

Abstract
Key points
In vitro evidence has identified that coagulation is activated by increased oxidative stress, though the link and underlying mechanism in humans have yet to be established.
We conducted the first randomised controlled trial in healthy participants to examine if oral antioxidant prophylaxis alters the haemostatic responses to hypoxia and exercise given their synergistic capacity to promote free radical formation.
Systemic free radical formation was shown to increase during hypoxia and was further compounded by exercise, responses that were attenuated by antioxidant prophylaxis.
In contrast, antioxidant prophylaxis increased thrombin generation at rest in normoxia, and this was normalised only in the face of prevailing oxidation.
Collectively, these findings suggest that human free radical formation is an adaptive phenomenon that serves to maintain vascular haemostasis.
Abstract
In vitro evidence suggests that blood coagulation is activated by increased oxidative stress although the link and underlying mechanism in humans have yet to be established. We conducted the first randomised controlled trial to examine if oral antioxidant prophylaxis alters the haemostatic responses to hypoxia and exercise. Healthy males were randomly assigned double‐blind to either an antioxidant (n = 20) or placebo group (n = 16). The antioxidant group ingested two capsules/day that each contained 500 mg of l‐ascorbic acid and 450 international units (IU) of dl‐α‐tocopherol acetate for 8 weeks. The placebo group ingested capsules of identical external appearance, taste and smell (cellulose). Both groups were subsequently exposed to acute hypoxia and maximal physical exercise with venous blood sampled pre‐supplementation (normoxia), post‐supplementation at rest (normoxia and hypoxia) and following maximal exercise (hypoxia). Systemic free radical formation (electron paramagnetic resonance spectroscopic detection of the ascorbate radical (A•−)) increased during hypoxia (15,152 ± 1193 AU vs. 14,076 ± 810 AU at rest, P < 0.05) and was further compounded by exercise (16,569 ± 1616 AU vs. rest, P < 0.05), responses that were attenuated by antioxidant prophylaxis. In contrast, antioxidant prophylaxis increased thrombin generation as measured by thrombin–antithrombin complex, at rest in normoxia (28.7 ± 6.4 vs. 4.3 ± 0.2 μg mL−1 pre‐intervention, P < 0.05) and was restored but only in the face of prevailing oxidation. Collectively, these findings are the first to suggest that human free radical formation likely reflects an adaptive response that serves to maintain vascular haemostasis.
Keywords: oxidative stress, haemostasis, activated coagulation, exercise, hypoxia
Key points
In vitro evidence has identified that coagulation is activated by increased oxidative stress, though the link and underlying mechanism in humans have yet to be established.
We conducted the first randomised controlled trial in healthy participants to examine if oral antioxidant prophylaxis alters the haemostatic responses to hypoxia and exercise given their synergistic capacity to promote free radical formation.
Systemic free radical formation was shown to increase during hypoxia and was further compounded by exercise, responses that were attenuated by antioxidant prophylaxis.
In contrast, antioxidant prophylaxis increased thrombin generation at rest in normoxia, and this was normalised only in the face of prevailing oxidation.
Collectively, these findings suggest that human free radical formation is an adaptive phenomenon that serves to maintain vascular haemostasis.
Introduction
It has been established that physical exercise (Powers & Jackson, 2008) and arterial hypoxaemia (Bailey et al. 2001) independently increase the local and systemic formation of ubisemiquinone, superoxide (O2 •−), hydroxyl and nitric oxide free radicals. Human studies in our laboratory have demonstrated that hypoxia promotes free radical‐mediated lipid peroxidation subsequent to the metal‐catalysed formation of intermediary lipid‐derived oxygen‐centred alkoxyl (LO•) and carbon‐centred alkyl (LC•) radicals that is further compounded with the superimposition of acute exercise (Bailey et al. 2004, 2009b, 2010, 2011b, 2018). Free radical accumulation reduces the endogenous levels of antioxidants, (Bakonyi & Radak, 2004) favouring free radical formation and oxidative stress that when in excess, can result in cellular membrane damage (Lipinski, 2011).
The nutritional antioxidants, ascorbate and α‐tocopherol are thermodynamically well suited to serve as effective aqueous/lipid soluble chain‐breaking two‐electron reductants given their thermodynamic hierarchy and corresponding low one‐electron reduction potentials (E°′ of 282 mV for the ascorbate radical (A•−)–ascorbate monoanion (AH−) couple (Williams & Yandell, 1982) and 480 mV for the α‐tocopheroxyl radical (TO•)–α‐tocopherol couple (Simic, 1990)) that are comfortably positioned above most oxidizing species. Furthermore, in combination, they can work synergistically to prevent lipid peroxidation given the thermodynamic capacity of ascorbate to repair (∆E°′ ≈ +200 mV) TO• (Tappel, 1968; Packer et al. 1979; Sharma & Buettner, 1993). In support, oral prophylaxis with ascorbate and α‐tocopherol has previously been shown by our group to attenuate systemic LO•–LC• formation in young participants free of any coagulopathy and improve vascular endothelial function subsequent to increased nitric oxide bioavailability (Richardson et al. 2007; Wray et al. 2009). While the precise scavenging mechanisms remain a matter of debate, ascorbate‐mediated suppression of O2 •− and hydrogen peroxide formation has been identified as the most thermodynamically cogent candidate though redox‐independent mechanisms also warrant consideration given that ascorbate can alter expression of α‐ketoglutarate‐dependent dioxygenases, hypoxia‐inducible factor‐α and enzymes involved in histone methylation (Cobley et al. 2015).
Inspiratory hypoxia incurred during exposure to terrestrial altitudes ranging from 4000–6542 m, which approximates to an equivalent to more severe stimulus incorporated in the present study ( of 12% ≈ 4600 m), has been shown to increase thrombin generation (Le Roux et al. 1992; Mannucci et al. 2002). In vitro evidence (Görlach et al. 2000; Görlach, 2004; Herkert et al. 2004) suggests that activation of coagulation is triggered by NADPH oxidase‐mediated O2 •− formation within the blood vessel wall (Görlach et al. 2002). This activates tissue factor in vivo (Herkert & Görlach, 2002), triggering thrombin generation. It has also been shown that hypoxia shifts the endothelial phenotype towards one in which anticoagulant properties are diminished and pro‐inflammatory features dominate the endovascular milieu (Ten & Pinsky, 2002; Foley & Conway, 2016). However, these studies have been confined to the in vitro setting and thus to what extent haemostasis is subject to redox regulation in the in vivo human remains to be established.
Thus for the first time, we conducted a randomised, double‐blind, placebo‐controlled antioxidant trial in young healthy participants that incorporated acute maximal exercise combined with inspiratory hypoxia as established ‘oxidative stressors’. We hypothesised that based on in vitro findings, hypoxia‐induced systemic free radical formation would be associated with activated coagulation and further compounded with the superimposition of acute exercise subsequent to more pronounced arterial hypoxaemia. We further hypothesised that these responses would be attenuated following antioxidant prophylaxis using a combination of aqueous/lipid‐soluble chain‐breaking antioxidants with an established capacity to attenuate systemic free radical‐mediated lipid peroxidation.
Methods
Ethical approval
The study was approved by the University of South Wales Human Research Ethics Committee (UK Ref: LF#27012011). All procedures were carried out in accordance with the Declaration of Helsinki of the World Medical Association (Williams, 2008) except for registration in a database, and written informed consent was obtained from all participants.
Participants
Forty healthy male participants were recruited into the study with a mean ± SEM age, stature and mass of 25 ± 1 years, 1.81 ± 0.01 m and 84.8 ± 2.1 kg, respectively. Participants were native lowlanders, free of any underlying cardiovascular, pulmonary and cerebrovascular disease and all were non‐smokers. They were asked to refrain from consuming any antioxidant supplements or anti‐inflammatory medications 8 weeks prior to experimentation and follow a low nitrate/nitrite diet for 4 days prior to testing, by avoiding fruits, salads and cured meats (Wang et al. 1997). They were also asked to refrain from caffeine and physical activity for 2 days prior to the study.
Design
The study adopted a randomised, double blind, placebo‐controlled antioxidant trial and consisted of three phases incorporating nutritional (antioxidant/placebo) and experimental (normoxia/hypoxia/exercise) interventions illustrated in Fig. 1 and outlined in detail below.
Figure 1. Experimental schema of the randomised double‐blind placebo‐controlled antioxidant study.
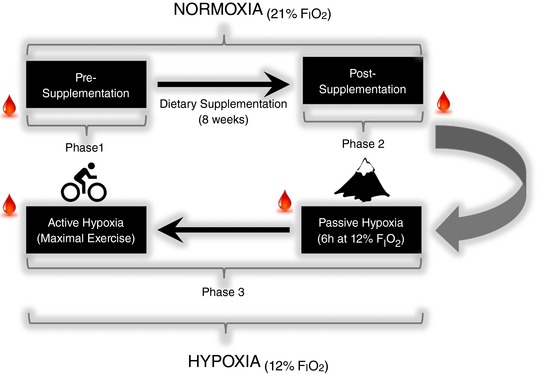
Blood sampling; , fraction of inspired oxygen. [Color figure can be viewed at http://wileyonlinelibrary.com]
Nutritional interventions
The nutritional interventions were as follows: (phase 1) pre‐supplementation, (phase 2) supplementation and (phase 3) post‐supplementation.
During phase 1, participants were tested under resting conditions and randomly assigned to a group. During phase 2, they consumed a daily dose of antioxidants or placebo control and in phase 3 they were exposed to inspiratory hypoxia and physical exercise.
Pre‐supplementation (phase 1)
For baseline pre‐supplementation testing, participants entered the laboratory at 09.00 h following a 12 h overnight fast and rested in a supine position for 30 min prior to blood sampling. Intravenous sampling without stasis was carried out via venepuncture from a prominent antecubital vein using an 18‐Gauge precision glide needle (BD Diagnostics, Plymouth, UK) into vacutainers (BD Diagnostics). Following baseline sampling, participants were randomly assigned to either an antioxidant or a placebo group.
Supplementation (phase 2)
The antioxidant group consumed 2 capules/day (during breakfast and evening meals) that each contained 500 mg of l‐ascorbic acid and 450 international units (IU) of dl‐α‐tocopherol acetate (RPL Ltd, Blackpool, UK) for 8 weeks. The placebo group consumed capsules of identical appearance, taste and smell that each contained an equal quantity of plant cellulose extract but of no nutritional value. They returned to the laboratory exactly 8 weeks after their initial visit for phase 3 of the experiment.
Post‐supplementation (phase 3)
This phase of the experiment involved baseline, post‐supplementation testing which served a dual purpose: (1) ascertaining the effectiveness of phase 2 by checking the blood‐borne levels of our target antioxidant vitamins and (2) serving as a new baseline before intervention with inspiratory hypoxia and physical exercise.
Experimental interventions
Resting measurements in normoxia and hypoxia
During the experimental day, water was available ad libitum and participants were fed a protein shake (1.25% cocoa powder, 0.16% chocolate flavouring, 0.1% aspartame and 98.49% pure whey isolate) made with water (700 mL water: 50 g powder) after 3 h. Participants were asked to wake no later than 07.00 h on their day of testing and to immediately consume their morning dose of supplement or placebo. Upon return to the laboratory at 09.30 h, the participants were once again venepunctured, and baseline sampling was carried out as in phase 1. After baseline measurements, they were fitted with a pulse oximeter (Nonin Onyx II 9550, Nonin, MN, USA) on their right index finger (Basaranoglu et al. 2015) and exposed to normobaric hypoxia ( = 12%) until completion of the experiment.
Hypoxia was induced using an environmental chamber (Weiss Technik, Ebbw Vale, UK), with controlled temperature (21°C) and relative humidity (50%). The passive exposure period lasted 6 h, during which time participants were asked to remain on a bed in either a supine or a seated position. At 3 h of exposure time, participants were given their normal dose of either intervention or placebo with water to ensure adequate circulating levels of target vitamins.
Exercise
Following passive exposure to hypoxia, participants were asked to undertake a standardised incremental exercise challenge to volitional exhaustion on an electromagnetically braked cycle ergometer (Lode Corvial, Lode, Groningen, Netherlands) in hypoxia. The test consisted of maintaining a cadence of 70 revolutions per minute (rpm) on the ergometer, with power output increasing by 35 W at the end of each minute of exercise. The test was complete when the participant fell below 60 rpm for more than 15 s or felt they could no longer continue. Expired gases were collected via an online metabolic cart (Medical Graphics UK, Gloucester, UK).
Metabolic measurements
Bloods were centrifuged at 600 g at 4°C for 10 min. Plasma and serum samples were decanted into cryogenic vials ((Nalgene® Labware, Thermo Fisher Scientific Inc., Waltham, MA, USA) and immediately snap‐frozen in liquid nitrogen (N2) and stored at −80°C. Samples were left to defrost at 37°C in the dark for 5 min before batch analysis. Unlike a previous publication (Fall et al. 2011), we specifically chose not to correct blood‐borne analytes for plasma volume shifts, notably hypoxia/exercise‐induced haemoconcentration, given that researchers do not traditionally account for this potential artefact.
Chemicals and reagents
These were of the highest available purity from Sigma‐Aldrich (UK).
Haemostasis
Plasma activated partial thromboplastin time (aPTT), prothrombin time (PT), fibrinogen (FB) and D‐dimer (DD) were measured using an ACL Futura Plus automated coagulometer (Instrumentation Laboratory, Warrington, UK). Blood was collected at each sampling point into sodium citrate Vacutainers (BD Diagnostics), spun and separated as described in phase 1 and stored at −80°C until analysis. aPTT was measured using the HemosIL APTT‐SP (liquid) assay, FB and PT were measured using a joint assay, HemosIL PT–Fibrinogen HS Plus, and DD was measured using the HemosIL D‐Dimer assay. Thrombin–antithrombin complex (T‐AT) was measured using Enzygnost TAT micro (Siemens Medical, Camberley, UK), a sandwich enzyme immunoassay for the in vitro determination of human T‐AT. Prothrombin fragment 1 and 2 (PF1+2) was measured using Enzygnost F1+2 (monoclonal) (Siemens Medical). F1+2 (monoclonal) is an enzyme immunoassay based on the sandwich principle in microtitre format, and based on monoclonal mouse antibodies for the in vitro determination of human PF1+2. The intra and inter‐assay coefficients of variation (CVs) were both <5% for all measurements.
Free radicals
Electron paramagnetic resonance (EPR) spectroscopy was used to directly measure A•− as a global biomarker of one‐electron chemistry in plasma and is compatible with an increased reactivity of systemic free radicals with ascorbate (see later) and/or change in the rate of its repair (Bailey et al. 2009a; Bain et al. 2018). Exactly 1 mL of EDTA plasma was injected into a high‐sensitivity multiple‐bore sample cell (AquaX, Bruker Daltonics, Billerica, MA, USA) housed within a TM110 cavity of an EPR spectrometer operating at X‐band (9.87 GHz). Samples were analysed using a modulation frequency of 100 kHz, modulation amplitude of 0.65 G, microwave power of 10 mW, receiver gain of 2 × 105 AU, time constant of 41 ms, magnetic field centre of 3477 G and scan width of ±50 G for three incremental scans. After identical baseline correction and filtering, each of the spectra was subject to double integration using graphical analysis software (OriginPro V.8.5, OriginLab Corp., Northampton, MA, USA). The intra‐ and interassay CVs were both <5%.
Antioxidants
Plasma was stabilised and deproteinated using 10% metaphosphoric acid and ascorbic acid assayed by fluorimetry based on the condensation of dehydroascorbic acid with 1,2‐phenylenediamine (Vuilleumier & Keck, 1989). Concentrations of the lipid‐soluble antioxidants including α/γ‐tocopherol, α/β‐carotene, retinol, lycopene, zexanthin, β‐cryptoxanthin and lutein were determined using HPLC (Catignani & Bieri, 1983; Thurnham et al. 1988). The intra‐ and interassay CVs were both <5%.
In‐Vitro study
Concept
In order to disassociate to what extent A•− reflected ‘authentic’ changes in systemic free radical formation rather than simply an increase in the circulating concentration of substrate (ascorbate) available for oxidation following supplementation, an in vitro study was performed to determine (and retrospectively) correct for the relationship between ascorbate and A•−.
Approach
Varying concentrations of ascorbate (0, 25, 50, 100, 250, 500, 750, 1000 μm) were added to phosphate buffer solution (50 mm, pH 7.4) that had been treated with chelating resin (Chelex 100) via the batch method and the absence of adventitious catalytic metals confirmed as previously outlined (Buettner, 1988). Changes in A•− were recorded as outlined above and the corresponding relationship determined between the percentage increases in ascorbate and A•−.
Statistical analysis
Following confirmation of distribution normality using the Shapiro–Wilks test, data were analysed using a two‐factor mixed ANOVA incorporating one between‐ (group: antioxidant vs. placebo) and one within‐ (condition: rest normoxia vs. rest hypoxia vs. exercise hypoxia) subjects factor. Following a significant main effect and interaction, a Bonferroni‐corrected paired samples Student's t test was employed to make post hoc comparisons at each level of the within‐subjects factor. Between‐group comparisons were assessed using an independent samples t test. Data are presented as mean ± standard error of mean (SEM) with significance for all two‐tailed tests set at P < 0.05.
Results
Compliance
Two participants withdrew from the study and two reported prior to phase 3 that they had not complied with their dosing regimen and were subsequently excluded from the overall analysis. Thus, the final data presented are based on a total of 36 participants. The antioxidant group (n = 20) were aged 25 ± 6 years, of stature 179 ± 5 cm, mass 84 ± 13 kg and BMI 26 ± 4 kg m−2, and the placebo group (n = 16) were aged 25 ± 5 years, of stature 184 ± 8 cm, mass 85 ± 12 kg and BMI 25 ± 4 kgm−2 (P > 0.05 for all variables).
In‐vitro study
The relationship between ascorbate and A•− was found to be logarithmic:
Pre‐supplementation
Antioxidants
There were no between‐group differences (P > 0.05) in any of the antioxidants (Table 1).
Table 1.
Effects of supplementation on antioxidants normoxia
| Placebo group (n = 16) | Antioxidant group (n = 20) | |||||
|---|---|---|---|---|---|---|
| Antioxidant | Pre | Post | Δ | Pre | Post | Δ |
| Ascorbate (ng mL−1) | 74.5 ± 4.0 | 68.8 ± 4.1 | −5.7 | 70.0 ± 4.1 | 104.2 ± 11.7*† | 34.2 |
| α‐Tocopherol (mg mL−1) | 33.3 ± 2.2 | 38.3 ± 3.1 | 5.0 | 31.4 ± 2.8 | 41.2 ± 3.5* | 9.8 |
| γ‐Tocopherol (mg mL−1) | 3.4 ± 0.3 | 1.9 ± 0.5* | 1.6 | 3.3 ± 0.4 | 1.6 ± 0.2 | −1.7 |
| Retinol (μmol L−1) | 1.11 ± 0.36 | 1.94 ± 0.92* | 0.83 | 1.18 ± 0.10 | 1.95 ± 0.17* | 0.77 |
| Lutein (μmol L−1) | 0.10 ± 0.02 | 0.14 ± 0.02 | 0.04 | 0.13 ± 0.02 | 0.15 ± 0.02 | 0.02 |
| Zeaxanthin (μmol L−1) | 0.05 ± 0.01 | 0.05 ± 0.005 | 0.00 | 0.04 ± 0.005 | 0.04 ± 0.004 | 0.00 |
| β‐cryptoxanthin (μmol L−1) | 0.09 ± 0.01 | 0.08 ± 0.01 | −0.01 | 0.09 ± 0.05 | 0.07 ± 0.03 | −0.02 |
| α‐Carotene (μmol L−1) | 0.39 ± 0.06 | 0.04 ± 0.08* | 0.35 | 0.38 ± 0.08 | 0.04 ± 0.01* | −0.34 |
| β‐Carotene (μmol L−1) | 0.80 ± 0.19 | 0.14 ± 0.03* | 0.66 | 0.73 ± 0.16 | 0.12 ± 0.02* | −0.61 |
| Lycopene (μmol L−1) | 3.82 ± 0.23 | 0.12 ± 0.01* | 3.70 | 3.38 ± 0.47 | 0.13 ± 0.02* | −3.25 |
Values are means ± SEM. *Different from pre‐supplementation for given group (P < 0.05); †different from placebo for given phase of supplementation (P < 0.05).
Free radicals
Figure 2 provides typical examples of observed differences in EPR spectral intensities between experimental states with corresponding hydrogen hyperfine coupling constants (a H) of ∼1.76 G (g = 2.0052, where g is the electron spin g factor). There were no between group differences (P > 0.05) in A•− corrected for de novo oxidation of supplemented ascorbate (Fig. 3).
Figure 2. Typical electron paramagnetic resonance (EPR) spectra of the ascorbate free radical.
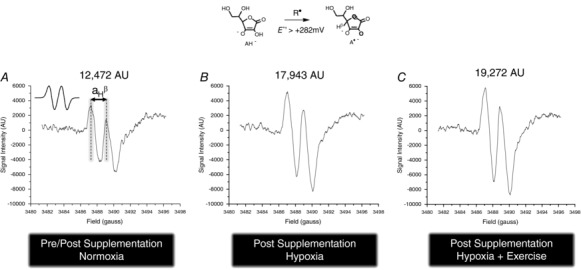
EPR spectra are from the venous circulation of a single participant from the placebo group in normoxia pre/post (placebo) supplementation, following 6 h passive exposure to hypoxia (12% O2) and maximal exercise in hypoxia. All spectra were filtered and scaled identically in arbitrary units (AU). A•− appears as a (filtered) doublet with a hydrogen hyperfine coupling constant (a H β) of ∼1.76 gauss (see inset of A for simulated spectrum).
Figure 3. Effects of supplementation, inspiratory hypoxia and exercise on systemic free radical formation.
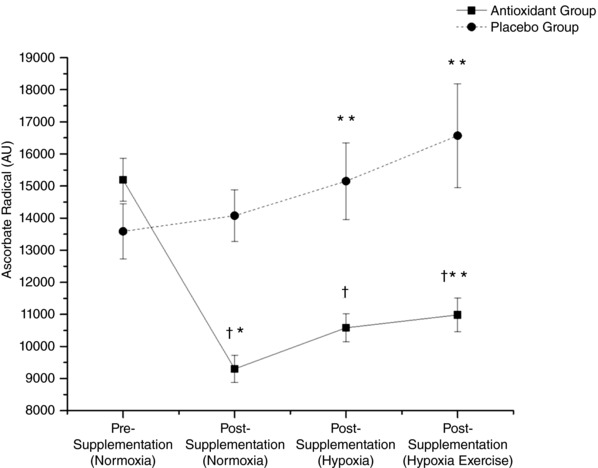
Values are corrected for de novo oxidation of supplemented ascorbate in vitro (see Methods). Values are means ± SEM. †Different vs. placebo group (P < 0.05); *different within group vs. pre‐supplementation normoxia (P < 0.05); **different within group vs. post‐supplementation normoxia (P < 0.05).
Haemostasis
There were differences in prothrombin time (PT) between groups (Fig. 4 D), with the antioxidant group having an elongated PT compared to the placebo group (P < 0.05). Other biomarkers of haemostasis showed no differences between groups (P > 0.05, Fig. 4 A–F).
Figure 4. Effects of supplementation, inspiratory hypoxia and exercise on coagulation and fibrinolysis.
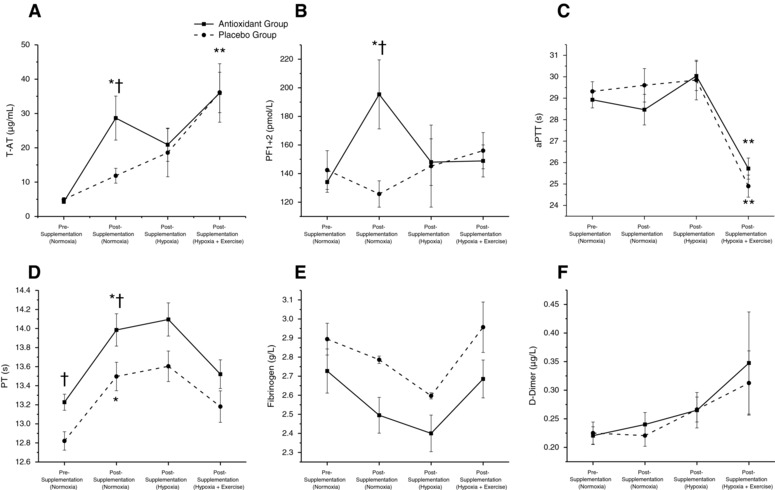
Values are mean ± SEM. T‐AT, thrombin–antithrombin complex; PF, prothrombin fragment; aPTT, activated partial thromboplastin time; PT, prothrombin time. *Different within groups vs. pre‐supplementation normoxia (P < 0.05); **different within groups vs. post supplementation hypoxia (P < 0.05); †different between groups (P < 0.05).
Post‐supplementation
Antioxidants
Compared with the placebo, supplementation increased ascorbate (69 vs.104 ng mL−1, P < 0.05). There was no difference in α‐tocopherol between groups (38.3 vs.41.2 mg mL−1, P > 0.05) but there was an increase in α‐tocopherol pre‐ to post‐supplementation in the antioxidant group (31.38 vs. 41.22 mg mL−1, P < 0.05). Retinol increased (P < 0.05) in both the antioxidant and placebo groups but there was no difference between groups (1.94 vs. 1.95 μmol L−1, P > 0.05). Supplementation decreased α‐carotene in both groups (P < 0.05), but there was no difference between groups (0.04 vs. 0.04 μmol L−1 P > 0.05). Other antioxidants were not affected by 8 weeks of supplementation (Table 1).
Free radicals
After correction for the increased circulating bioavailability of the antioxidant ascorbate, A•− was shown to decrease in the antioxidant group (9300 ± 422 vs.14,076 ± 810 AU, P < 0.05). The decrease in A•− was different compared to pre‐supplementation (15,190 ± 670 vs. 9300 ± 422 AU, P < 0.05) whereas the placebo control group showed no changes relative to pre‐supplementation (13,586 ± 854 vs.14,076 ± 810 AU, P > 0.05, Fig. 3).
Haemostasis
After 8 weeks of intervention, plasma thrombin–antithrombin complex (T‐AT) increased in both groups (P < 0.05), but more so in the antioxidant group compared with placebo (11.8 vs.28.7 μg mL−1, P < 0.05, Fig. 4 A). Prothrombin fragments 1 + 2 (PF1+2) also increased (P < 0.05) post‐intervention (Fig. 4 B); the antioxidant group had significantly greater concentrations of PF1+2 post‐intervention than the placebo group (126 vs.198 pmol L−1, P < 0.05) and compared to pre‐intervention, where the placebo group did not change (P > 0.05). PT elongated in both groups post‐intervention (P < 0.05) and the antioxidant group remained elongated compared to placebo (P < 0.05, Fig. 4 D). There were no differences in other markers of haemostasis post‐intervention.
Hypoxia
Cardiorespiratory measurements
There were no differences in heart rate or blood oxygen saturation between supplementation groups. After 6 h exposure to hypoxia, resting heart rate increased in both the antioxidant and placebo groups (66 ± 2 vs. 77 ± 2 beats min−1, P < 0.05). After hypoxic exposure, arterial oxyhaemoglobin saturation () decreased as expected in both groups (98 ± 1 vs. 82 ± 1 %, P < 0.05).
Antioxidants
Ascorbate did not increase following exposure to hypoxia vs. normoxia post‐supplementation in either group. However, the antioxidant group had significantly higher levels of ascorbate than the placebo group (114 vs.70 ng mL−1, P < 0.05, Table 2). None of the lipid‐soluble antioxidants were affected by 6 h of hypoxia and there were no differences either within or between groups.
Table 2.
Effects of hypoxia and exercise on antioxidants following supplementation
| Placebo group (n = 16) | Antioxidant group (n = 20) | |||||
|---|---|---|---|---|---|---|
| Normoxia | Hypoxia | Hypoxia | Normoxia | Hypoxia | Hypoxia | |
| Antioxidant | Rest | Rest | Exercise | Rest | Rest | Exercise |
| Ascorbate (ng mL−1) | 68.8 ± 4.1 | 70.1 ± 4.9 | 71.9 ± 5.1 | 104.2 ± 11.7 | 113.8 ± 7.4 | 111.0 ± 8.5† |
| α‐Tocopherol (mg mL−1) | 38.3 ± 3.1 | 39.0 ± 3.2 | 42.8 ± 3.8 | 41.2 ± 3.5 | 44.4 ± 4.4 | 46.0 ± 3.3 |
| γ‐Tocopherol (mg mL−1) | 1.90 ± 0.52 | 2.05 ± 0.45 | 1.57 ± 0.28 | 1.58 ± 0.20 | 2.89 ± 1.0 | 3.05 ± 0.50 |
| Retinol (μmol L−1) | 1.94 ± 0.10 | 1.69 ± 0.12 | 2.16 ± 0.08 | 1.95 ± 0.17 | 1.87 ± 0.12 | 2.09 ± 0.27 |
| Lutein (μmol L−1) | 0.14 ± 0.02 | 0.13 ± 0.02 | 0.15 ± 0.02 | 0.15 ± 0.02 | 0.14 ± 0.01 | 0.15 ± 0.02 |
| Zeaxanthin (μmol L−1) | 0.05 ± 0.005 | 0.05 ± 0.005 | 0.05± 0.005 | 0.04 ± 0.004 | 0.04 ± 0.003 | 0.05 ± 0.005 |
| β‐Cryptoxanthin (μmol L−1) | 0.08 ± 0.01 | 0.07 ± 0.01 | 0.09 ± 0.12 | 0.07 ± 0.03 | 0.06 ± 0.05 | 0.07 ± 0.04 |
| α‐Carotene (μmol L−1) | 0.04 ± 0.08 | 0.04 ± 0.07 | 0.05 ± 0.07 | 0.04 ± 0.007 | 0.03 ± 0.05 | 0.03 ± 0.003 |
| β‐Carotene (μmol L−1) | 0.13 ± 0.03 | 0.12 ± 0.02 | 0.15 ± 0.03 | 0.12 ± 0.02 | 0.09 ± 0.02 | 0.07 ± 0.01 |
| Lycopene (μmol L−1) | 0.12 ± 0.01 | 0.11 ± 0.01 | 0.15 ± 0.02 | 0.13 ± 0.02 | 0.11 ± 0.02 | 0.11 ± 0.02 |
Values are means ± SEM. †Different from placebo group for given inspirate and state (P < 0.05).
Free radicals
A•− was lower in the antioxidant compared with the placebo group (10,583 ± 443 vs.15,152 ± 1193 AU, P < 0.05). The increase in A•− in the antioxidant group compared with normoxia, post‐supplementation was not significant (9300 ± 422 vs.10,583 ± 443 AU, P > 0.05). Hypoxia increased A•− in the placebo group (14,076 ± 810 vs. 15,152 ± 1193 AU, P < 0.05, Fig. 2).
Haemostasis
Plasma T‐AT did not change during hypoxia in the placebo or antioxidant group. However, the difference between the groups following 8 weeks of intervention was eliminated (Fig. 4 A). The plasma levels of PF1+2 did not alter after exposure to hypoxia, but as with T‐AT, the difference between groups following intervention was returned to zero, following exposure to hypoxia (Fig. 3 B). Plasma aPTT was not altered by hypoxia in either group (Fig. 4 C), PT did not alter through any time point, but as with T‐AT and PF1+2, post‐intervention differences in PT were again eliminated by hypoxia. Plasma fibrinogen concentrations were unchanged by hypoxia (Fig. 4 E) and there were no differences between the intervention groups. D‐dimer, the biomarker of blood fibrinolysis, also remained unaltered by this stimulus and between groups (Fig. 4 F).
Exercise
Cardiorespiratory measurements
Immediately following exercise, remained lower compared to normoxia (98 ± 1% vs. 86 ± 6%, P < 0.05). Exercise increased heart rate vs. normoxia and vs. resting hypoxia (66 ± 2 vs. 77 ± 2 vs. 115 ± 3 beats min−1, P < 0.05).
Antioxidants
As with hypoxia, there were no changes in antioxidants following maximal exercise. Ascorbate remained unchanged compared to normoxia, post‐supplementation and in resting exposure to hypoxia, but remained elevated (P < 0.05) compared to placebo (Table 2).
Free radicals
Normalised A•− remained lower in the antioxidant compared to the placebo group (10,984 ± 529 vs.16,569 ± 1618 AU, P< 0.05, Fig. 3). In the antioxidant group, normalised A•− increased vs. normoxia, post‐supplementation (10,984 ± 529 vs. 9300 ± 529 AU, P < 0.05) and placebo group experienced a similar increase (16,569 ± 1616 vs.15,993 ± 1193 AU, P < 0.05).
Haemostasis
Exercise increased T‐AT in the placebo group (P < 0.05), but not in the antioxidant group (Fig. 4 A). While PF1+2 did not change following exercise (Fig. 4 B) aPTT shortened in both groups (P < 0.05, Fig. 4 C). PT, fibrinogen and D‐dimer were unaltered by exercise (Fig. 4 D–F).
Discussion
The present study has revealed two primary findings. First, and as anticipated, hypoxia and, to a greater extent, exercise were shown to promote systemic free radical formation, responses that were attenuated by antioxidant prophylaxis. In contrast, and indeed contrary to our original hypothesis, antioxidant prophylaxis increased thrombin generation at rest in normoxia, and was restored but only in the face of prevailing oxidation. Collectively, these findings are the first to suggest that human free radical formation likely represents an adaptive phenomenon that serves to maintain vascular haemostasis.
Supplementation
The combined increases in the systemic concentration of ascorbate and α‐tocopherol confirm that our participants adhered to the supplementation regimen. This was associated with a corresponding reduction in A•− (corrected for de novo oxidation of supplemented ascorbate) highlighting a genuine reduction in systemic free radical formation. Following supplementation, the increase in both PF1+2 and T‐AT implies increased thrombin generation (Chandler & Velan, 2003) and antithrombin activity (Mannucci, 1994). These data suggest that increased thrombin generation was accompanied by a concomitant increase in the enzyme–inhibitor complex. These data also indicate that global coagulation cascade times and fibrinolysis, as measured by D‐dimer, remained unaltered. Collectively, these findings challenge the prior research of Wang et al. (2009) who found that pre‐treatment with chain breaking antioxidant vitamins suppressed thrombin generation albeit within a thrombophilic setting.
Hypoxia
The concentration of ascorbate in human plasma is orders of magnitude greater than any number of free radicals that exhibit E°′ > 282 mV (i.e. comparatively more oxidised in terms of thermodynamic hierarchy or ‘pecking order’) associated with the A•−/AH− couple (Buettner, 1993). Thus, when these species are generated within the systemic circulation, they have the thermodynamic potential to react endogenously with this terminal small‐molecule antioxidant to form the distinctive A•− doublet (R• + AH− → A•− + R‐H) that is readily observable using EPR spectroscopy (Buettner, 1993). Thus, the elevation in A•− combined with an inverse relationship with provides direct evidence that hypoxaemia increased systemic free radical formation consistent with our previous findings (Bailey et al., 2009a, b).
Hypoxia was also associated with normalisation of the supplementation‐mediated increase in thrombin generation, disagreeing with the current literature. For example, chronic obstructive pulmonary disease (Donaldson et al. 2010) and obstructive sleep apnoea (Yaggi et al. 2005) predispose patients to stroke and it is currently thought that this is attributable to systemic oxidation (Donaldson et al. 2010). Notwithstanding other, potentially confounding co‐morbidities, the increased risk of atherothrombotic events associated with arterial hypoxaemia may thus be linked to free radical‐mediated activation of coagulation.
Previous data from our laboratory also suggest that hypoxia alone and in combination with acute exercise, known to compound systemic oxidative stress, activate coagulation by shortening activated partial thromboplastin time (Fall et al. 2011). However, the present data fail to support this. The reduction in PF1+2 and T‐AT suggests that in contrast to what we originally expected, free radical formation during hypoxia normalised the previously thrombophilic profiles of the antioxidant group yet failed to affect the placebo group. Therefore, the data suggest that free radical formation decreases (and not increases, as we expected) thrombin generation, contrary to the limited findings reported in vitro.
Previously, authors have suggested that the hypoxia‐induced increase in thrombin generation was associated with oxidised low density lipoprotein (LDL‐OX)‐mediated increased assembly of the prothrombinase complex (Rota et al., 1998) and oxidative stress increased the bioactivity of factor VIII by an as of yet unknown mechanism (Koprivica et al. 2011). The existing literature regarding the thrombogenicity of hypoxia remains conflicting and our findings collectively support those that state that hypoxia alone fails to alter blood coagulation (Andrew et al. 1987; Crosby et al. 2003; Hodkinson et al. 2003; Fall et al. 2011, 2015).
Exercise
We have consistently shown that exercise in hypoxia increases A•−, LO• and LC• (Davison et al. 2006; Bailey et al. 2011a, 2018; Woodside et al. 2014). In the present study, T‐AT increased following exercise in hypoxia in the placebo group but remained unchanged in the antioxidant group. The activation of antithrombin and subsequent generation of T‐AT can be interpreted as a consequence of thrombin generation (Burns et al. 2003). Collectively, we interpret these findings to reflect exercise‐induced activation of coagulation in the placebo group only, and that antioxidant prophylaxis negated this. PF1+2 remained unchanged following the exercise challenge and therefore these data suggest that the activity of factor X (the principal agent in the conversion of prothrombin to thrombin) was likely unaffected by exercise. This leads to the conclusion that T‐AT is elevated because of an increase in the activity of antithrombin (Chandler & Velan, 2003) per se and not as a consequence of increased thrombin generation. To the authors’ knowledge, this is a novel finding and could possibly explain previous studies that have shown attenuation of coagulation during hypoxic exercise (DeLoughery et al. 2004). The half‐life of PF1+2 is ∼90 min (Mannucci, 1994) and thus it would be interesting to extend the time course in follow‐up studies or incorporate an alternative biomarker such as fibrinopeptide A given its shorter half‐life (∼5 mins) (Mannucci, 1994).
Activated partial thromboplastin time was shortened post‐exercise in both groups, reflecting increased activity of the contact factor pathway of coagulation consistent with our previous research (Fall et al. 2011) and PT remained unchanged, which suggests the tissue factor pathway was unaltered. This finding corresponds with research suggesting that exercise promotes expression of vWF (Stakiw et al., 2008), leading to an increase in the vWF–FVIII complex, increasing the activity of the intrinsic coagulation pathway. Furthermore, activation of the coagulation cascade coincided with the increase in A•−. Recent research in animal models also shows that NADPH oxidases generate O2 •− promoting platelet activation (Delaney et al. 2016).
It is also known that oxidised lipoproteins support the assembly of the Va/Xa (prothrombinase) complex (Rota et al. 1998) and as such increase thrombin generation, as indeed our findings corroborate. Furthermore, studies in patients with antiphospholipid syndrome, which promotes a hypercoagulable state, have demonstrated that coagulation is activated by oxidative stress via overexpression of monocyte tissue factor (Ferro et al. 2003). Without the analysis of PF1+2 and T‐AT, these data would suggest increased coagulation activation and support the hypoxic findings of Wang and colleagues (2009). Interestingly, the conversion of prothrombin to thrombin is controlled by both pathways and suggests that despite the shortening of aPTT, that there is an inhibitory mechanism that free radicals exert over coagulation and this lies in the activation of antithrombin per se.
In contrast, fibrinolysis as measured by D‐dimer remained unaltered throughout the course of the study. This is of interest because acute exercise and exercise training have been shown to increase fibrinolysis in healthy men and in patients with vascular disease (Womack et al. 2003; Killewich et al. 2004). The present data suggest that hypoxia suppresses this, but interestingly Womack and colleagues (2003) argue that the fibrinolytic response to exercise is governed by a participant's baseline ‘fibrinolytic potential’, i.e. the ability to increase fibrinolysis in response to a pro‐coagulant stimulus. It is possible that hypoxia is suppressing the fibrinolytic potential of our participants (Mannucci et al. 2002) and that this phenomenon is equally subject to redox regulation in vivo, most likely via a free radical‐mediated increase in PAI‐1 (the principle inhibitor of fibrinolysis) expression.
Conclusions
The combined application of state‐of‐the art analytical techniques with an experimental model of hypoxia and exercise known to increase free radical formation has identified that antioxidant prophylaxis increased resting thrombin generation in normoxia and this was restored only in the face of prevailing oxidation. These findings are the first to suggest that human free radical formation likely represents an adaptive phenomenon that serves to maintain vascular haemostasis, reflecting the hormetic–haemostatic benefits of oxidative stress as we have recently postulated (Bain et al. 2018). Since arterial hypoxaemia in human circulatory disease is associated with oxidative stress and thrombophilia, further research is required to differentiate physiologically adaptive from pathologically maladaptive oxidation.
Additional information
Competing interests
None.
Author contributions
Intellectual content and study design: L.F. and D.M.B. Collection, analysis and interpretation of data: L.F., J.V.B., C.J.M., D.D., K.J.N., J.M.E., I.S.Y. and D.M.B. Drafting manuscript and graphical representation of data: L.F. and D.M.B. Critical evaluation of manuscript: L.F., B.D. and D.M.B. All authors have read and approved the final version of this manuscript and agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
D.M.B. is a Royal Society Wolfson Research Fellow (No. WM170007). This study was funded by a grant from the Higher Education Funding Council for Wales (D.M.B.) to fund a PhD studentship (L.F.).
Acknowledgements
We appreciate the cheerful cooperation of all study participants and the technical support of Dr Dean Whitcombe, Faculty of Life Sciences and Education, University of South Wales.
Biographies
Lewis Fall studied for a PhD in Health and Exercise Sciences under D.M.B.’s supervision while at the University of Glamorgan. He has since been appointed Senior Lecturer in Human Physiology and Course Leader for Medical Science at the University of South Wales. He is interested in the redox regulation of haemostasis and application to clinical pathology.

Damian M. Bailey is a Royal Society Wolfson Research Fellow and Professor of Physiology & Biochemistry at the University of South Wales where he leads the Neurovascular Research Laboratory. His research focuses on understanding the source, mechanisms and consequences of free radical formation across the clinical spectrum of human health and disease with a specific focus on the cerebrovasculature. He is a Fellow of the Physiological Society, Royal Society of Chemistry and American College of Sports Medicine for contributions to clinical vascular physiology.
Edited by: Scott Powers & Karyn Hamilton
Linked articles This article is highlighted in a Perspectives article by Crabtree et al. To read this article, visit https://doi.org/10.1113/JP276786.
This is an Editor's Choice article from the 15 October 2018 issue.
References
- Andrew M, O'Brodovich H & Sutton J (1987). Operation Everest II: coagulation system during prolonged decompression to 282 Torr. J Appl Physiol 63, 1262–1267. [DOI] [PubMed] [Google Scholar]
- Bailey DM, Davies B & Young IS (2001). Intermittent hypoxic training: implications for lipid peroxidation induced by acute normoxic exercise in active men. Clin Sci 101, 465–475. [PubMed] [Google Scholar]
- Bailey DM, Dehnert C, Luks AM, Menold E, Castell C, Schendler G, Faoro V, Gutowski M, Evans KA, Taudorf S, James PE, McEneny J, Young IS, Swenson ER, Mairbaurl H, Bartsch P & Berger MM (2010). High‐altitude pulmonary hypertension is associated with a free radical‐mediated reduction in pulmonary nitric oxide bioavailability. J Physiol 588, 4837–4847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey DM, Evans KA, James PE, McEneny J, Young IS, Fall L, Gutowski M, Kewley E, McCord JM, Moller K & Ainslie PN (2009a). Altered free radical metabolism in acute mountain sickness: implications for dynamic cerebral autoregulation and blood–brain barrier function. J Physiol 587, 73–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey DM, Evans KA, McEneny J, Young IS, Hullin DA, James PE, Ogoh S, Ainslie PN, Lucchesi C, Rockenbauer A, Culcasi M & Pietri S (2011a). Exercise‐induced oxidative‐nitrosative stress is associated with impaired dynamic cerebral autoregulation and blood–brain barrier leakage. Exp Physiol 96, 1196–1207. [DOI] [PubMed] [Google Scholar]
- Bailey DM, Rasmussen P, Evans KA, Bohm AM, Zaar M, Nielsen HB, Brassard P, Nordsborg NB, Homann PH, Raven PB, McEneny J, Young IS, McCord JM & Secher NH (2018). Hypoxia compounds exercise‐induced free radical formation in humans; partitioning contributions from the cerebral and femoral circulation. Free Radic Biol Med 124, 104–113. [DOI] [PubMed] [Google Scholar]
- Bailey DM, Taudorf S, Berg RM, Lundby C, McEneny J, Young IS, Evans KA, James PE, Shore A, Hullin DA, McCord JM, Pedersen BK & Moller K (2009b). Increased cerebral output of free radicals during hypoxia: implications for acute mountain sickness? Am J Physiol Regul Integr Comp Physiol 297, R1283–R1292. [DOI] [PubMed] [Google Scholar]
- Bailey DM, Taudorf S, Berg RMG, Lundby C, Pedersen BK, Rasmussen P & Møller K (2011b). Cerebral formation of free radicals during hypoxia does not cause structural damage and is associated with a reduction in mitochondrial PO2; evidence of O2‐sensing in humans? J Cereb Blood Flow Metab 31, 1020–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey DM, Young IS, McEneny J, Lawrenson L, Kim J, Barden J & Richardson RS (2004). Regulation of free radical outflow from an isolated muscle bed in exercising humans. Am J Physiol Heart Circ Physiol 287, H1689–H1699. [DOI] [PubMed] [Google Scholar]
- Bain AR, Ainslie PN, Hoiland RL, Barak OF, Drvis I, Stembridge M, MacLeod DM, McEneny J, Stacey BS, Tuaillon E, Marchi N, De Maudave AF, Dujic Z, MacLeod DB & Bailey DM (2018). Competitive apnea and its effect on the human brain: focus on the redox regulation of blood‐brain barrier permeability and neuronal‐parenchymal integrity. FASEB J 32, 2305–2314. [DOI] [PubMed] [Google Scholar]
- Bakonyi T & Radak Z (2004). High altitude and free radicals. J Sports Sci Med 3, 64–69. [PMC free article] [PubMed] [Google Scholar]
- Basaranoglu G, Bakan M, Umutoglu T, Zengin SU, Idin K & Salihoglu Z (2015). Comparison of SpO2 values from different fingers of the hands. SpringerPlus 4, 561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buettner GR ( 1988). In the absence of catalytic metals ascorbate does not autoxidize at pH 7: ascorbate as a test for catalytic metals. J Biochem Biophys Methods 16, 27–40. [DOI] [PubMed] [Google Scholar]
- Buettner GR ( 1993). The pecking order of free radicals and antioxidants: lipid peroxidation, α‐tocopherol, and ascorbate. Arch Biochem Biophys 300, 535–543. [DOI] [PubMed] [Google Scholar]
- Burns P, Wilmink T, Fegan C & Bradbury AW (2003). Exercise in claudicants is accompanied by excessive thrombin generation. Eur J Vasc Endovasc Surg 26, 150–155. [DOI] [PubMed] [Google Scholar]
- Catignani GL & Bieri JG (1983). Simultaneous determination of retinol and alpha‐tocopherol in serum or plasma by liquid chromatography. Clin Chem 29, 708–712. [PubMed] [Google Scholar]
- Chandler WL & Velan T (2003). Estimating the rate of thrombin and fibrin generation in vivo during cardiopulmonary bypass. Blood 101, 4355–4362. [DOI] [PubMed] [Google Scholar]
- Cobley JN, McHardy H, Morton JP, Nikolaidis MG & Close GL (2015). Influence of vitamin C and vitamin E on redox signaling: implications for exercise adaptations. Free Radic Biol Med 84, 65–76. [DOI] [PubMed] [Google Scholar]
- Crosby A, Talbot NP, Harrison P, Keeling D & Robbins PA (2003). Relation between acute hypoxia and activation of coagulation in human beings. Lancet 361, 2207–2208. [DOI] [PubMed] [Google Scholar]
- Davison GW, Morgan RM, Hiscock N, Garcia JM, Grace F, Boisseau N, Davies B, Castell L, McEneny J, Young IS, Hullin D, Ashton T & Bailey DM (2006). Manipulation of systemic oxygen flux by acute exercise and normobaric hypoxia: implications for reactive oxygen species generation. Clin Sci 110, 133–141. [DOI] [PubMed] [Google Scholar]
- Delaney MK, Kim K, Estevez B, Xu Z, Stojanovic‐Terpo A, Shen B, Ushio‐Fukai M, Cho J & Du X (2016). Differential roles of the NADPH‐oxidase 1 and 2 in platelet activation and thrombosis. Arterioscler Thromb Vasc Biol 36, 846–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLoughery T, Robertson D, Smith C & Sauer D (2004). Moderate hypoxia suppresses exercise‐induced procoagulant changes. Br J Haematol 125, 369–372. [DOI] [PubMed] [Google Scholar]
- Donaldson GC, Hurst JR, Smith CJ, Hubbard RB & Wedzicha JA (2010). Increased risk of myocardial infarction and stroke following exacerbation of COPD. Chest 137, 1091–1097. [DOI] [PubMed] [Google Scholar]
- Fall L, Evans KA, Lewis MH & Bailey DM (2011). Haemostatic response to hypoxaemic/exercise stress: the dilemma of plasma volume correction. J Clin Pathol 64, 269–271. [DOI] [PubMed] [Google Scholar]
- Fall L, New KJ, Evans KA & Bailey DM (2015). Arterial hypoxaemia and its impact on coagulation: significance of altered redox homeostasis. J Clin Pathol 68, 752–754. [DOI] [PubMed] [Google Scholar]
- Ferro D, Saliola M, Meroni PL, Valesini G, Caroselli C, Praticò D, Fitzgerald GA, Shoenfeld Y & Violi F (2003). Enhanced monocyte expression of tissue factor by oxidative stress in patients with antiphospholipid antibodies: effect of antioxidant treatment. J Thromb Haemost 1, 523–531. [DOI] [PubMed] [Google Scholar]
- Foley JH & Conway EM (2016). Cross talk pathways between coagulation and inflammation. Circ Res 118, 1392–1408. [DOI] [PubMed] [Google Scholar]
- Görlach A ( 2004). Redox control of blood coagulation. Antioxid Redox Signal 6, 687–690. [DOI] [PubMed] [Google Scholar]
- Görlach A, Brandes RP, Bassus S, Kronemann N, Kirchmaier CM, Busse R & Schini‐Kerth VB (2000). Oxidative stress and expression of p22phox are involved in the up‐regulation of tissue factor in vascular smooth muscle cells in response to activated platelets. FASEB J 14, 1518–1528. [PubMed] [Google Scholar]
- Görlach A, Kietzmann T & Hess J (2002). Redox signaling through NADPH oxidases: involvement in vascular proliferation and coagulation. Ann N Y Acad Sci 973, 505–507. [DOI] [PubMed] [Google Scholar]
- Herkert O, Djordjevic T, Belaiba RS & Görlach A (2004). Insights into the redox control of blood coagulation: role of vascular NADPH oxidase‐derived reactive oxygen species in the thrombogenic cycle. Antioxid Redox Signal 6, 765–776. [DOI] [PubMed] [Google Scholar]
- Herkert O & Görlach A (2002). Redox control of tissue factor expression in smooth muscle cells and other vascular cells. Methods Enzymol 352, 220–231. [DOI] [PubMed] [Google Scholar]
- Hodkinson PD, Hunt BJ, Parmar K & Ernsting J (2003). Is mild normobaric hypoxia a risk factor for venous thromboembolism? J Thromb Haemost 1, 2131–2133. [DOI] [PubMed] [Google Scholar]
- Killewich LA, Macko RF, Montgomery PS, Wiley LA & Gardner AW (2004). Exercise training enhances endogenous fibrinolysis in peripheral arterial disease. J Vasc Surg 40, 741–745. [DOI] [PubMed] [Google Scholar]
- Koprivica Z, Djordjevic D, Vuletic M, Zivkovic V, Barudzic N, Andjelkovic N, Djuric D, Iric‐Cupic V, Krkeljic J & Jakovljevic V (2011). Von Willebrand factor and oxidative stress parameters in acute coronary syndromes. Oxid Med Cell Longev 2011, 918312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Roux G, Larmignat P, Marchal M & Richalet JP (1992). Haemostasis at high altitude. Int J Sports Med 13, S49–S51. [DOI] [PubMed] [Google Scholar]
- Lipinski B ( 2011). Hydroxyl radical and its scavengers in health and disease. Oxid Med Cell Longev 2011, 809696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mannucci PM ( 1994). Mechanisms, markers and management of coagulation activation. Br Med Bull 50, 851–870. [DOI] [PubMed] [Google Scholar]
- Mannucci PM, Gringeri A, Peyvandi F, Paolantonio TD & Mariani M (2002). Short‐term exposure to high altitude causes coagulation activation and inhibits fibrinolysis. Thromb Haemost 87, 342–343. [PubMed] [Google Scholar]
- Packer JE, Slater TF & Willson RL (1979). Direct observation of a free radical interaction between vitamin E and vitamin C. Nature 278, 737–738. [DOI] [PubMed] [Google Scholar]
- Powers SK & Jackson MJ (2008). Exercise‐induced oxidative stress: cellular mechanisms and impact on muscle force production. Physiol Rev 88, 1243–1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson RS, Donato AJ, Uberoi A, Wray DW, Lawrenson L, Nishiyama S & Bailey DM (2007). Exercise‐induced brachial artery vasodilation: role of free radicals. Am J Physiol Heart Circ Physiol 292, H1516–H1522. [DOI] [PubMed] [Google Scholar]
- Rota S, McWilliam NA, Baglin TP & Byrne CD (1998). Atherogenic lipoproteins support assembly of the prothrombinase complex and thrombin generation: modulation by oxidation and vitamin E. Blood 91, 508–515. [PubMed] [Google Scholar]
- Sharma M & Buettner G (1993). Interaction of vitamin C and vitamin E during free radical stress in plasma: an ESR study. Free Radic Biol Med 14, 649–653. [DOI] [PubMed] [Google Scholar]
- Simic MG (1990). Pulse radiolysis in study of oxygen radicals. Methods Enzymol 186, 89–100. [DOI] [PubMed] [Google Scholar]
- Stakiw J, Bowman M, Hegadorn C, Pruss C, Notley C, Groot E, Lenting PJ, Rapson D, Lillicrap D & James P (2008). The effect of exercise on von Willebrand factor and ADAMTS‐13 in individuals with type 1 and type 2B von Willebrand disease. J Thromb Haemost 6, 90–96. [DOI] [PubMed] [Google Scholar]
- Tappel AL (1968). Will antioxidant nutrients slow aging processes? Geriatrics 23, 97–105. [PubMed] [Google Scholar]
- Ten VS & Pinsky DJ (2002). Endothelial response to hypoxia: physiologic adaptation and pathologic dysfunction. Curr Opin Crit Care 8, 242–250. [DOI] [PubMed] [Google Scholar]
- Thurnham DI, Smith E & Flora PS (1988). Concurrent liquid‐chromatographic assay of retinol, α‐tocopherol, β‐carotene, α‐carotene, lycopene, and β‐cryptoxanthin in plasma, with tocopherol acetate as internal standard. Clin Chem 34, 377–381. [PubMed] [Google Scholar]
- Vuilleumier J & Keck E (1989). Fluorometric assay of vitamin C in biological materials using a centrifugal analyser with fluorescence attachment. J Micronut Anal 5, 25–34. [Google Scholar]
- Wang J, Brown MA, Tam SH, Chan MC & Whitworth JA (1997). Effects of diet on measurement of nitric oxide metabolites. Clin Exp Pharmacol Physiol 24, 418–420. [DOI] [PubMed] [Google Scholar]
- Wang JS, Cheng ML, YEN HC, Lou BS & Liu HC (2009). Vitamin Esuppresses enhancement of factor VIII‐dependent thrombin generation by systemichypoxia. Stroke. 40, 656–659. [DOI] [PubMed] [Google Scholar]
- Williams JR (2008). The Declaration of Helsinki and public health. Bull World Health Organ 86, 650–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams NH & Yandell JK (1982). Outer‐sphere electron‐transfer reactions of ascorbic anions. Aust J Chem 35, 1133–1144. [Google Scholar]
- Womack CJ, Nagelkirk PR & Coughlin AM (2003). Exercise‐induced changes in coagulation and fibrinolysis in healthy populations and patients with cardiovascular disease. Sports Med 33, 795–807. [DOI] [PubMed] [Google Scholar]
- Woodside JD, Gutowski M, Fall L, James PE, McEneny J, Young IS, Ogoh S & Bailey DM (2014). Systemic oxidative‐nitrosative‐inflammatory stress during acute exercise in hypoxia; implications for microvascular oxygenation and aerobic capacity. Exp Physiol 99, 1648–1662. [DOI] [PubMed] [Google Scholar]
- Wray DW, Uberoi A, Lawrenson L, Bailey DM & Richardson RS (2009). Oral antioxidants and cardiovascular health in the exercise‐trained and untrained elderly: a radically different outcome. Clin Sci 116, 433–441. [DOI] [PubMed] [Google Scholar]
- Yaggi HK, Concato J, Kernan WN, Lichtman JH, Brass LM & Mohsenin V (2005). Obstructive sleep apnea as a risk factor for stroke and death. N Engl J Med 353, 2034–2041. [DOI] [PubMed] [Google Scholar]