

Abstract
Key Points
Accumulating evidence indicates that passive heat therapy (chronic use of hot tubs or saunas) has widespread physiological benefits, including enhanced resistance against novel stressors (‘stress resistance’).
Using a cell culture model to isolate the key stimuli that are likely to underlie physiological adaptation with heat therapy, we showed that both mild elevations in temperature (to 39°C) and exposure to serum from human subjects who have undergone 8 weeks of heat therapy (i.e. altered circulating factors) independently prevented oxidative and inflammatory stress associated with hypoxia‐reoxygenation in cultured endothelial cells.
Our results elucidate some of the mechanisms (i.e. direct effects of temperature vs. circulating factors) by which heat therapy seems to improve resistance against oxidative and inflammatory stress.
Heat therapy may be a promising intervention for reducing cellular damage following ischaemic events, which has broad implications for patients with cardiovascular diseases and conditions characterized by ‘chronic’ ischaemia (e.g. peripheral artery disease, metabolic diseases, obesity).
Abstract
Repeated exposure to passive heat stress (‘heat therapy’) has widespread physiological benefits, including cellular protection against novel stressors. Increased heat shock protein (HSP) expression and upregulation of circulating factors may impart this protection. We tested the isolated abilities of mild heat pretreatment and serum from human subjects (n = 10) who had undergone 8 weeks of heat therapy to protect against cellular stress following hypoxia‐reoxygenation (H/R), a model of ischaemic cardiovascular events. Cultured human umbilical vein endothelial cells were incubated for 24 h at 37°C (control), 39°C (heat pretreatment) or 37°C with 10% serum collected before and after 8 weeks of passive heat therapy (four to five times per week to increase rectal temperature to ≥ 38.5°C for 60 min). Cells were then collected before and after incubation at 1% O2 for 16 h (hypoxia; 37°C), followed by 20% O2 for 4 h (reoxygenation; 37°C) and assessed for markers of cell stress. In control cells, H/R increased nuclear NF‐κB p65 protein (i.e. activation) by 106 ± 38%, increased IL‐6 release by 37 ± 8% and increased superoxide production by 272 ± 45%. Both heat pretreatment and exposure to heat therapy serum prevented H/R‐induced NF‐κB activation and attenuated superoxide production; by contrast, only exposure to serum attenuated IL‐6 release. H/R also decreased cytoplasmic haemeoxygenase‐1 (HO‐1) protein (known to suppress NF‐κB), in control cells (−25 ± 8%), whereas HO‐1 protein increased following H/R in cells pretreated with heat or serum‐exposed, providing a possible mechanism of protection against H/R. These data indicate heat therapy is capable of imparting resistance against inflammatory and oxidative stress via direct heat and humoral factors.
Keywords: endothelial function, stress resistance, ischemia reperfusion, hot water immersion, sauna, heat shock proteins
Key Points
Accumulating evidence indicates that passive heat therapy (chronic use of hot tubs or saunas) has widespread physiological benefits, including enhanced resistance against novel stressors (‘stress resistance’).
Using a cell culture model to isolate the key stimuli that are likely to underlie physiological adaptation with heat therapy, we showed that both mild elevations in temperature (to 39°C) and exposure to serum from human subjects who have undergone 8 weeks of heat therapy (i.e. altered circulating factors) independently prevented oxidative and inflammatory stress associated with hypoxia‐reoxygenation in cultured endothelial cells.
Our results elucidate some of the mechanisms (i.e. direct effects of temperature vs. circulating factors) by which heat therapy seems to improve resistance against oxidative and inflammatory stress.
Heat therapy may be a promising intervention for reducing cellular damage following ischaemic events, which has broad implications for patients with cardiovascular diseases and conditions characterized by ‘chronic’ ischaemia (e.g. peripheral artery disease, metabolic diseases, obesity).
Introduction
Cardiovascular diseases (CVDs) remain the leading cause of death in the developed world. Of CVD‐related deaths, > 60% are attributable to fatal cardiovascular (CV) ischaemic events, including myocardial infarction and stroke (Benjamin et al. 2017). Tissue damage associated with ischaemic events occurs due to a combination of ischaemia and the significant inflammatory and oxidative stress induced by subsequent reperfusion, referred to as ischaemia‐reperfusion injury (Yang et al. 2016). Endothelial cells are particularly vulnerable (Gori et al. 2007; Jabs et al. 2010), as they are at the interface of the blood and the smooth muscle, are one of the first cell types to be exposed to the potentially damaging stress, and rapidly synthesize and release pro‐inflammatory cytokines and oxygen‐derived free radicals, propagating damage to nearby cells. Furthermore, endothelial cell injury and swelling can impede blood flow upon reperfusion, resulting in greater damage to cell types such as myocardial cells (Chan et al. 2012) and neurons (Asiedu‐Gyekye & Vaktorovich, 2003). Thus, interventions aimed at improving resistance against inflammatory and oxidative stress, in particular in endothelial cells, may protect against tissue damage and reduce mortality associated with ischaemic events.
One such protective intervention may be passive heat therapy, the repeated use of hot baths or saunas. Indeed, lifelong habitual sauna use has been shown to considerably reduce CV‐related and all‐cause mortality, including a 48% reduction in fatal events associated with coronary heart disease (Laukkanen et al. 2015). Studies in animals have demonstrated that 4 weeks of continuous heat exposure reduces infarct size following myocardial ischaemia‐reperfusion in isolated hearts (Eynan et al. 2002) and reduces neuronal damage and cognitive deficits following cerebral hypoxia‐reoxygenation (H/R) (Yacobi et al. 2014). In humans, we showed that acute hot water immersion protects against reductions in brachial artery endothelial function induced by whole‐arm ischaemia‐reperfusion (Brunt et al. 2016 c), a validated in vivo model of endothelial ischaemia‐reperfusion (Kharbanda et al. 2001; Loukogeorgakis et al. 2010).
The mechanisms behind the protective effects of heat therapy are probably multifold. Elevations in body core temperature upregulate heat shock proteins (HSPs), a class of proteins that stabilize and activate a variety of other proteins important to the cardiovascular system, including anti‐inflammatory and anti‐oxidative proteins (Baek et al. 2000; Van Molle et al. 2002; Choi et al. 2005; Kim et al. 2005). In addition, as heat stress is a systemic stimulus, it is likely that humoral factors play a large role in adaptation. The independent effects of these two key stimuli can be isolated using cell culture models.
Therefore, in the present study, we investigated whether heat therapy could induce cellular stress resistance, and the key associated mechanisms, using an H/R model in cultured endothelial cells. H/R, which is the ex vivo equivalent of ischaemia‐reperfusion injury, induces inflammatory and oxidative stress primarily through activation of the transcription factor nuclear factor‐kappa B (NF‐κB) (Natarajan et al. 2002; Xie et al. 2005; Zhang et al. 2017). Using this model, we first investigated whether mild heat pretreatment at 39°C for 24 h or exposure to 10% serum from human subjects who had undergone 8 weeks of passive heat therapy could protect endothelial cells against H/R‐induced inflammatory and oxidative stress and hypothesized that both would reduce NF‐κB activation, release of pro‐inflammatory cytokines [e.g. interleukin (IL)‐6] and oxidative stress (e.g. superoxide production).
Secondly, we investigated potential mechanisms behind heat therapy‐mediated protection against H/R. Haemeoxygenase‐1 (HO‐1), which is also an HSP (Hsp32), is capable of suppressing NF‐κB activation (Chi et al. 2014) and has been shown to protect against H/R‐induced inflammation (Park et al. 2013; Chen et al. 2016). We hypothesized that both heat pretreatment and exposure to heat therapy serum would upregulate HO‐1 and that it would remain elevated following H/R.
Methods
Ethical approval
All subjects provided oral and written informed consent prior to participation in the study, as set forth by the Declaration of Helsinki. All experimental procedures were approved by the Institutional Review Board at the University of Oregon (protocol number: 09272013.25). The study was registered on ClinicalTrials.gov (identifier: NCT02518399).
Human subjects
Subject characteristics are provided in Table 1. Twenty young (22 ± 1 years), healthy (no history of CV‐related diseases), inactive (<2 h aerobic exercise per week) male and female subjects who were non‐smokers and were not taking any prescription medications except contraceptives participated in 8 weeks of either hot (40.5°C; heat therapy group) or thermoneutral (36°C; sham group) water immersion (n = 10 per group) 4–5 times per week (36 sessions total), as described in detail elsewhere (Brunt et al. 2016 a,b). Heat therapy was sufficient to maintain rectal temperature (Tre) between 38.5 and 39.0°C for 60 min per session; thermoneutral water immersion mimicked the hydrostatic effects of heat therapy while maintaining rectal temperature within 0.2°C of resting (Table 1).
Table 1.
Subject characteristics
| Heat therapy group (n = 10) | Sham group (n = 10) | |||
|---|---|---|---|---|
| 0wk | 8wk | 0wk | 8wk | |
| Male/female | 4/6 | – | 4/6 | – |
| Age (years) | 22 ± 1 | – | 22 ± 1 | – |
| Height (cm) | 173 ± 4 | – | 172 ± 3 | – |
| Weight (kg) | 67 ± 3 | – | 66 ± 3 | – |
| Body mass index (kg/m2) | 22.4 ± 0.6 | – | 22.5 ± 0.6 | – |
| Resting blood pressure (mmHg) | ||||
| Systolic | 112 ± 2 | 108 ± 2 | 110 ± 3 | 107 ± 3 |
| Diastolic | 69 ± 1 | 65 ± 2* | 67 ± 1 | 68 ± 2 |
| Mean arterial | 83 ± 1 | 79 ± 1* | 81 ± 1 | 81 ± 2 |
| Resting heart rate (beats/min) | 59 ± 3 | 59 ± 3 | 62 ± 3 | 61 ± 2 |
| Rectal temperature (°C) | ||||
| Resting | 37.3 ± 0.1 | 36.9 ± 0.1*† | 37.2 ± 0.1 | 37.2 ± 0.1 |
| Peak | 39.0 ± 0.03† | 38.8 ± 0.04*† | 37.5 ± 0.1 | 37.4 ± 0.1 |
Data are shown as mean ± SEM. * P < 0.05 vs. 0wk within group, † P < 0.05 vs. sham group at the same time point. Data are also published elsewhere (Brunt et al. 2016 a,b).
Venous blood was drawn at baseline (prior to the first water immersion session, ‘0wk’), acutely 1 h after the first session (‘Acute HT’) and chronically following 8 weeks of heat therapy (‘8wk’) into serum‐separating vacutainers and Ficoll Hypaque‐containing cell preparation tubes with sodium citrate (CPT Vacutainer, BD, Franklin Lakes, NJ, USA) for the separation of peripheral blood mononuclear cells (PBMCs) from whole blood. The timing of acute blood collection was selected based on pilot data indicating that Hsp70 protein peaks in PBMCs 1 h after hot water immersion. At 8 weeks, blood was collected 24–48 h after the last water immersion session in order to capture the chronic effects of heat therapy rather than the acute effects of the last session (the acute increase in Hsp70 protein in PBMCs completely subsided by 24 h in our pilot work). Subjects reported to the laboratory for these two blood draw visits at approximately the same time of day (±1 h), fasted from food for ≥ 2 h, and having abstained from exercise for ≥ 24 h. Subjects were asked to maintain food and caffeine intake during the previous 24 h similarly between the two visits. Women were studied under the same hormonal conditions (i.e. same phase of their menstrual cycle if naturally menstruating or in the same phase of contraceptives). After collection, venous blood was kept at room temperature for 30 min and then separated by centrifugation. Serum and PBMCs were stored at –80°C until analysis or use in cell culture experiments. We chose to perform cell culture experiments using only sera from heat therapy subjects (not sham serum), as we observed no changes in in vivo vascular function outcomes in sham subjects (Brunt et al. 2016 a,b) and as our model allowed for comparison of responses to H/R between before and after heat therapy within subjects. However, sera and PBMCs from both groups were analysed for Hsp70 (see below).
Cell culture experimental protocol
Human umbilical vein endothelial cells (HUVECs; ATCC, Manassas, VA, USA) were cultured under standard conditions (37°C, 5% CO2, 20% O2) in vascular cell basal medium supplemented with an endothelial growth kit (ATCC). Cells were used for experiments after 2–3 passages. Following a 4‐h serum starve, cells were incubated for 24 h under one of the following conditions (all with 5% CO2 and 20% O2): (1) to investigate the direct effects of elevations in temperature, cells were incubated in supplemented media at 37°C (control) or at 39°C (‘heat pre‐treatment’); and (2) to investigate the effects of humoral factors, cells were incubated under standard conditions in non‐supplemented media with 10% serum from human subjects in the heat therapy group collected at each time point (0wk, Acute HT, and 8wk).
Following 24 h of pre‐incubation, a subset of cells and conditioned media from each condition were collected (referred to as ‘basal’ or ‘pre‐H/R’). The remaining cells were then moved to a 1% oxygen chamber for 16 h of hypoxia, followed by 4 h of reoxygenation at 20% oxygen (all cells maintained at 37°C), after which cells and conditioned media were collected (‘H/R’). We conducted extensive pilot work to optimize the durations of hypoxia and reoxygenation. These were selected as the shortest durations that were still long enough to consistently induce detectible cellular stress using our approaches (see Discussion).
Protein extraction and Western blotting
Cells collected before and after H/R were separated into cytoplasmic and nuclear components for assessment of nuclear NF‐κB p65 and cytoplasmic HO‐1 using the Rockland Nuclear/Cytoplasmic Extract Isolation protocol (https://rockland-inc.com/NuclearExtract.aspx), as previously described (Kiemer et al. 2000). Additional whole cells collected following 24 h of pre‐incubation (pre‐H/R) and PBMCs collected from human subjects were combined with radioimmunoprecipitation assay buffer plus protease inhibitor and lysed via sonication.
Protein from nuclear extracts (10–25 μg), cytoplasmic extracts (10–25 μg), cell lysates (20–50 μg) and PBMC lysates (50 μg) were loaded onto 4–20% SDS polyacrylamide separating gels (Life Technologies, Grand Island, NY, USA), separated by electrophoresis, transferred onto nitrocellulose membranes (Amersham Protran; GE Life Sciences, Marlborough, MA, USA), and Ponceau‐stained to assess transfer across each gel. Nitrocellulose membranes were incubated overnight at 4°C in blocking buffer (Odyssey Blocking buffer; LI‐COR Biosciences, Lincoln, NE, USA) containing primary antibodies, followed by incubation for 1 h at room temperature with the appropriate secondary antibodies. Immunoreactive bands were digitized using a LI‐COR Odyssey infrared imaging system and digitized images were quantified using LI‐COR Image Studio software. Nuclear extract blots were probed for NF‐κB p65 (Cell Signaling Technologies, Danvers, MA, USA, 8242: 1:500; RRID:AB_10859369), lamin A/C (Sigma‐Aldrich, St Louis, MO, USA, SAB4200421; 1:500; RRID:AB_2728671) and β‐actin (to confirm appropriateness of normalizing to lamin A/C; Cell Signaling Technology, 8457; 1:1000; RRID:AB_10950489). Cytoplasmic extract blots were probed for HO‐1 (Assay Designs, Enzo Life Sciences, Farmingdale, NY, USA, SPA‐895; 1:200; RRID:AB_2314637) and β‐actin. Cell lysate blots were probed for superoxide dismutase 2 (MnSOD/SOD2; Sigma‐Aldrich, S5569; 1:1,000; RRID:AB_261921), Hsp70 (Abcam, Cambridge, MA, USA, ab6535; 1:5000; RRID:AB_305549) and vinculin (loading control; Cell Signaling Technology, 4650; 1:1000; RRID:AB_10559207). PBMC lysates were probed for Hsp70 and β‐actin.
Superoxide production
Real‐time superoxide production was assessed in live HUVECs using a cellular superoxide detection assay (Abcam; ab139476), which reacts rapidly and specifically with intracellular superoxide to generate a fluorescent product with excitation/emission of 550/620 nm. Following the 24 h of heat pre‐treatment or serum exposure, HUVECs plated in 96‐well plates were incubated for 1 h with 1 μM superoxide detection stain. Cell fluorescence was determined with a fluorescence microscope with Cy3 filter at 10× optical zoom (Axio Observer.D1; Zeiss, Oberkochen, Germany) immediately following the 1 h incubation with the stain (basal conditions), following 2 h of 1% O2 hypoxia, and following 15, 30, 60, 90 and 120 min of reoxygenation at 20% O2. The duration of H/R was shortened compared to other experiments to prevent decay of the fluorescent stain. Total cell fluorescence per frame was determined using ImageJ analysis software (National Institutes of Health, Bethesda, MD, USA). Total fluorescence of blank un‐stained wells was subtracted from treated wells to obtain values of corrected total cell fluorescence and values from triplicate wells per condition were averaged.
Enzyme‐linked immunosorbent assays
Concentrations of IL‐6 were measured in serum and conditioned media collected before and after H/R using a commercial enzyme linked immunosorbent assay (ELISA) kit (R&D Quantikine 6050; R&D Systems, Minneapolis, MN, USA) according to the manufacturer's instructions and, for conditioned media, were then normalized to total protein in the media (Bio‐Rad Protein Assay; Bio‐Rad, Hercules, CA, USA). Concentrations of Hsp70 in serum from human subjects in both the heat therapy and sham groups were also measured using a high‐sensitivity commercial ELISA kit (Enzo ADI‐EKS‐715; Enzo Life Sciences) according to the manufacturer's instructions.
Statistics
Statistical analyses were performed in R version 3.4.1. Subject characteristics and physiological variables (T re, blood pressure and heart rate) were compared across time into the intervention and across groups using a two‐way mixed design ANOVA with within factor of group and between factor of time into the intervention (0wk vs. 8wk) or using Student's unpaired t test (i.e. characteristics measured only at baseline). For heat pre‐treatment, basal protein expression (MnSOD, Hsp70), superoxide production AUC (area under the curve), and fold changes (pre‐ to post‐H/R) in protein (nuclear NF‐κB, HO‐1) and media [IL‐6] between 37°C and 39°C pre‐treatment conditions were compared using Student's unpaired two‐tailed t test. For serum exposure, basal protein expression (MnSOD, Hsp70), superoxide production AUC, and fold changes (pre‐ to post‐H/R) in protein (nuclear NF‐κB, HO‐1) and media [IL‐6] between cells exposed to 0wk, Acute HT and 8wk serum were compared using one‐way repeated measures ANOVA. For both sets of experiments, media [IL‐6] and superoxide production was compared across time into H/R (pre‐ to post‐H/R for [IL‐6] or across the seven time points for superoxide production) and across conditions (pre‐incubation temperatures or serum treatment) and using two‐way repeated measures ANOVA. Unfortunately, we were underpowered to compare PBMC Hsp70 expression using two‐way ANOVA. Thus, we used Student's paired t tests to compare 0wk vs. Acute HT and 0wk vs. 8wk within groups.
Relationships between post‐H/R HO‐1 protein (fold change from pre‐H/R) and H/R‐induced NF‐κB p65 activation (fold change from pre‐H/R), peak superoxide production and media [IL‐6] were investigated using linear regression, combining data across temperature conditions or serum exposure time points.
For all analyses, when a significant main effect was detected, pairwise comparisons were made with Tukey's post hoc test. Significance was set to an alpha level of P < 0.05. Data are presented as mean ± SEM.
Results
Subject characteristics and rectal temperature responses to heat therapy/thermoneutral water immersion are included in Table 1. Twenty subjects completed 8 weeks of heat therapy or thermoneutral water immersion (n = 10 per group). Subjects were well matched across both groups for age, height, weight, body mass index and resting blood pressure, as reported elsewhere (Brunt et al. 2016 a,b) (Table 1). Heat therapy sessions resulted in a peak rectal temperature of 39.0 ± 0.1°C during the first session, which only decreased slightly as subjects became acclimated to 38.8 ± 0.1°C during the last session (P < 0.01). Peak rectal temperature attained by subjects during the first and last sessions in the sham group was 37.4 ± 0.2°C and 37.3 ± 0.3°C, respectively (P = 0.50). As such, the temperature conditions for heat pretreatment cell culture experiments were chosen to be 39°C and 37°C.
Heat pretreatment and heat therapy serum prevented H/R‐induced NF‐κB activation
NF‐κB activation was quantified as the fold change in NF‐κB p65 protein in nuclear extracts (i.e. nuclear translocation) from pre‐ to post‐H/R (Ghosh et al. 1998; Han et al. 2013; Zhu et al. 2015), measured using Western blot and normalized to lamin A/C. Results are summarized in Figure 1. In cells incubated under control 37°C conditions, H/R induced NF‐κB activation, as indicated by a 2.0 ± 0.3‐fold increase in nuclear NF‐κB p65 protein. Heat pretreatment prevented H/R‐induced NF‐κB activation (1.2 ± 0.1‐fold change from pre‐H/R, P = 0.03 vs. 37°C) as did exposing cells to serum from human subjects collected after 8 weeks of heat therapy (0.9 ± 0.2‐fold change from pre‐H/R, P = 0.007 vs. 0wk serum; main effect of serum condition: P = 0.03). Exposing cells to serum collected following acute hot water immersion only tended to attenuate NF‐κB activation (1.4±.0.2‐fold change from pre‐H/R, P = 0.06 vs. 0wk serum). Normalization of NF‐κB data to β‐actin (instead of lamin A/C) yielded similar results (37°C: 2.0 ± 0.3 vs. 39°C: 1.1 ± 0.1, P = 0.03; 0wk: 2.5 ± 0.2 vs. 8wk: 1.0 ± 0.4, P = 0.02).
Figure 1. Nuclear factor kappa‐B (NF‐κB) activation.
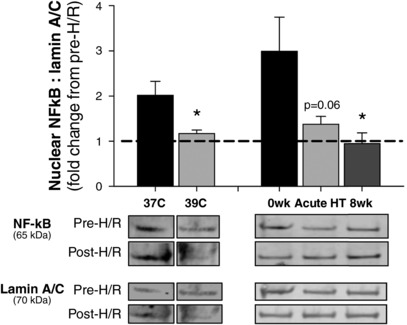
Fold change from pre‐ to post‐hypoxia reoxygenation (H/R) in NF‐κB p65 protein in endothelial cell nuclear extracts, indicative of NF‐κB activation, with representative Western blot images shown below. H/R‐induced NF‐κB activation was prevented in cells pre‐heated at 39°C for 24 h or exposed to sera collected from human subjects who underwent 8 weeks of passive heat therapy. Data are mean ± S.E. * P < 0.05 vs. control conditions (37°C or 0wk).
Heat therapy serum, but not heat pretreatment, prevented H/R‐induced pro‐inflammatory cytokine release
To assess H/R‐induced inflammation, IL‐6 concentrations were measured in conditioned media by ELISA and normalized to total protein concentration in the media. Results are summarized in Figure 2. Both heat pretreatment (P = 0.02) and exposure to serum collected following acute hot water immersion (P = 0.03) increased basal (pre‐H/R) IL‐6 concentration, possibly reflecting acute stress. H/R induced a 1.37 ± 0.08‐fold increase in IL‐6 release into the media (P = 0.01 vs. pre‐H/R). Interestingly, H/R induced a comparable increase in media [IL‐6] in heat pretreated cells (pre‐ to post‐H/R fold change in 39°C cells: 1.40 ± 0.14 vs. 37°C cells, P = 0.85). Although exposure to serum collected following acute hot water immersion increased pre‐H/R media [IL‐6], we observed no further increase in media [IL‐6] following H/R (pre‐ vs. post‐H/R in acute HT cells: P = 0.19). Similarly, the H/R‐mediated increase in media [IL‐6] was prevented by exposure to serum collected following 8 weeks of heat therapy (pre‐ vs. post‐H/R in 8wk cells: P = 0.22). Media concentrations of pro‐inflammatory tumour necrosis factor (TNF)α were also measured, but concentrations were under levels detectible by ELISA.
Figure 2. Interleukin (IL)‐6 concentrations.
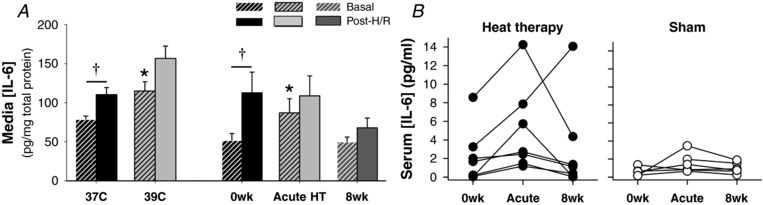
A, concentrations of IL‐6 normalized to total protein in conditioned media collected following 24 h of incubation (‘Basal’; dashed bars) and subsequent hypoxia‐reoxygenation (‘H/R’; solid bars) of endothelial cells incubated at 37°C, at 39°C (heat pretreatment), or with 10% serum from heat therapy subjects collected before (0wk) and after (Acute HT) the first hot water immersion session and after 8 weeks of heat therapy (8wk). Data are mean ± SEM. * P < 0.05 vs. control conditions (37°C or 0wk). † P < 0.05 vs. basal. B, concentrations of IL‐6 in serum from individual heat therapy (n = 7) and sham (n = 6) subjects.
To investigate whether increases in basal media [IL‐6] at the ‘acute HT’ time point were due to increased IL‐6 released from cells following serum exposure vs. greater levels of IL‐6 present in the serum (i.e. increased in vivo IL‐6 release during hot water immersion), we measured serum [IL‐6] by ELISA in a subset of subjects. No significant differences were observed due to high variability in [IL‐6] (range at 0wk: 0.2–8.6 pg/ml); however, the individual data indicated that acute hot water immersion increased serum [IL‐6] in all seven subjects analysed (Fig. 2 B, left) and, on average, serum [IL‐6] increased from 2.3±1.1 (0wk) to 4.9±1.8 (Acute HT; P = 0.30 vs. 0wk). No changes were observed between 0wk and 8wk or in sham subjects (Fig. 2 B, right).
Heat pretreatment and heat therapy serum reduced basal and H/R‐induced oxidative stress
Superoxide production was assessed using a fluorescent cell‐permeable superoxide stain. Cells were stained following 24 h of heat pretreatment or serum exposure and then imaged under basal (pre‐H/R) conditions, following 2 h of 1% hypoxia, and at multiple time points during 2 h of reoxygenation (time durations shortened to prevent decay of the fluorescent stain). Both heat pretreatment (P = 0.002) and serum exposure (main effect: P < 0.001; 0wk vs. Acute HT: P = 0.002, vs. 8wk: P = 0.01) reduced basal (pre‐H/R) superoxide production. In control cells (37°C), H/R induced a 2.9 ± 0.2‐fold peak increase in superoxide production. In cells pre‐heated at 39°C, superoxide production was lower throughout H/R compared to control cells (main effect of temperature: P < 0.001) (Fig. 3 A). Exposure to serum collected following both acute hot water immersion and 8 weeks of heat therapy significantly reduced superoxide production throughout H/R compared to 0wk serum (main effect of serum condition: P < 0.001) (Fig. 3 B). However, these reductions occurred without a reduction in area under the curve (above basal) superoxide production (37°C: 408 ± 18 vs. 39°C: 439 ± 22 A.U.min, P = 0.43; 0wk: 102 ± 34, Acute HT: 115 ± 41, 8wk: 109 ± 47 A.U.min, P = 0.90), indicating protection during H/R is probably due to the reduction in basal superoxide production.
Figure 3. Superoxide production and manganese superoxide dismutase (MnSOD) protein.
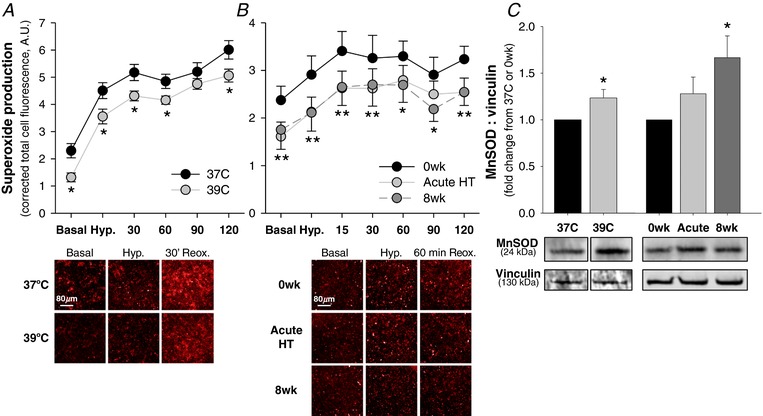
A and B, superoxide production in cells pre‐incubated (A) at 37°C vs. 39°C or (B) with 10% serum from human subjects collected before (0wk) and after the first hot water immersion session (Acute HT) and following 8 weeks of heat therapy (8wk), with representative images taken at 10× optical zoom with Cy3 fluorescent filter shown below (brightness and contrast adjusted uniformly across all images). Cells were stained for superoxide following pre‐incubation and imaged under basal conditions, following 2 h of 1% O2 hypoxia, and during 2 h of reoxygenation (20% O2). Total cell fluorescence per well was quantified and corrected for blank (unstained) wells. Data are mean ± SEM for n = 15 (heat pretreatment) or n = 10 (serum exposure) data points per condition, with wells averaged in triplicate per data point per condition. A, * P < 0.05 vs. 37°C; B, *8wk P < 0.05 vs. 0wk, **both Acute HT and 8wk P < 0.05 vs. 0wk. C, manganese superoxide dismutase (MnSOD) protein in cell lysates collected following 24 h pre‐incubation at 37°C, at 39°C or exposed to 10% serum from human subjects. Representative Western blot images are shown below. Data are mean ± SEM fold change from 37°C (heat pretreatment) or 0wk (serum exposure). * P < 0.05 vs. 37°C (heat pretreatment) or 0wk (serum exposure). [Color figure can be viewed at http://wileyonlinelibrary.com]
To investigate a potential mechanism by which heat pretreatment and heat therapy serum reduce superoxide production, we measured manganese superoxide dismutase (MnSOD) protein in cell lysates collected following heat pretreatment or serum incubation (pre‐H/R) via Western blot (Fig. 3 C). Heat pretreatment significantly increased MnSOD protein, compared to control cells (P = 0.02). Exposure to serum collected following 8 weeks of heat therapy significantly increased MnSOD protein (P = 0.02 vs. 0wk; main effect of time point: P = 0.03), whereas exposure to serum collected following acute hot water immersion only tended to increase MnSOD protein (P = 0.15 vs. 0wk).
Heat pretreatment and heat therapy serum reversed H/R‐induced suppression of HO‐1
To investigate a potential mechanism by which heat pretreatment and heat therapy serum protect against H/R, we measured HO‐1 (Hsp32) in cytoplasmic extracts by Western blot. Neither heat pretreatment (P = 0.52) nor exposure to heat therapy serum (P = 0.83) affected HO‐1 protein under basal conditions (pre‐H/R; Fig. 4 A). In control cells (37°C), H/R suppressed HO‐1 protein expression (reduced down to 0.69 ± 0.07‐fold of pre‐H/R, P < 0.001 vs. pre‐H/R). However, HO‐1 was instead upregulated post‐H/R following heat pretreatment (1.28 ± 0.25‐fold, P = 0.03) and exposure to 8 weeks of heat therapy serum (P = 0.03 vs. 0wk; main effect: P = 0.03) (Fig. 4 B). There was no effect of Acute HT serum on H/R‐induced HO‐1 expression (P = 0.57 vs. 0wk).
Figure 4. Haemeoxygenase‐1 (HO‐1) protein.
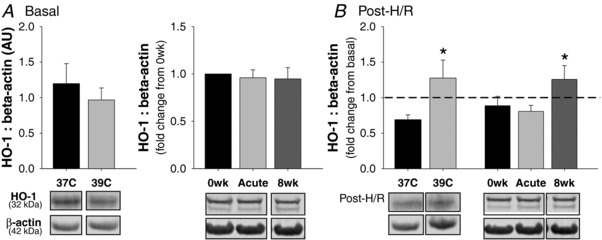
A, HO‐1 protein in cytoplasmic extracts collected following 24 h of pre‐incubation at 37°C, at 39°C (heat pretreatment), left panel, or exposed to serum collected from human subjects before (0wk) and after (Acute HT) the first hot water immersion session and following 8 weeks of heat therapy (8wk), right panel. B, fold change in HO‐1 protein from before (basal) to after hypoxia‐reoxygenation. Representative Western blot images are shown below (panel B shows post‐H/R only; refer to representative blots for panel A to determine fold change from basal depicted on the graph). Data are mean ± SEM. * P < 0.05 vs. 37°C or 0wk.
For direct heating experiments, post‐H/R HO‐1 (fold change from pre‐H/R) was significantly inversely related to H/R‐induced IL‐6 release (fold change from pre‐H/R; r 2 = 0.29, P = 0.04), but not NF‐κB activation (r 2 = 0.08, P = 0.25). For serum exposure experiments, post‐H/R HO‐1 was significantly inversely related to peak H/R‐induced superoxide production (r 2 = 0.33, P = 0.003), but was not related to NF‐κB activation (r 2 = 0.10, P = 0.11) or IL‐6 release (r 2 = 0.03, P = 0.42).
Effects of heat therapy on Hsp70
To investigate the role of Hsp70 in mediating protection against H/R, we first measured intracellular Hsp70 by Western blot in cell lysates collected following 24 h of heating or serum exposure (but pre‐H/R). Hsp70 was increased in cells that were heat pretreated (P < 0.001 vs. 37°C) but was unchanged in cells exposed to heat therapy serum (P = 0.66) (Fig. 5 A).
Figure 5. Heat shock protein (Hsp) 70.
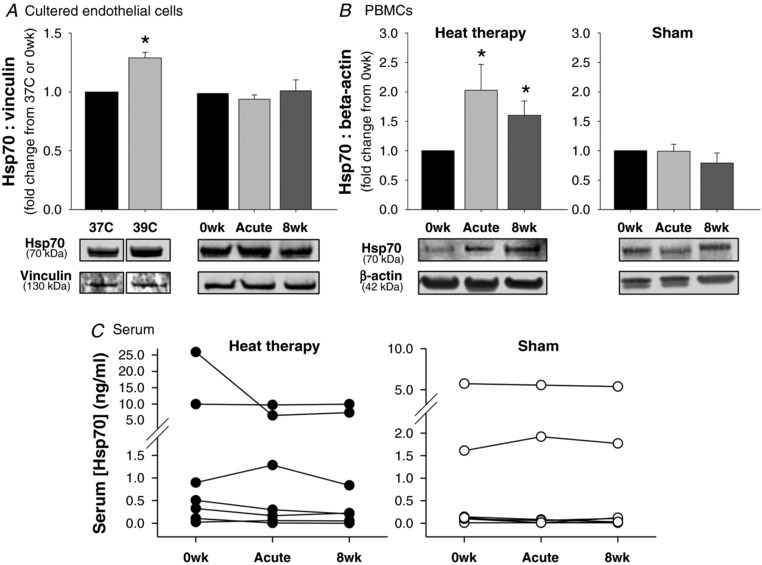
A and B, Hsp70 in (A) cultured endothelial cells pre‐incubated at 37°C, at 39°C (heat pretreatment) or with serum from human subjects collected before (0wk) and after (Acute HT) the first hot water immersion session or following 8 weeks of heat therapy (8wk) and in (B) peripheral blood mononuclear cells collected at the same time points from human subjects who participated in 8 weeks of heat therapy (left) or 8 weeks of thermoneutral water immersion (right). Representative Western blot images are shown below. Data are mean ± SEM. C, free Hsp70 concentrations in serum collected from human subjects at the same time points (n = 6‐7 per group; individual data shown). [Hsp70] was below detection in several subjects (excluded from analysis; n = 3 in heat therapy group, n = 4 in sham group). Despite high variability across individuals, [Hsp70] concentrations were relatively stable within individuals across the interventions.
To further investigate whether circulating Hsp70 may have played a role in protecting serum‐exposed cells against H/R, despite no change in intracellular Hsp70, we measured extracellular Hsp70 in serum from human subjects by ELISA. However, there were also no differences in free circulating extracellular Hsp70 concentrations in serum with heat therapy (P = 0.35) (Fig. 5 C). Of note, there was large intra‐individual variability in serum [Hsp70], and multiple samples were under detection; however, the individual data clearly show no change across heat therapy within individuals (Fig. 5 D). There were also no changes in serum [Hsp70] in sham subjects (P = 0.75) (Fig. 5 D).
We additionally measured intracellular Hsp70 protein in PBMCs isolated from venous blood from human subjects by Western blot (Fig. 5 B). Although PBMCs were not present in serum that cultured endothelial cells were exposed to, signalling related to increases in PBMC Hsp70 may have played a role in protection against H/R. Relative to 0wk, Hsp70 was significantly increased in PBMCs following both acute hot water immersion (P = 0.04) and chronically following 8 weeks of heat therapy (P = 0.04). There were no changes in Hsp70 protein in PBMCs from human subjects in the sham group.
Discussion
In the present study, we investigated the isolated effects of elevated temperature (heat pretreatment) and circulating factors upregulated by heat therapy (serum exposure) on endothelial cell stress resistance against H/R, a pro‐inflammatory and pro‐oxidative stimulus that mimics in vivo ischaemia‐reperfusion. Our major findings are that: (1) heat pretreatment prevented H/R‐induced activation of NF‐κB, reduced superoxide production under basal conditions and during H/R, but did not affect H/R‐induced IL‐6 production; and (2) exposing cells to serum collected from inactive human subjects following both acute hot water immersion and following 8 weeks of heat therapy prevented H/R‐induced NF‐κB activation and IL‐6 release, and reduced superoxide production under basal conditions and during H/R.
In addition, we investigated potential mechanisms behind heat therapy‐mediated protection against H/R and observed that: (1) haemeoxygenase‐1 (Hsp32) was upregulated in heat‐pretreated cells and cells exposed to 8 weeks of heat therapy serum; (2) Hsp70 was upregulated in heat‐pretreated cells, but not in cells exposed to serum from human heat therapy subjects; and (3) MnSOD was upregulated in heat‐pretreated cells and cells exposed to 8 weeks of heat therapy serum. Combined, these observations provide potential mechanisms by which elevations in endothelial cell temperature and circulating factors following heat therapy impart protection against inflammatory and oxidative stress induced by H/R, as summarized in Fig. 6.
Figure 6. Summary of the protective effects of heat therapy on cellular hypoxia‐reoxygenation (H/R).
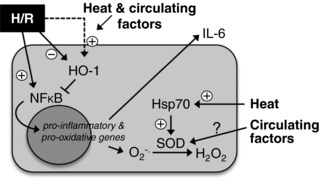
Both direct heat and circulating factors in serum from subjects who have undergone heat therapy suppress H/R‐induced activation of nuclear factor kappa‐B (NF‐κB) and downstream oxidative stress (i.e. superoxide [O2 −] production) and inflammation (i.e. interleukin [IL]‐6 release), probably via upregulation of haemeoxygenase‐1 (HO‐1), heat shock proteins (Hsp) and/or anti‐oxidant enzymes (e.g. superoxide dismutase [SOD]).
Establishment of cellular hypoxia‐reoxygenation model
Cellular H/R has been used fairly extensively in previous studies as a model of in vivo ischaemia‐reperfusion. However, there has been great variation in the protocols used. For example, within studies that have used endothelial cells, protocols range from a short period of H/R (e.g. 1–2 h) (Zhu et al. 2015) to multiple repetitive cycles of H/R over a similar duration as employed in the present study (Han et al. 2013). We therefore conducted pilot work to optimize the duration of both hypoxia and reoxygenation used in the present study with the aim of identifying durations that induced sufficient inflammatory and oxidative stress to detect potential protection with heat therapy, while being as short as possible to be comparable to durations of H/R that may occur in vivo. Based on these pilot data, we settled on using 16 h of hypoxia followed by 4 h of reoxygenation, with the exception of experiments assessing real‐time superoxide production. These experiments were shortened in order to prevent decay of the fluorescent superoxide stain, but due to the sensitivity of this stain, we had no trouble detecting substantial increases in superoxide production with this shortened protocol of H/R. Overall, we observed in control cells (incubated under standard conditions at 37°C) that H/R induced a 2.0‐fold increase in nuclear NF‐κB p65 protein, indicative of increased NF‐κB activation (Ghosh et al. 1998; Han et al. 2013), a 1.4‐fold increase in IL‐6 release, a 2.9‐fold increase in peak superoxide production and a reduction in HO‐1 protein to 0.7‐fold of pre‐H/R levels.
As discussed above, cellular H/R is a good model to investigate mechanisms (e.g. heat vs. circulating factors) by which interventions may impart protection against in vivo ischaemia‐reperfusion. In addition, pairing this model with serum exposure may prove particularly helpful for non‐invasively assessing generalized/systemic stress resistance. Impaired stress resistance is a hallmark of disease and dysfunction in many clinical populations, including primary ageing and cardiovascular diseases (Lomeli et al. 2017). By contrast, enhanced stress resistance (specifically, ‘adaptation to stress’) has been identified as one of the ‘seven pillars of ageing’, recognizing its important contribution to optimized longevity (Kennedy et al. 2014). Thus, developing bioassays to investigate stress resistance and secondly investigating the utility of interventions that may improve stress resistance is of great biomedical importance. In particular, impaired resistance against inflammatory and oxidative stress are key in the pathophysiology of many prevalent diseases, including cardiovascular, metabolic and neurogenic diseases, and so H/R represents a clinically relevant stressor to use for such investigations into stress resistance.
Basal effects of heat pretreatment and serum exposure
For heat pretreatment, endothelial cells were incubated for 24 h at 39°C. A temperature of 39°C was chosen to be comparable to body core temperatures attained by human subjects during hot water immersion sessions. We chose to heat cells for 24 h to ensure a sufficient effect to detect differences (based on pilot data) and to represent a more chronic period of heating relative to the timeline of the cell life cycle. For serum exposure, endothelial cells were exposed to 10% sera collected from human subjects before, 1 h following acute hot water immersion and 24–48 h following the last session of 8 weeks of heat therapy. We chose to collect sera 1 h following the first hot water immersion session as intracellular Hsp70 expression peaked in PBMCs at this time point in our pilot data. Sera were collected 24–48 h following the last session of heat therapy to reflect the chronic effects of heat therapy rather than the acute effects of the last hot water immersion session.
Both heat pretreatment and exposure to heat therapy serum reduced basal superoxide production and increased MnSOD protein expression. The majority of reactive oxygen species produced in endothelial cells is produced in the mitochondria (Widlansky & Gutterman, 2011); therefore, basal reductions in superoxide were probably attributable to upregulation of MnSOD (the mitochondrial isoform of SOD). With heat pretreatment, upregulation of MnSOD probably occurred due to increased intracellular Hsp70 protein, which has been shown to directly upregulate MnSOD (Suzuki et al. 2002). Additionally, although not measured in the present study, MnSOD activity may also have been elevated, as Hsp70 knockout in rodent models has been shown to suppress MnSOD activity (Choi et al. 2005). Intracellular Hsp70 protein was not increased by exposure to heat therapy serum, and nor were extracellular Hsp70 concentrations in the serum. Thus, upregulation of MnSOD and subsequent suppression of oxidative stress with serum exposure appear to be due to HSP‐independent pathways.
Concentrations of the inflammatory cytokine, IL‐6, were increased following heat pretreatment. Although chronic inflammation is generally damaging for endothelial cells and a target for age‐ and disease‐related therapies, acute inflammation may instead help mediate cellular adaptation. For example, IL‐6 is elevated acutely following exercise and is thought to mediate adaptation to exercise training (Pedersen & Fischer, 2007). Similarly, IL‐6 has been shown to be elevated following acute hot water immersion (Leicht et al. 2015; Faulkner et al. 2017), which was also observed in the present study (on average, an ∼2‐fold increase). [IL‐6] was also increased following exposure to sera collected following the first hot water immersion session (Acute HT). Importantly, it is possible this increase could be attributable to the increase in serum [IL‐6] with acute HT, as 10% serum was present in the cell culture media. However, it seems unlikely that such a low concentration of serum could have accounted for the entirety of the increase in media [IL‐6] we observed. Thus, we believe this increase reflects acute cellular stress. Exposure to 8 weeks of heat therapy serum did not affect basal [IL‐6], and [IL‐6] was also unchanged in the serum following 8 weeks of heat therapy.
The protective effects we observed with exposure to heat therapy serum raise the question of what changed in the serum. Unfortunately, conducting an extensive search of circulating proteins and/or metabolites in order to determine which were altered in the serum and therefore may have mediated our observed effects was outside the scope of this investigation; however, we speculate that heat therapy probably upregulated circulating anti‐inflammatory and/or anti‐oxidative proteins/metabolites (suppression of pro‐inflammatory/oxidative agents is less likely in a healthy population, but a possible mechanism to consider in patient populations). In addition, a recent study reported that acute passive heat stress reduces circulating levels of endothelial‐ and platelet‐derived microparticles (Bain et al. 2017). Elevated circulating levels of these particles are known to promote endothelial oxidative stress (Burger et al. 2011) and inflammation (Melki et al. 2017) and are thought to play a pathogenic role in the progression of vascular disease (Barteneva et al. 2013). Thus, reduced levels in serum collected following heat therapy could have mediated our observed effects. Lastly, we cannot completely rule out HSP‐mediated effects, as Hsp70 protein was upregulated intracellularly in PBMCs. Although PBMCs were not present in the serum, it is possible microvesicles/exosomes derived from PBMCs or other tissues were present. Microvesicles contain similar contents as their parent cells (Lovren & Verma, 2013), but as these contents are not freely circulating, they are not detectible by ELISA. Furthermore, microvesicles can fuse with and/or enter other cell types where they can act as signalling molecules (Lovren & Verma, 2013), and therefore may have mediated the suppression of oxidative stress observed with serum exposure.
Effects of heat pretreatment and serum exposure on H/R
Both heat pretreatment and exposure to heat therapy serum suppressed H/R‐induced NF‐κB activation to an extent that post‐H/R nuclear (i.e. activated) NF‐κB was not different from pre‐H/R levels. Our results are consistent with previous studies that have shown heat pretreatment suppresses NF‐κB activation (Frossard et al. 2001; Chen et al. 2004; Nakabe et al. 2015) and downstream cytokine production (Ensor et al. 1994; Kim et al. 2005) in response to various pro‐inflammatory stimuli; however, to our knowledge, we are the first to show that heat pretreatment suppresses NF‐κB activation specifically in response to H/R. Our finding that protection can be conferred via circulating factors upregulated by heat therapy is indeed novel.
Although both heat pretreatment and heat therapy serum suppressed NF‐κB activation, they had varying effects on downstream cytokine production and oxidative stress. Superoxide production was reduced during H/R by both heat pretreatment and serum exposure; in addition to suppression of NF‐κB activation, this was due in part to upregulation of MnSOD, as discussed above. H/R‐induced IL‐6 release was attenuated by exposure to heat therapy serum, but not by heat pretreatment, although pretreatment at higher temperatures and/or for > 24 h may have also attenuated IL‐6 release, as shown in response to other pro‐inflammatory stimuli following heat shock of ≥ 42°C (Kim et al. 2005). A limitation of our study is that we did not measure other cytokines (e.g. IL‐1β and TNFα), although we attempted to measure TNFα in conditioned media and unfortunately all concentrations were under the detection limit. We speculate that production and release of such pro‐inflammatory cytokines were probably also attenuated by heat pretreatment and exposure to heat therapy serum. Indeed, heat pretreatment at higher temperatures has previously been shown to attenuate cytokine release in response to other pro‐inflammatory stimuli, e.g. endotoxin‐induced TNFα and IL‐1 release (Snyder et al. 1992; Kluger et al. 1997).
One mechanism by which these effects may have been mediated is through upregulation of HO‐1. HO‐1, the inducible form of haemeoxygenase, was first discovered and named for its ability to degrade haem (Maines et al. 1986), but has since been shown to be identical to Hsp32 (Keyse & Tyrrell, 1989) and to confer cellular protection, particularly against oxidative stress (Stocker, 1990). Although heat pretreatment alone (i.e. pre‐H/R) did not upregulate HO‐1 protein expression, we did observe that the combination of heat and H/R induced a robust upregulation of HO‐1. Furthermore, HO‐1 was upregulated after H/R in cells exposed to heat therapy serum, a novel finding. Importantly, H/R‐induced changes in HO‐1 were significantly related to post‐H/R media IL‐6 concentrations in the heat pretreatment experiments and to H/R‐induced superoxide production in the serum exposure experiments, suggesting upregulation of HO‐1 may have contributed to the protective effects of heat therapy against H/R.
Of course, other mechanisms probably exist that were outside the scope of this investigation. For example, the transcription factor Nrf2 (nuclear factor erythroid 2‐related factor 2) is an established mediator of cellular resistance against oxidative stress (Nguyen et al. 2009) with known cross‐talk between its signalling pathways and that of NF‐κB (Wardyn et al. 2015). Additionally, both MnSOD and HO‐1 are downstream targets of Nrf2 (Howden, 2013). Nrf2 has been shown to be upregulated following heat pretreatment and to mediate the protective effects of heat pretreatment against heat shock‐induced apoptosis (Glory & Averill‐Bates, 2016). Furthermore, hypoxic pre‐conditioning (another proposed intervention for protecting against in vivo ischaemia‐reperfusion injury) attenuates cellular H/R‐induced oxidative stress via Nrf2 activation (Huang et al. 2014). Thus, it seems plausible that heat therapy‐induced upregulation of Nrf2 could have contributed upstream to the protective effects of heat therapy we observed in the present study. Upregulation of anti‐inflammatory IL‐10, which has been shown to directly suppress NF‐κB activation (Clarke et al. 1998), is another possibility. It is also possible that the short bout of hypoxia directly upregulated Nrf2. However, as we observed differences in downstream Nrf2 targets (e.g. HO‐1) across experimental conditions, there were likely to be effects of heat pretreatment and heat therapy serum independent of hypoxia‐induced Nrf2 upregulation. In addition, we observed changes in other Nrf2 downstream targets (e.g. MnSOD) under basal conditions, further suggesting that heat therapy had cellular effects independent of hypoxia itself. Future studies should investigate the role of these mechanisms in heat therapy‐mediated stress resistance.
Conclusion/perspectives
In this study, we sought to isolate the effects of elevations in temperature and circulating factors altered by heat therapy on cellular stress resistance. We observed that both mild elevations in temperature (to 39°C) and exposure to serum from human subjects who have undergone 8 weeks of heat therapy independently prevented oxidative and inflammatory stress associated with H/R in cultured endothelial cells. Specifically, both heat pretreatment and exposure to heat therapy serum prevented H/R‐induced NF‐κB activation and attenuated superoxide production, whereas only exposure to serum attenuated IL‐6 release. The finding that heat therapy beneficially alters the extracellular milieu presents a novel mechanism of heat therapy‐induced physiological adaptation that should be further explored. Future studies should also aim to identify which factors in the serum were responsible for beneficial adaptation, as doing so was outside of the scope of this study. In vivo, the effects of elevated temperature and circulating factors would presumably be combined and amplified, suggesting heat therapy is a promising intervention for reducing cellular damage following ischaemic events. These findings suggest protection by heat therapy may be beneficial for patients at risk for ischaemic cardiovascular events (e.g. myocardial infarction and stroke), and may also have the potential to reduce symptoms and pathology related to frequent smaller‐scale ischaemia, such as diabetic neuropathy, pressure ulcer formation in individuals with limited mobility (e.g. spinal cord injury), limb claudication related to peripheral artery disease, or metabolic dysfunction associated with obesity (Ely et al. 2018).
In addition, we have established a model to assess stress resistance across intervention studies. This is particularly helpful as cellular stress resistance is difficult to assess in such studies – either cells must be obtained via invasive procedures or more technically challenging procedures must be used to assess in vivo ischaemia‐reperfusion, neither of which is generally feasible for large‐scale clinical trials. On the other hand, serum can be easily obtained and cell culture experiments can be performed using serum from multiple subjects at once.
Additional information
Competing interests
None.
Author contributions
Experiments were conducted at the University of Oregon in the laboratories of Drs Christopher Minson and Hans Dreyer. V.E.B., K.W.‐N. and C.T.M. conceived of and designed experiments. V.E.B., K.W.‐N. and L.N.C. collected and analysed data. V.E.B. and C.T.M. interpreted data. V.E.B. prepared figures and drafted the manuscript. All authors revised the manuscript critically for intellectual content. All authors approved the final version of the manuscript and agree to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
This study was supported by American Heart Association Grant 14PRE20380300, the Eugene and Clarissa Evonuk Memorial Foundation, and the Kenneth and Kenda Singer Endowment. V.E.B. is supported by F32 HL140875. C.T.M. is supported by the Kenneth and Kenda Singer Endowment and AHA Grant 160016.
Acknowledgements
The authors would like to thank the subjects who participated in this study and Taylor M. Eymann, Matthew J. Howard, Michael A. Francisco, Brett R. Ely and Andrew J. Jeckell for assistance with data collection. The authors also thank Dr Hans C. Dreyer and Dr Jonathan Muyskens for providing laboratory space and support, and Dr Pope Moseley for conceptual contribution.
Biography
Vienna E. Brunt received her PhD from the University of Oregon in 2016. The work contained in this manuscript represents follow‐up experiments from her dissertation work investigating the effects of 8 weeks of heat therapy on vascular function in young, sedentary adults. Dr Brunt is currently a postdoctoral fellow at the University of Colorado Boulder. Her long‐term research goals are to investigate the efficacy of novel lifestyle interventions for preserving vascular function with ageing, thereby preventing and/or delaying the progression of cardiovascular diseases.

Edited by: Scott Powers & Karyn Hamilton
This is an Editor's Choice article from the 15 October 2018 issue.
References
- Asiedu‐Gyekye IJ & Vaktorovich A (2003). The “no‐reflow” phenomenon in cerebral circulation. Med Sci Monit 9, BR394–BR397. [PubMed] [Google Scholar]
- Baek SH, Min JN, Park EM, Han MY, Lee YS, Lee YJ & Park YM (2000). Role of small heat shock protein HSP25 in radioresistance and glutathione‐redox cycle. J Cell Physiol 183, 100–107. [DOI] [PubMed] [Google Scholar]
- Bain AR, Ainslie PN, Bammert TD, Hijmans JG, Sekhon M, Hoiland RL, Flück D, Donnelly J & DeSouza CA (2017). Passive heat stress reduces circulating endothelial and platelet microparticles. Exp Physiol 102, 663–669. [DOI] [PubMed] [Google Scholar]
- Barteneva NS, Fasler‐Kan E, Bernimoulin M, Stern JNH, Ponomarev ED, Duckett L & Vorobjev IA (2013). Circulating microparticles: square the circle. BMC Cell Biol 14, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamin EJ, Blaha MJ, Chiuve SE, Cushman M, Das SR, Deo R, de Ferranti SD, Floyd J, Fornage M, Gillespie C, Isasi CR, Jiménez MC, Jordan LC, Judd SE, Lackland D, Lichtman JH, Lisabeth L, Liu S, Longenecker CT, Mackey RH, Matsushita K, Mozaffarian D, Mussolino ME, Nasir K, Neumar RW, Palaniappan L, Pandey DK, Thiagarajan RR, Reeves MJ, Ritchey M, Rodriguez CJ, Roth GA, Rosamond WD, Sasson C, Towfighi A, Turner MB, Virani SS, Voeks JH, Wiley JZ, Wilkins JT, Wu JHY, Alger HM, Wong SS (2017). Heart disease and stroke statistics—2017 update: a report from the American Heart Association. Circulation 135, e146–e603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunt VE, Eymann TM, Francisco MA, Howard MJ & Minson CT (2016. a). Passive heat therapy improves cutaneous microvascular function in sedentary humans via improved nitric oxide‐dependent dilation. J Appl Physiol 121, 716–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunt VE, Howard MJ, Francisco MA, Ely BR & Minson CT (2016. b). Passive heat therapy improves endothelial function, arterial stiffness, and blood pressure in sedentary humans. J Physiol 594, 5329–5342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunt VE, Jeckell AT, Ely BR, Howard MJ, Thijssen DHJ & Minson CT (2016. c). Acute hot water immersion is protective against impaired vascular function following forearm ischemia‐reperfusion in young healthy humans. Am J Physiol Regul Integr Comp Physiol 311, R1060–R1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burger D, Montezano AC, Nishigaki N, He Y, Carter A & Touyz RM (2011). Endothelial microparticle formation by angiotensin II is mediated via Ang II receptor type I/NADPH oxidase/Rho kinase pathways targeted to lipid rafts. Arterioscler Thromb Vasc Biol 31, 1898–1907. [DOI] [PubMed] [Google Scholar]
- Chan W, Stub D, Clark DJ, Ajani AE, Andrianopoulos N, Brennan AL, New G, Black AD, Shaw JA, Reid CM, Dart AM & Duffy SJ (2012). Usefulness of transient and persistent no reflow to predict adverse clinical outcomes following percutaneous coronary intervention. Am J Cardiol 109, 478–485. [DOI] [PubMed] [Google Scholar]
- Chen D, Jin Z, Zhang J, Jiang L, Chen K, He X, Song Y, Ke J & Wang Y (2016). HO‐1 protects against hypoxia/reoxygenation‐induced mitochondrial dysfunction in H9c2 cardiomyocytes. PLoS ONE 11, e0153587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Arrigo A‐P & Currie RW (2004). Heat shock treatment suppresses angiotensin II‐induced activation of NF‐κB pathway and heart inflammation: a role for IKK depletion by heat shock? Am J Physiol Heart Circ Physiol 287, H1104–H1114. [DOI] [PubMed] [Google Scholar]
- Chi P‐L, Liu C‐J, Lee I‐T, Chen Y‐W, Hsiao L‐D & Yang C‐M (2014). HO‐1 induction by CO‐RM2 attenuates TNF‐α‐induced cytosolic phospholipase A2 expression via inhibition of PKCα‐dependent NADPH oxidase/ROS and NF‐κB. Mediators Inflamm 2014, 279171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi S, Park KA, Lee HJ, Park MS, Lee JH, Park KC, Kim M, Lee S‐H, Seo J‐S & Yoon B‐W (2005). Expression of Cu/Zn SOD protein is suppressed in hsp 70.1 knockout mice. J Biochem Mol Biol 38, 111–114. [DOI] [PubMed] [Google Scholar]
- Clarke CJ, Hales A, Hunt A & Foxwell BM (1998). IL‐10‐mediated suppression of TNF‐α production is independent of its ability to inhibit NF κB activity. Eur J Immunol 28, 1719–1726. [DOI] [PubMed] [Google Scholar]
- Ely BR, Clayton ZS, McCurdy CE, Pfeiffer J & Minson CT (2018). Meta‐inflammation and cardiometabolic disease in obesity: can heat therapy help? Temperature 5, 9–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ensor JE, Wiener SM, McCrea KA, Viscardi RM, Crawford EK & Hasday JD (1994). Differential effects of hyperthermia on macrophage interleukin‐6 and tumor necrosis factor‐α expression. Am J Physiol Cell Physiol 266, C967–C974. [DOI] [PubMed] [Google Scholar]
- Eynan M, Knubuvetz T, Meiri U, Navon G, Gerstenblith G, Bromberg Z, Hasin Y & Horowitz M (2002). Heat acclimation‐induced elevated glycogen, glycolysis, and low thyroxine improve heart ischemic tolerance. J Appl Physiol 93, 2095–2104. [DOI] [PubMed] [Google Scholar]
- Faulkner SH, Jackson S, Fatania G & Leicht CA (2017). The effect of passive heating on heat shock protein 70 and interleukin‐6: a possible treatment tool for metabolic diseases? Temperature 4, 292–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frossard JL, Pastor CM & Hadengue A (2001). Effect of hyperthermia on NF‐κB binding activity in cerulein‐induced acute pancreatitis. Am J Physiol Gastrointest Liver Physiol 280, G1157–G1162. [DOI] [PubMed] [Google Scholar]
- Ghosh S, May MJ & Kopp EB (1998). NF‐κB and Rel proteins: evolutionarily conserved mediators of immune responses. Annu Rev Immunol 16, 225–260. [DOI] [PubMed] [Google Scholar]
- Glory A & Averill‐Bates DA (2016). The antioxidant transcription factor Nrf2 contributes to the protective effect of mild thermotolerance (40°C) against heat shock‐induced apoptosis. Free Radic Biol Med 99, 485–497. [DOI] [PubMed] [Google Scholar]
- Gori T, Di Stolfo G, Sicuro S, Dragoni S, Lisi M, Forconi S & Parker JD (2007). Nitroglycerin protects the endothelium from ischaemia and reperfusion: human mechanistic insight. Br J Clin Pharmacol 64, 145–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Q, Yeung SC, Ip MSM & Mak JCW (2013). Intermittent hypoxia‐induced NF‐κB and HO‐1 regulation in human endothelial EA.hy926 cells. Cell Biochem Biophys 66, 431–441. [DOI] [PubMed] [Google Scholar]
- Howden R (2013). Nrf2 and cardiovascular defense. Oxid Med Cell Longev 2013, 104308–104310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X‐S, Chen H‐P, Yu H‐H, Yan Y‐F, & Huang Q‐R (2014). Nrf2‐dependent upregulation of antioxidative enzymes: a novel pathway for hypoxic preconditioning‐mediated delayed cardioprotection. Mol Cell Biochem 385, 33–41. [DOI] [PubMed] [Google Scholar]
- Jabs A, Fasola F, Muxel S, Münzel T & Gori T (2010). Ischemic and non‐ischemic preconditioning: endothelium‐focused translation into clinical practice. Clin Hemorheol Microcirc 45, 185–191. [DOI] [PubMed] [Google Scholar]
- Kennedy BK, Berger SL, Brunet A, Campisi J, Cuervo AM, Epel ES, Franceschi C, Lithgow GJ, Morimoto RI, Pessin JE, Rando TA, Richardson A, Schadt EE, Wyss‐Coray T & Sierra F (2014). Geroscience: linking aging to chronic disease. Cell 159, 709–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keyse SM & Tyrrell RM (1989). Heme oxygenase is the major 32‐kDa stress protein induced in human skin fibroblasts by UVA radiation, hydrogen peroxide, and sodium arsenite. Proc Natl Acad Sci U S A 86, 99–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kharbanda RK, Peters M, Walton B, Kattenhorn M, Mullen M, Klein N, Vallance P, Deanfield J & MacAllister R (2001). Ischemic preconditioning prevents endothelial injury and systemic neutrophil activation during ischemia‐reperfusion in humans in vivo . Circulation 103, 1624–1630. [DOI] [PubMed] [Google Scholar]
- Kiemer AK, Vollmar AM, Bilzer M, Gerwig T & Gerbes AL (2000). Atrial natriuretic peptide reduces expression of TNF‐α mRNA during reperfusion of the rat liver upon decreased activation of NF‐κB and AP‐1. J Hepatol 33, 236–246. [DOI] [PubMed] [Google Scholar]
- Kim I, Shin H‐M & Baek W (2005). Heat‐shock response is associated with decreased production of interleukin‐6 in murine aortic vascular smooth muscle cells. Naunyn Schmiedebergs Arch Pharmacol 371, 27–33. [DOI] [PubMed] [Google Scholar]
- Kluger MJ, Rudolph K, Soszynski D, Conn CA, Leon LR, Kozak W, Wallen ES & Moseley PL (1997). Effect of heat stress on LPS‐induced fever and tumor necrosis factor. Am J Physiol Regul Integr Comp Physiol 273, R858–R863. [DOI] [PubMed] [Google Scholar]
- Laukkanen T, Khan H, Zaccardi F & Laukkanen JA (2015). Association between sauna bathing and fatal cardiovascular and all‐cause mortality events. JAMA Intern Med 175, 542–548. [DOI] [PubMed] [Google Scholar]
- Leicht CA, Kouda K, Umemoto Y, Banno M, Kinoshita T, Moriki T, Nakamura T, Bishop NC, Goosey‐Tolfrey VL & Tajima F (2015). Hot water immersion induces an acute cytokine response in cervical spinal cord injury. Eur J Appl Physiol 115, 2243–2252. [DOI] [PubMed] [Google Scholar]
- Lomeli N, Bota DA & Davies KJA (2017). Diminished stress resistance and defective adaptive homeostasis in age‐related diseases. Clin Sci 131, 2573–2599. [DOI] [PubMed] [Google Scholar]
- Loukogeorgakis SP, van den Berg MJ, Sofat R, Nitsch D, Charakida M, Haiyee B, de Groot E, MacAllister RJ, Kuijpers TW & Deanfield JE (2010). Role of NADPH oxidase in endothelial ischemia/reperfusion injury in humans. Circulation 121, 2310–2316. [DOI] [PubMed] [Google Scholar]
- Lovren F & Verma S (2013). Evolving role of microparticles in the pathophysiology of endothelial dysfunction. Clin Chem 59, 1166–1174. [DOI] [PubMed] [Google Scholar]
- Maines MD, Trakshel GM & Kutty RK (1986). Characterization of two constitutive forms of rat liver microsomal heme oxygenase. Only one molecular species of the enzyme is inducible. J Biol Chem 261, 411–419. [PubMed] [Google Scholar]
- Melki I, Tessandier N & Boilard E (2017). Platelet microvesicles in health and disease. Platelets 28, 214–221. [DOI] [PubMed] [Google Scholar]
- Nakabe N, Kokura S, Shimozawa M, Katada K, Sakamoto N, Ishikawa T, Handa O, Takagi T, Naito Y, Yoshida N & Yoshikawa T (2015). Hyperthermia attenuates TNF‐α‐induced up regulation of endothelial cell adhesion molecules in human arterial endothelial cells. Int J Hyperthermia 23, 217–224. [DOI] [PubMed] [Google Scholar]
- Natarajan R, Fisher BJ, Jones DG & Fowler AA (2002). Atypical mechanism of NF‐κB activation during reoxygenation stress in microvascular endothelium: a role for tyrosine kinases. Free Radic Biol Med 33, 962. [DOI] [PubMed] [Google Scholar]
- Nguyen T, Nioi P & Pickett CB (2009). The Nrf2‐antioxidant response element signaling pathway and its activation by oxidative stress. J Biol Chem 284, 13291–13295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J, Kang J‐W & Lee S‐M (2013). Activation of the cholinergic anti‐inflammatory pathway by nicotine attenuates hepatic ischemia/reperfusion injury via heme oxygenase‐1 induction. Eur J Pharmacol 707, 61–70. [DOI] [PubMed] [Google Scholar]
- Pedersen BK & Fischer CP (2007). Physiological roles of muscle‐derived interleukin‐6 in response to exercise. Curr Opin Clin Nutr Metab Care 10, 265–271. [DOI] [PubMed] [Google Scholar]
- Snyder YM, Guthrie L, Evans GF & Zuckerman SH (1992). Transcriptional inhibition of endotoxin‐induced monokine synthesis following heat shock in murine peritoneal macrophages. J Leukoc Biol 51, 181–187. [DOI] [PubMed] [Google Scholar]
- Stocker R (1990). Induction of haem oxygenase as a defence against oxidative stress. Free Radic Res Commun 9, 101–112. [DOI] [PubMed] [Google Scholar]
- Suzuki K, Murtuza B, Sammut IA, Latif N, Jayakumar J, Smolenski RT, Kaneda Y, Sawa Y, Matsuda H & Yacoub MH (2002). Heat shock protein 72 enhances manganese superoxide dismutase activity during myocardial ischemia‐reperfusion injury, associated with mitochondrial protection and apoptosis reduction. Circulation 106, I270–I276. [PubMed] [Google Scholar]
- Van Molle W, Wielockx B, Mahieu T, Takada M, Taniguchi T, Sekikawa K & Libert C (2002). HSP70 protects against TNF‐induced lethal inflammatory shock. Immunity 16, 685–695. [DOI] [PubMed] [Google Scholar]
- Wardyn JD, Ponsford AH & Sanderson CM (2015). Dissecting molecular cross‐talk between Nrf2 and NF‐κB response pathways. Biochem Soc Trans 43, 621–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Widlansky ME & Gutterman DD (2011). Regulation of endothelial function by mitochondrial reactive oxygen species. Antioxid Redox Signal 15, 1517–1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie H, Ray PE & Short BL (2005). NF‐κB activation plays a role in superoxide‐mediated cerebral endothelial dysfunction after hypoxia/reoxygenation. Stroke 36, 1047–1052. [DOI] [PubMed] [Google Scholar]
- Yacobi A, Bach YS & Horowitz M (2014). The protective effect of heat acclimation from hypoxic damage in the brain involves changes in the expression of glutamate receptors. Temperature 1, 57–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Q, He G‐W, Underwood MJ & Yu C‐M (2016). Cellular and molecular mechanisms of endothelial ischemia/reperfusion injury: perspectives and implications for postischemic myocardial protection. Am J Transl Res 8, 765–777. [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Du Q, Yang Y, Wang J, Dou S, Liu C & Duan J (2017). The protective effect of Luteolin on myocardial ischemia/reperfusion (I/R) injury through TLR4/NF‐κB/NLRP3 inflammasome pathway. Biomed Pharmacother 91, 1042–1052. [DOI] [PubMed] [Google Scholar]
- Zhu M, Ding J, Jiang H, Kong L, Sun Z, Chen J & Miao C (2015). Propofol ameliorates endothelial inflammation induced by hypoxia/reoxygenation in human umbilical vein endothelial cells: role of phosphatase A2. Vascul Pharmacol 1–9. [DOI] [PubMed] [Google Scholar]