department of Health and Human Services, Centers for Disease Control and Prevention, National Center for Chronic Disease Prevention and Health Promotion, Office on Smoking and Health; Atlanta, GA: 2014. The Health Consequences of Smking-50 Years of Progress: A Report of the Surgeon General, 2014. [Google Scholar]
- 7.Trpkov K., Smith S.C., Patel P., Amin M.B. Uper urinary tract urothelial carcinoma pathogy. In: Shariat S.F., Xylinas E., editors. Upper Tract Urothelial Carcinoma. Springer; New York: 2015. pp. 55–71. [Google Scholar]
- 8.Chen Q., Espey M.G., Sun A.Y., Lee J.H., Krishna M.C., Shacter E., Choyke P.L., Pooput C., Kirk K.L., Buettner G.R., Levine M. Ascorbate in pharmacologic concentrations selectively generates ascorbate radical and hydrogen peroxide in extracellular fluid in vivo. Proc. Natl. Acad. Sci. U. S. A. 2007;104:8749–8754. doi: 10.1073/pnas.0702854104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen Q., Espey M.G., Sun A.Y., Pooput C., Kirk K.L., Krishna M.C., Khosh D.B., Drisko J., Levine M. Pharmacologic doses of ascorbate act as a pro oxidant and decreased growth of aggressive tumor xenografts in mice. Proc. Natl. Acad. Sci. U. S.A. 2008;105:11105–11109. doi: 10.1073/pnas.0804226105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yun J., Mullarky E., Lu C., Bosch K.N., Kavalier A., Rivera K. Vitamin C selectively kills KRAS and BRAF mutant colorectal cancer cells by targeting GAPDH. Science. 2015;350:1391–1396. doi: 10.1126/science.aaa5004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Frei B., England L., Ames B.N. Ascorbate is an outstanding antioxidant in human blood plasma. Proc. Natl. Acad. Sci. U. S. A. 1989;86:6377–6381. doi: 10.1073/pnas.86.16.6377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ghosh A., Ganguly S., Dey N., Banerjee S., Das A., Chattopadhyay D.J. Causation of cigarette smoke-induced emphysema by p-benzoquinone and its prevention by vitamin C. Am. J. Respir. Cell Mol. Biol. 2015;52:315–322. doi: 10.1165/rcmb.2013-0545OC. [DOI] [PubMed] [Google Scholar]
- 13.Panda K., Chattopadhyay R., Ghosh M.K., Chattopadhyay D.J., Chatterjee I.B. Vitamin C prevents cigarette smoke induced oxidative damage of proteins and increased proteolysis. Free Radic. Biol. Med. 1999;27(November (9-10)):1064–1079. doi: 10.1016/s0891-5849(99)00154-9. [DOI] [PubMed] [Google Scholar]
- 14.Panda K., Chattopadhyay R., Chattopadhyay D.J., Chatterjee I.B. Vitamin C prevents cigarette smoke-induced oxidative damage in vivo. Free Radic. Biol. Med. 2000;29(July (2)):115–124. doi: 10.1016/s0891-5849(00)00297-5. [DOI] [PubMed] [Google Scholar]
- 15.Banerjee S., Chattopadhyay R., Ghosh A., Koley H., Panda K., Roy S., Chattopadhyay D., Chatterjee I.B. Cellular and molecular mechanisms of cigarette smoke-induced lung damage and prevention by vitamin C. J. Inflamm. (Lond.) 2008;5:21. doi: 10.1186/1476-9255-5-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Banerjee S., Maity P., Mukherjee S., Sil A.K., Panda K., Chattopadhyay D., Chatterjee I.B. Black tea prevents cigarette smoke-induced apoptosis and lung damage. J. Inflamm. (Lond.) 2007;14:4:3. doi: 10.1186/1476-9255-4-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Starke R.M., Thompson J.W., Ali M.S., Pascale C.L., Lege A.M., Ding D., Chalouhi N., Hasan D.M., Jabbour P., Owens G.K., Toborek M., Hare J.M., Dumont A.S. Cigarette smoke initiates oxidative stress-induced cellular phenotypic modulation leading to cerebral aneurysm pathogenesis. Arteriosclerosis Thrombosis Vasc. Biol. 2018;38(2018):610–621. doi: 10.1161/ATVBAHA.117.310478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Asami S., Manabe H., Miyake J., Tsurudome Y., Hirano T., Yamaguchi R., Itoh H., Kasai H. Cigarette smoking induces an increase in oxidative DNA damage,8 -hydroxydeoxyguanosine, in a central site of the human lung. Carcinogenesis. 1997;8:1763–1766. doi: 10.1093/carcin/18.9.1763. [DOI] [PubMed] [Google Scholar]
- 19.Halliwell B., Poulsen H.E. Springer Press; 2006. Cigarette Smoke and Oxidative Stress. ISBN 3-540-31410-31415. [Google Scholar]
- 20.Kannan K., Jain S.K. Oxidative stress and apoptosis. Pathophysiology. 2000;7:153–163. doi: 10.1016/s0928-4680(00)00053-5. [DOI] [PubMed] [Google Scholar]
- 21.Ryoo H.D., Bergmann A. The role of apoptosis-induced proliferation for regeneration and cancer. CSH Perspect. Biol. 2012;4 doi: 10.1101/cshperspect.a008797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hanahan D., Weinberg R.A. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 23.Frenald K., Kurokawa M. Evading apoptosis in cancer. Trends Cell Biol. 2013;23(12):620–633. doi: 10.1016/j.tcb.2013.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Masuda M., Takano Y., Iki M., Makiyama K., Noguchi S., Hosaka M. Expression and prognostic value of CD44 isoforms in transitional cell carcinoma of renal pelvis and ureter. J. Urol. 1999;161:805–808. [PubMed] [Google Scholar]
- 25.Krabbe L.M., Bagrodia A., Lotan Y., Gayed B.A., Darwish O.M., Youssef R.M. Prospective analysis of Ki-67 as an independent predictor of oncologic outcomes in patients with high-grade upper tract urothelial carcinoma. J. Urol. 2014;191(11):28–34. doi: 10.1016/j.juro.2013.07.012. [DOI] [PubMed] [Google Scholar]
- 26.Yemelyanova A.R., Vang M., Kshirsagar D., Lu M.A., Marks M., Shih I. Immunohistochemical staining patterns of p53 can serve as a surrogate marker for TP53 mutations in ovarian carcinoma, an immunohistochemical and nucleotide sequencing analysis. Mod. Pathol. 2011;24:1248–1253. doi: 10.1038/modpathol.2011.85. [DOI] [PubMed] [Google Scholar]
- 27.Desai S., Lim S.D., Jimenez R.E., Chun T., Keane T.E., McKenney J.K. Relationship of cytokeratin 20 and CD44 protein expression with WHO/ISUP grade in pTa and pT1 papillary urothelial neoplasia. Mod. Pathol. 2000;13(12):1315–1323. doi: 10.1038/modpathol.3880241. [DOI] [PubMed] [Google Scholar]
- 28.Furihata M., Ohtsuki Y., Sonobe H., Shuin T., Yamamoto A., Terao N. Prognostic significance of cyclin E and p53 protein over expression in carcinoma of the renal pelvis and ureter. Br. J. Cancer. 1998;77:783–788. doi: 10.1038/bjc.1998.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mitra A.P., Hansel D.E., Cote R.J. Prognostic value of cell-cycle regulation biomarkers in bladder cancer. Semin. Oncol. 2012;39:524–533. doi: 10.1053/j.seminoncol.2012.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tut V.M., Braithwaite K.L., Angus B., Neal D.E., Lunec J., Mellon J.K. Cyclin D1 expression in transitional cell carcinoma of the bladder, correlation with p53, waf1, pRb, and Ki67. Br. J. Cancer. 2001;84(2):270–275. doi: 10.1054/bjoc.2000.1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Leibl S., Zigeuner R., Hutterer G., Chromecki T., Rehak P., Langner C. EGFR expression in urothelial carcinoma of the upper urinary tract is associated with disease progression and metaplastic morphology. APMIS. 2008;116(2008):27–32. doi: 10.1111/j.1600-0463.2008.00859.x. [DOI] [PubMed] [Google Scholar]
- 32.Yarden Y. The EGFR family and its ligands in human cancer:signalling mechanisms and therapeutic oprtunities. Eur. J. Cancer. 2001;37:S3–S8. doi: 10.1016/s0959-8049(01)00230-1. [DOI] [PubMed] [Google Scholar]
- 33.Santarpia Libero, Lippman Scott L., E-Naggar Adel K. Targeting the MAPK–RAS–RAF signaling pathway in cancer therapy. Expert Opin. Ther. Targets. 2012;16(1):103–119. doi: 10.1517/14728222.2011.645805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dey N., Chattopadhyay D.J., Chatterjee I.B. Molecular mechanisms of cigarette smoke-induced proliferation of lung cells and prevention by vitamin C. J. Oncol. 2011;2011:561862. doi: 10.1155/2011/561862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dunn K.L., Espino P.S., Drobic B., He S., Davie J.R. The Ras-MAPK signal transduction pathway, cancer and chromatin remodeling. Biochem. Cell Biol. 2005;83:1–14. doi: 10.1139/o04-121. [DOI] [PubMed] [Google Scholar]
- 36.Schmidt E.V. The role of c-myc in cellular growth control. Oncogene. 1999;18(19):2988–2996. doi: 10.1038/sj.onc.1202751. [DOI] [PubMed] [Google Scholar]
- 37.Sears R., Nuckolls F., Haura E., Taya Y., Tamai K., Nevins J.R. Multiple Ras dependent phosphorylation pathways regulate c-Myc protein stability. Genes Dev. 2000;14:2501–2514. doi: 10.1101/gad.836800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lee C.C.R., Yamamoto S., Morimura K., Wanibuchi H., Nishisaka N., Ikemoto S., Nakatani T., Wada S., Kishimoto T., Fukushima S. Significance of cyclin D1 overexpression in transitional cell carcinomas of the urinary bladder and its correlationwith histopathologic features. Cancer. 1997;79(4):780–789. doi: 10.1002/(sici)1097-0142(19970215)79:4<780::aid-cncr15>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- 39.Bortner D.M., Rosenberg M.P. Induction of mammary gland hyperplasia and carcinomas in transgenic mice expressing human cyclin E. Mol. Cell Biol. 1997;17:453–459. doi: 10.1128/mcb.17.1.453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Donnellan R., Chetty R. Cyclin E in human cancers. FASEB J. 1999;13:773–780. doi: 10.1096/fasebj.13.8.773. [DOI] [PubMed] [Google Scholar]
- 41.Sandhu C., Slingerland J. Deregulation of the cell cycle in cancer. Cancer Detect Prev. 2000;24:107–118. [PubMed] [Google Scholar]
- 42.Sherr C.J. Cancer cell cycles. Science. 1996;274:1672–1677. doi: 10.1126/science.274.5293.1672. [DOI] [PubMed] [Google Scholar]
- 43.Li Y., Schellhorn H.E. New developments and novel therapeutic perspectives for vitamin C. J. Nutr. 2007;137:2171–2184. doi: 10.1093/jn/137.10.2171. [DOI] [PubMed] [Google Scholar]
- 44.Luo J., Shen L., Zheng D. Association between vitamin C intake and lung cancer: a dose-response meta-analysis. Sci. Rep. 2014;4:6161. doi: 10.1038/srep06161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hoffer L.J., Levine M., Assouline S., Melnychuk D., Padayatty S.J., Rosadiuk K., Rousseau C., Robitaille L., Miller W.H. Phase I clinical trial of i.v. ascorbic acid in advanced malignancy. Ann. Oncol. 2008;19(11):1969–1974. doi: 10.1093/annonc/mdn377. [DOI] [PubMed] [Google Scholar]
- 46.Stephenson C.M., Levin R.D., Spector T., Lis C.G. Phase I clinical trial to evaluate the safety, tolerability, and pharmacokinetics of high-dose intravenous ascorbic acid in patients with advanced cancer. Cancer Chemother. Pharmacol. 2013;72:139–146. doi: 10.1007/s00280-013-2179-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bortner D.M., Rosenberg M.P. Induction of mammary gland hyperplasia and carcinomas in transgenic mice expressing human cyclin E. Mol Cell Biol. 1997;17:453–459. doi: 10.1128/mcb.17.1.453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sgambato A., Camerini A., Pani G., Cangiano R., Faraglia B., Bianchino G., Bari B.D., Galeotti T., Cittadini A. Increased expression of cyclin E is associated with an increased resistance to doxorubicin in rat fibroblasts. Br. J. Cancer. 2003;88(June (12)):1956–1962. doi: 10.1038/sj.bjc.6600970. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.