

Abstract
Up to 40% of esophageal carcinomas have a biallelic intact TP53 gene. It is largely unclear how these carcinoma cells prevent apoptosis, what is the kind of pathway alterations, or whether therapeutically relevant alterations exist in this subgroup. We evaluated The Cancer Genome Atlas (TCGA) data to compare TP53-mutated with TP53–wild-type tumors regarding copy number variations, gene mutations, and expression patterns of protein-coding genes and miRNAs. Additionally, we analyzed up to 428 esophageal adenocarcinomas (EACs) in total using an ultra-deep parallel sequencing panel, immunohistochemistry, as well as fluorescence in situ hybridization. In the TCGA cohort, 17.3% has a biallelic intact TP53 gene. This group has a smaller average total size of somatic copy number variations. Some protein coding genes and miRNAs were differentially expressed between the TP53-wild-type and TP53-mutated group to emphasize mdm2, CCND2, TP73, or miRNA 150, 488, or 4662a. In addition, 50% of the TP53–wild-type tumors carry somatic mutations in at least one of the genes involved in the TP53 pathway. Our patient cohort revealed 41.3% TP53–wild-type tumors; 5.6% were MDM2 amplified. In accordance with the TCGA data, we did not find a prognostic relevance of TP53 in our tumor cohort as well. The mutation status of TP53 defines an important subtype in esophageal carcinoma. Our comprehensive molecular analysis revealed important and potentially therapeutically relevant genomic alterations in this subgroup.
Introduction
Esophageal carcinoma is the seventh most common cancer worldwide and shows strong differences in global distribution. The incidence rate for squamous cell carcinoma is highest in East Asia, inducing enormous social-economic problems, whereas esophageal adenocarcinomas (EACs) present with the highest prevalence rates in Northern Europe and the USA with still increasing incidence. The overall survival is dismal, with a 5-year survival rate of about 20% [1].
The tumor protein 53 (TP53) is the most commonly mutated and deleted gene across all carcinoma types. Due to the main function of TP53 as a gatekeeper in controlling and maintaining genomic integrity and inducing apoptosis in response to genomic alterations, a loss of function is crucial for cancer cells [2]. This is especially true for both main types of esophageal carcinomas, which are squamous cell carcinoma and adenocarcinoma, both of which show a TP53 gene alteration in 50% to 80% of the cases [3].
Most recently, the absence of relevant molecular tumor subtypes in esophageal carcinoma was confirmed. For example, EAC shows a molecular profile similar to the so-called chromosomal instable subtype of gastric cancer (characterized by a TP53 mutation and in up to 20% by an ERBB2 amplification), but the absence of Epstein-Barr virus–related tumors and the rarity of microsatellite instability were highlighted, two important subtypes described earlier in gastric carcinoma [1], [4]. Furthermore, EACs characteristically show a high content of copy number variations (CNVs) leading to chromosomal instability (the so-called genomic catastrophe in EAC) and squamous cell carcinomas showing molecular similarities to non–HPV-related head-neck carcinomas [5], [6], [7].
On the other hand, according to the patient cohort analyzed by the The Cancer Genome Atlas (TCGA), about 17% of esophageal carcinomas have a biallelic intact TP53 gene [9 out of 96 analyzed squamous cell carcinomas (9.3%) and 23 out of 89 analyzed adenocarcinomas (25.8%)].
It is largely unclear how these carcinoma cells prevent apoptosis, what kind of pathway alterations do exist up- or downstream of TP53, and whether therapeutically relevant alterations exist in this group. Likely, a functional loss of TP53 exists in all of the TP53 nonmutated tumors mediated by pathway alterations located up- or downstream of TP53. In this study, we used the existing TCGA data on esophageal carcinoma to compare TP53-mutated with TP53–wild-type tumors regarding somatic CNVs and gene mutations as well as the expression patterns of protein-coding genes and micro-RNAs (miRNAs) and correlated some of these aspects with our larger patient cohort of EAC. To the best of our knowledge, this is the first comprehensive study focusing on TP53 nonmutated esophageal carcinoma.
Material and Methods
TCGA Data Analysis
Data Access and Data Types
All data were obtained as level 3 data from the Legacy Archive of TCGA data portal. Data on somatic mutations were based on whole-exome sequencing processed through the Firehose pipeline of the Genome Data Analysis Center at the Broad Institute. Data on somatic copy number alterations were based on the SNP 6.0 microarray platform (Affymetrix Inc., Santa Clara, CA) and given as genomic segments of equal copy number derived from the Circular Binary Segmentation algorithm [8]. Data on gene and miRNA expression were obtained as raw RNA-Seq read counts. Genes in the TP53 pathway were selected according to the Kyoto Encyclopedia of Genes and Genomes pathway database.
Data Analysis
All statistical analyses were based on the functional statistics programming language R, version 3.2.3. RNA-Seq read count data were processed through the DESeq2 algorithm and its implementation as an R package, version 1.10.1 [9]. Survival analysis of TP53–wild-type and mutant patients as well as comparative analysis of somatic copy number alterations was performed through analysis workflows on the Cancer Systems Biology Database [10]. For the analysis of somatic copy number alterations, the cohort was split according to the TP53 mutation status of the patients and processed separately through the GISTIC2 algorithm, version 2.0.23 [11].
Patients and Tumor Samples
We analyzed formalin-fixed and paraffin-embedded material of 428 patients with EACs that underwent primary surgical resection or resection after neoadjuvant therapy between 1999 and 2013 at the Department of General, Visceral and Cancer Surgery, University of Cologne, Germany. Standard surgical procedure was laparotomic or laparoscopic gastrolysis and right transthoracic en bloc esophagectomy including two-field lymphadenectomy of mediastinal and abdominal lymph nodes. Reconstruction was performed by high intrathoracic esophagogastrostomy as described previously [12]. Patients with advanced esophageal cancer (cT3, cNx, M0) received preoperative chemoradiation (5-FU, cisplatin, 40 Gy as treated in the area prior the CROSS trial) or chemotherapy alone. Follow-up data were available for all patients. Patient characteristics are given in Table 1. Depending on the effect of neoadjuvant chemo- or radiochemotherapy, there is a preponderance of minor responders, defined as histopathological residual tumor of ≥10% [13]. (See Table 2.)
Table 1.
Fold-Changes Between the TP53-Mutated Versus -Nonmutated Esophageal Carcinoma
| Molecule | Fold-Change | P Value | P Value (Adjusted) | |
|---|---|---|---|---|
| Considering Protein-Coding Genes | ||||
| Induced | MDM2 | 2.94 | <.0001 | <.0001 |
| CCND2 | 1.79 | .0016 | .0409 | |
| SESN2 | 1.51 | .0003 | .0139 | |
| Repressed | CCND1 | −1.89 | .0002 | .0111 |
| RPRM | −2.29 | .0008 | .0259 | |
| ADGRB1 | −2.1 | .0041 | .079 | |
| TP53AIP1 | −2.01 | .008 | .1048 | |
| IGFBP3 | −1.61 | .0062 | .0617 | |
| TP73 | −1.74 | .0042 | .0724 | |
| Considering miRNAs | ||||
| Induced | miR-150 | 2.31 | <.0001 | .0012 |
| miR-5090 | 2.15 | .001 | .0536 | |
| miR-3667 | 2.09 | .0116 | .1631 | |
| Repressed | miR-4423 | −3.11 | <.0001 | <.0001 |
| miR-488 | −4.09 | .0018 | .0725 | |
| miR-1269a | −3.56 | .0004 | .0308 | |
| miR-466 | −3.51 | .0018 | .0725 | |
| miR-6512 | −3.24 | .0027 | .0725 | |
| miR-4662a | −3.11 | <.0001 | <.0001 | |
| miR-612 | −3.11 | .0026 | .0725 | |
Table 2.
Characteristics of Our Patient Cohort (n = 387)
| TP53 Mutational Status |
||||||||
|---|---|---|---|---|---|---|---|---|
| Total |
Wild Type |
Mutated |
P Value | |||||
| No. | % | No. | % | No. | % | |||
| Sex | Female | 37 | 9.6% | 16 | 43.2% | 21 | 56.8% | |
| Male | 350 | 90.4% | 144 | 41.1% | 206 | 58.9% | .468 | |
| Tumor stage | pT 1 | 42 | 10.9% | 13 | 31.00% | 29 | 69.00% | |
| pT 2 | 34 | 8.8% | 25 | 73.5% | 9 | 26.5% | ||
| pT 3 | 301 | 77.9% | 120 | 39.7% | 182 | 60.3% | ||
| pT 4 | 9 | 2.3% | 2 | 22.2% | 7 | 77.8% | <.001 | |
| Lymph node status | pN 0 | 147 | 38.1% | 62 | 42.2% | 85 | 57.8% | |
| pN 1 | 151 | 39.1% | 54 | 35.8% | 97 | 64.2% | ||
| pN 2 | 46 | 11.9% | 20 | 43.5% | 26 | 56.5% | ||
| pN 3 | 42 | 10.9% | 23 | 54.8% | 19 | 45.2% | .158 | |
| UICC stage | I | 78 | 20.1% | 38 | 48.3% | 40 | 51.7% | |
| II | 71 | 18.3% | 24 | 34.00% | 47 | 66.00% | ||
| III | 163 | 42.2% | 63 | 38.5% | 100 | 61.5% | ||
| IV | 75 | 19.4% | 30 | 39.3% | 46 | 60.7% | .461 | |
| Total | 328 | 100% | 138 | 42.1% | 190 | 57.9% | ||
| mdm2 FISH | Wild type | 309 | 94.2% | 119 | 38.5% | 190 | 61.5% | |
| Amplified | 19 | 5.8% | 19 | 100% | 0 | 0 | <.001 | |
For tissue microarrays (TMAs), one tissue core from each tumor was punched out and transferred into a TMA recipient block. TMA construction was performed as previously described [14], [15]. In brief, tissue cylinders with a diameter of 1.2 mm each were punched from selected tumor tissue blocks using a self-constructed semiautomated precision instrument and embedded in empty recipient paraffin blocks. Four-micrometer–thick sections of the resulting TMA blocks were transferred to an adhesive-coated slide system (Instrumedics Inc., Hackensack, NJ) for fluorescence in situ hybridization (FISH) and immunohistochemistry (IHC).
Immunohistochemistry
For both TP53 and MDM2, IHC was performed using the primary antibody specific for TP53 (DAKO, clone DO-7, 1:1200, citrate-buffer) and MDM2 (Invitrogen, clone IF2, 1:100, EDTA-buffer) using a Bond Max automated system (Leica). A total of 387 EACs were analyzable on TMA slides.
The intensity of the TP53 staining was scored manually by two pathologists (A.Q. and H.L.) according to a three-tier scoring system. We defined score 0 as an absence of any TP53 staining within the tumor cell nucleus when compared with the faint nuclear staining in the surrounding fibroblasts. This faint nuclear staining was interpreted as the physiological amount of TP53 protein within nonmutated, nontumor cells. Score 0 was interpreted as a mutation of TP53 hindering the formation of TP53 protein. Score 1 was interpreted as the physiological amount of TP53 protein. The staining intensity was similar to the intensity of the surrounding stromal cells, interpreted as a nonmutated TP53 gene (wild-type TP53). Score 2 was strong staining of ≥30% of tumor cells or moderate staining of ≥70% and was interpreted as a TP53 mutation hinders a functionally active TP53 protein. Discrepant results were resolved by consensus review.
Fluorescence In Situ Hybridization
FISH for evaluation of MDM2 gene copy numbers was performed using the ZytoLight SPEC MDM2/CEN 12 Dual Color Probe (Zytomed Systems GmbH, Germany). Two-micrometer–thick tissue sections on suitable slides (SuperFrost Plus) were mounted by baking, followed by half-automated deparaffinization, protease digestion, and washing steps (VP2000 processor system, Abbott Molecular, Wiesbaden, Germany), and hybridized at 37°C overnight with the FISH probe. The slides were stained with DAPI before analysis. Cases were only further evaluated if control tissue nuclei displayed one or two clearly distinct signals of each color (MDM2 green, centromere chromosome 12 orange). The evaluation strategy followed that of MDM2 amplification in liposarcomas [16].
The tumor tissue was scanned for high-level cluster amplifications in 60 tumor cells using ×63 objective (DM5500 fluorescent microscope; Leica). Furthermore, the grade of polysomy was estimated.
Parallel Sequencing
Out of the cohort of 428 patients described above, we analyzed 39 EACs using formalin-fixed and paraffin-embedded material. The patients were selected regarding their immunohistochemical TP53 status representing the scores from 0 to 2 to correlate these immunohistochemically based data with the real TP53 mutational status. Tumor areas were marked on H&E-stained tissue slides by a pathologist, and DNA was extracted from corresponding unstained 10-μm–thick slides by manual macrodissection. After proteinase K treatment, the DNA was automatically purified using the Maxwell 16 FFPE Tissue LEV DNA Purification Kit (Promega, Madison, WI) on the Maxwell 16 Instrument (Promega) following the manufacturers' protocol. The DNA concentration was measured using a real-time qPCR-based method.
A panel of 12 genes was used (TP53, KRAS, NRAS, HRAS, BRAF, DDR2, ERBB2, KEAP1, NFE2L2, PIK3CA, PTEN, RHOA). For TP53, we analyzed exons 5-8.
Isolated DNA (10 ng each) was amplified with a customized GeneRead DNAseq Targeted Panel V2 (Qiagen) containing gastrointestinal cancer–related genes and the GeneRead DNAseq Panel PCR Kit V2 (Qiagen) according to the GeneRead DNASeq Gene Panel Handbook (Qiagen). Libraries were constructed using the Gene Read DNA Library I Core Kit and the Gene Read DNA I Amp Kit (Qiagen). For adapter ligation, the NEXTflex DNA Barcodes (Bio Scientific, Austin, TX) were used. Library products were quantified with Qubit dsDNA HS Assay Kit (Thermo Fisher Scientific, Waltham, MA) on the Qubit 2.0 Fluorometer (Thermo Fisher Scientific), diluted, and pooled in equal amounts. A total of 12 pM was sequenced on the MiSeq (Illumina) with a MiSeq reagent Kit V2 (300-cycles) (Illumina). Data were exported as FASTQ files. Alignment and annotation were done using a modified version of a previously described method [17]. Resulting BAM files were visualized using the Integrative Genomics Viewer (IGV; http://www.broadinstitute.org/igv/, Cambridge; USA). A 5% cutoff for variant calls was used, and results were only interpreted if the coverage was >200×.
Statistical Analysis of Patients' Samples
Clinical data were collected prospectively according to a standardized protocol. SPSS Statistics for Mac (Version 21, SPSS) was used for statistical analysis. Interdependence between stainings and clinical data was calculated using the chi-squared and Fisher's exact test, and displayed by cross-tables. Survival curves were plotted using the Kaplan-Meier method and analyzed using the log-rank test. Univariate and multivariate analyses were performed for prognostic factors of overall survival using the Cox regression model. All tests were two-sided and based on a significance level of P = .05.
Results
TCGA Data
We analyzed publicly available TCGA data of 185 esophageal carcinomas including 96 squamous cell carcinomas and 89 adenocarcinomas. Thirty-two of these have a biallelic intact TP53 gene [9 out of 96 analyzed squamous cell carcinomas (9.3%) and 23 out of 89 analyzed adenocarcinomas (25.8%)].
Genomic Complexity
In order to judge the effect of TP53 on the number and size of genomic CNVs, we summed up the overall size of all CNVs in base pairs for amplifications with CN > =3 and deletions with CN < =1 in each tumor. We consider this sum as a measure for the integrity or complexity of the respective genome (Figure 1a). A comparison between TP53–wild-type and TP53-mutant tumors exhibits an average total size of CNVs of 21.6 Mb in the wild-type patients compared to 45.5 Mb in the mutated patients (P = .0096). An analysis with the GISTIC2 algorithm revealed 3 amplification peaks and 7 deletion peaks in the TP53–wild-type patients compared to 25 amplification peaks and 38 deletion peaks in the mutant patients. When CNVs are compared in more detail, TP53–wild-type tumors mainly show amplifications in the regions of chromosomes 12p13.3, 12q15, and 17q21.1 and deletions in 5q12.1, 7q36.6, and 9p21.3 as well as single-gene deletions in the loci of 4q22.1, 16q23.1, 20p12.1, and 21q22.12 (Figure 1; Supplementary Table 1, online).
Figure 1.

Somatic CNVs in TP53–wild-type (negative) and TP53-mutated (positive) esophageal carcinomas (n = 185 including 96 SCCs and 89 EACs) according to the TCGA data.
Transcriptome Analysis
A comparative analysis of gene expression using RNA-Seq data revealed that 2272 genes were differentially expressed between the TP53–wild-type and the mutant patients at a cutoff of 1.5 for absolute linear fold enrichment and .01 for significance level. Among these genes, nine overlapped with genes known to be essential players up- and downstream of TP53 (Table 1, Figure 2). Subsequent analysis of data on miRNA expression revealed a set of 32 miRNAs differentially expressed between the two groups at a cutoff of 3 for linear fold enrichment and .01 for the significance level. Among these miRNAs, 10 have been previously reported to interact with members of the TP53 pathway (Table 1).
Figure 2.

Important altered genes located up- and downstream of TP53 when considering all of the 32 analyzed TP53–wild-type tumors (according to TCGA). In black: somatic point mutations and small indels. In red: somatic copy number gain (amplifications). In blue: somatic copy number loss (deletions).
Somatic Alterations in the TP53 Pathway
Sixteen of the TP53–wild-type tumors (50.0%) showed somatic mutations in at least one of the selected 60 genes which are functionally important in the up−/downstream pathways of TP53. Eight tumors (25.0%) had more than one mutation. Moreover, 11 of the patients (34.4%) carried somatic copy number alterations affecting at least 1 of the 60 genes relevant in the TP53 pathway; more than 1 gene was affected in 8 cases (25.0%).
There are only 5 TP53–wild-type patients which carried somatic mutations and copy number aberrations, and none of the patients carried more than one somatic mutation and more than one somatic copy number alteration in the TP53 pathway. In addition, among the 8 patients with expression of miR-150 above the upper quartile across all 32 TP53–wild-type tumors, there was only 1 patient with at least 1 somatic copy number alteration in the TP53 pathway.
Patients' Data
Patients' characteristics are given in Table 1. A total of 387 patients with EACs that underwent surgical tumor resection were interpretable on the single-spot TMA. Operative procedures were either thoracoabdominal en bloc esophagectomy (n = 268, 63.2%) with intrathoracic anastomosis or transhiatal esophagectomy with transabdominal or cervical anastomosis (n = 156, 36.8%). On the TMA, 37 patients (9.6%) were female and 350 (90.4%) were male. The median age of the entire patient cohort was 65.2 years (range 33.6-85.6 years) at time of diagnosis. Neoadjuvant treatment (chemo- or radiochemotherapy) was administered in 58.9% before operation. The median follow-up for the entire cohort was 52.0 months.
Immunohistochemistry (TP53 and MDM2)
A total of 387 patients with EACs were immunohistochemically interpretable for both TP53 and MDM2. In total, 227 revealed a positive (score 2) or completely negative (score 1) immunostaining for TP53 [both staining pattern interpreted as TP53- mutated tumors (58.7%)], and 160 showed only weak nuclear staining pattern (score 1) interpreted as TP53–wild-type tumors (41.3%). There was no survival difference between patients with TP53–wild-type tumors and those with TP53 mutations. TP53–wild-type tumors showed a median overall survival of 32.5 months (95% confidence interval (CI) 22.8-42.3 months) vs. 25.9 months in TP53-mutated tumors (95% CI 19.1-32.7 months), P = .657 (log-rank test).
FISH for MDM2
A total of 257 cases of EACs were interpretable by FISH. There were 20 cases with high-level amplification of MDM2 according to the evaluation criteria (5.6%) and 237 tumors without elevated MDM2 signals (94.4%). All the MDM2-amplified cases were TP53–wild-type tumors (P < .001).
Parallel Sequencing
We analyzed 39 EACs using the gene panel described above. Patients were preselected following determination of the TP53 protein status in three unequal groups of TP53 immunohistochemistry-based score (0 to 2) in order to compare these data with the real TP53 mutation status. The main focus was the TP53–wild-type group (TP53 protein score 1). Twenty-seven of the analyzed tumors were TP53 wild type, well corresponding to the protein score 1. Twenty-one of these TP53–wild-type tumors did not bear any further mutations; 6 tumors showed mutations in PTEN (2×), KRAS, KEAP1 (2×), and PIK3CA (compare Table 3). Twelve tumors showed a TP53 mutation of which 10 (83.3%) resulted in a truncated protein and 2 (16.7%) in a nonfunctional protein. All truncated proteins showed an immunohistochemistry-based score of 0 and the two cases with nonfunctional protein showed a score of 2. Ten out of 12 TP53 mutated cases did not show any further mutations (considering our 12-gene panel), one case demonstrated additional activating BRAF (exon 15; V600E) as well as KEAP1 gene mutation, and one case showed an additional activating KRAS mutation (compare Table 3).
Table 3.
Data of Parallel Sequencing of Our Patient Cohort (n = 39)
| Other Mutated Genes | TP53 Wild Type n = 27 | TP53 Mutated n = 12 |
|---|---|---|
| No other mutated genes | 21 | 10 |
| BRAF | 0 | p.V600E activating (AF 40.5%)* |
| KRAS | p.G12D activating (AF 27.0%) |
p.G12D activating (AF 40.3%) |
| NRAS | 0 | 0 |
| HRAS | 0 | 0 |
| KEAP1 | 1 × p.V369A unknown (AF 35.7%) 2 × p.Q620del unknown (AF 27.6% | 27.2%) |
p.E343* activating (AF 36.1%)* |
| PIK3CA | p.E542K activating (AF 2.7%) |
0 |
| PTEN | 1 × p.R47S unknown (AF 14.3%) 1 × p.D24Gfs*20 truncated protein (AF 4.4%) |
0 |
| DDR2 | 0 | 0 |
| ERBB2 | 0 | 0 |
| NFE2L2 | 0 | 0 |
| RHOA | 0 | 0 |
AF, allele frequency.
Additional mutations in the same tumor.
Discussion
We evaluated publically available TCGA data of 96 esophageal squamous cell carcinomas as well as 89 adenocarcinomas to compare genomic alterations of TP53-mutated with TP53–wild-type tumors because no comprehensive analysis of molecular alterations in TP53 esophageal wild-type tumors exists, and furthermore, there is a reasonable assumption to believe that TP53-nonmutated carcinomas react differentially in respect to treatment options in comparison to TP53-mutated tumors.
In our patient cohort, we focused on EAC because this subtype is by far the most common and is still showing an increasing incidence in Northern Europe.
The number and size of somatic CNVs (amplifications and deletions) in cancer can serve as an indicator for its genetic integrity [5]. According to the TCGA data, TP53-mutated tumors show twice the amount of CNVs as do TP53–wild-type tumors. In a set of 19 genes known to be important in the up- and downstream pathways of TP53, 20 of the TP53–wild-type tumors (62.5%) do not show any CNVs, supporting the hypothesis of a higher genetic stability in the TP53–wild-type tumor group. TP53–wild-type tumors mainly show amplifications in the regions of chromosomes 12p13.3, 12q15, and 17q21.1 and deletions in 5q12.1, 7q36.6, and 9p21.3 as well as single-gene deletions in the loci of 4q22.1, 16q23.1, 20p12.1, and 21q22.12. We identified some CNVs which might be of particular importance. The chromosome locus 12p13.33 is well known to show alterations in different malignant tumors including adenocarcinomas and squamous cell carcinomas. This region includes, e.g., gene amplifications of RAD51, ERC1, ADIPOR2, and WNK1 (Table 1). The latter encodes the WNK lysine-deficient protein kinase 1. This gene is related to the ERK-MAPK pathway, and there is growing evidence that WNK1 plays an important role in cancer via alterations of different signaling pathways and serves as an inhibitor of autophagy [18], [19]. ADIPOR2 encodes for the adiponectin receptor 2 important for the intake of adiponectin as well as fatty acid oxidation and glucose uptake [20], [21].
PTPRP and PTPRR are the two genes amplified and located on chromosome 12q15. PTPRP belongs to the tyrosine phosphatase family and is important for vasculogenesis and specifically expressed in endothelial cells. PTPRR encodes for a receptor-type tyrosine phosphatase and inhibits the MAP kinase pathway via epigenetic silencing. Furthermore, downregulation of PTPRR promotes the RAS-ERK pathway [22], [23], [24], [25], [26]. Elevation of the genomic copy number of PTPRP presumably promotes the vascular network of the tumor, but the exact role in esophageal carcinoma is unclear [27]. Co-deletions of the 9p21 loci are well-known genomic alterations mainly due to the loss of the important tumor suppressor gene CDK2NA and deletions of genes for different interferons, which can be linked to reduced survival and inflammatory reaction in melanoma [28], [29], [30].
The three most typically mutated genes located in the TP53 up-/downstream pathway are CDKN2A, ATM, and TSC2. Cyclin-dependent kinase inhibitor 2 (CDKN2A) encodes for p16, a well-known protein with tumor suppressor function via inhibition of CDK4/6, and for p14ARF, which activates TP53 and inhibits MDM2 [31]. The ataxia-telangiectasia mutated serine/threonine kinase (ATM) is activated by DNA double-strand breaks and activates different proteins involved in DNA repair, cell cycle stop, or apoptosis. TP53 is one important effector protein of ATM. The frequency of ATM mutation in sporadic cancer is usually low, but first evidence suggests that an inhibition of poly(ADP-ribose) polymerase, for example, using olaparib, is more effective in ATM-deficient tumors. TSC2 is believed to be a tumor suppressor gene and interacts with the GTPases RAP1A, RAB5, and RHEB. Mutations in the TSC2 gene cause diseases like tuberous sclerosis or lymphangioleiomyomatosis. Mutated TSC2 can cause a dysregulation of signaling including an activation of RHEB. RHEB activates mTORC1 and triggers the de novo pyrimidine nucleotide synthesis via CAD enzyme complex. Both effects (activation of mTOC1 and nucleotide synthesis) can support cell division and are of great advantage for carcinoma cells. On the other hand, this opens the treatment option of an mTOR pathway inhibition using commercially available mTOR inhibitors.
A higher genetic stability could also indicate a lesser content of tumor subclones, and a specific antitumor drug could be more effective due to the absent/lower content of preresistant subclones. There is growing evidence that the higher mutational load of tumor cells and the number of different tumor clones correlate with a better response to checkpoint inhibitors. Under these circumstances, one could speculate that the TP53–wild-type esophageal carcinomas do react less effectively to checkpoint inhibition, but further studies are needed to support this hypothesis.
The well-known TP53 inhibitor MDM2 showed the highest upregulation of its expression in the TP53–wild-type group. Inhibition of TP53 protein expression via MDM2 can help TP53-nonmutated tumors to escape from TP53-induced apoptosis. In contrast, a further MDM2 upregulation is not required in the TP53-mutated carcinomas, as these do already survive through a loss of function of the TP53 protein. This offers therapeutic options using MDM2 inhibitors especially effective in TP53–wild-type tumors. Copy number elevation is one important reason for an increase of MDM2 expression [16]. In the TCGA cohort, 3 out of 32 TP53–wild-type carcinomas (9.4%) have an MDM2 amplification. Additionally, we analyzed 257 EACs and found 5.6% of these to be MDM2 amplified and TP53 nonmutated, qualifying for a potential individualized treatment option in the future.
Using parallel sequencing, in our patient cohort, we found gene alterations in PIK3CA, KEAP1, KRAS, and PTEN in the group of TP53-nonmutated tumors, indicating that the AKT-mTOR pathway via activating PIK3CA mutations or inactivating PTEN mutations is important for these tumors. We found an activation of the RAS-RAF-ERK pathway (including simultaneously activating mutations in BRAF and KRAS) in the TP53-mutated group (compare Table 3).
Interestingly, the TP53 mutation status in esophageal carcinoma did not correlate with survival in our cohort as well as in the TCGA patient cohort (See Fig. 3). This could be explained by the functional loss of TP53 due to alterations of molecules located up- or downstream of TP53 like for instance MDM2 or miRNA-150. The product of CCND2, which also exhibits increased expression in the TP53–wild-type tumors, forms a complex with CDK4 and CDK6, together required for the G1/S-cell cycle activity. Furthermore, CCND2 inhibits the tumor suppressor RB1. Thus, an increase in CCND2 activity as shown by the TCGA data is meaningful for tumor cells. An inhibition of CCND2 using fangchinoline is correlated with a decrease in tumor growth. CCND2 inhibition could be another therapeutic option especially effective in TP53–wild-type tumors with upregulation of CCND2. Surprisingly, CCND1, a paralog of CCND2 with a common mode of function, is downregulated, indicating that an upregulation of CCND2 alone is acceptable for the tumor cells. Important further downregulated genes include RPRM, ADGRB1, and TP53AIP1, which are all strongly related to the TP53 pathway. In tumors with intact TP53 gene, a repression of effector proteins is meaningful.
Figure 3.
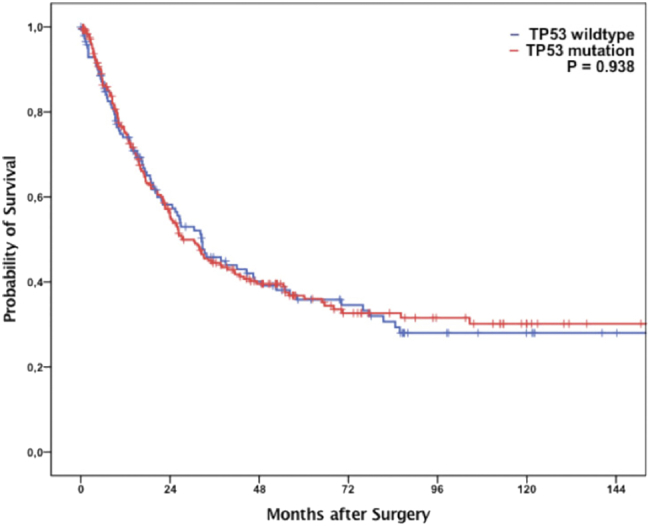
According to TCGA data, patients exhibit no survival differences between TP53–wild-type and TP53-mutated tumors (n = 185 including 96 SCCs and 89 EACs).
miRNAs are important posttranscriptional regulators. In tumor cells, most of them fulfill a tumor-suppressive function, for instance, miR-34, a downstream effector of TP53, or miR-193a, which is known to downregulate MDM2. Our data analyses show a downregulation of miR-193b in TP53–wild-type tumors which presumably triggers MDM2. On the other hand, for instance, miR-150 behaves in a tumor-supporting fashion mainly due to its capacity to downregulate TP53. These circumstances prevent the translation of the TP53 protein even though the TP53 gene itself is intact. Interestingly, in the small subset of TP53–wild-type esophageal tumors where miR-150 showed an intense upregulation, no copy number alterations and only very few numbers of mutations exist in the TP53-related pathway genes, suggesting that the tumors could in fact rely on miR-150 overexpression and its inhibition could be a relevant therapeutic option. Some miRNAs are known to be activated by TP53-like miRNA-34 or miRNA-149. [32], [33]
Our data analyses revealed that the miRNA-34 is downregulated in comparison to TP53-mutated esophageal carcinoma. The downregulation of miR-34 reflects its role as a tumor suppressor in many cancer cells via cell growth arrest and induction of apoptosis. This effect was shown to be relevant at least for miR-34a but is believed to also exist for miR-34b [34], [35], [36]. Its downregulation is further related to a so-called multi-anticancer drug resistance. miRNA-34 mimetics can attenuate this drug resistance in cell lines [37].
The most relevant downregulated miRNAs in comparison to the TP53-mutated tumors are the miRNA-488 and miRNA-4423. miRNA-488 is known to suppress cell migration and cell proliferation which was most recently described for non–small cell lung cancer. In addition, miRNA-488 has been shown to be a tumor suppressor that acts via targeting eIF3a [38]. Conversely, a downregulation of miRNA-488 is of great advantage for tumor cells. miRNA-4423 was found to be important in the airway epithelial cell differentiation and is likely to be important in lung carcinogenesis.
Conclusions
Our study revealed important and potentially therapeutically relevant molecular mechanisms in TP53-nonmutated esophageal carcinoma. We could demonstrate the rationale for a corresponding subtyping of esophageal carcinoma in the future.
The following are the supplementary data related to this article.
Genomic Alterations in the TP53 WT Group According to TCGA.
Fold-Change of miRNAs and Coding Genes.
Footnotes
Funding: This work was supported by the Deutsche Forschungsgemeinschaft (grant FR 3313/2-1 to PF).
References
- 1.Integrated genomic characterization of oesophageal carcinomaNature. 2017;541:169–175. doi: 10.1038/nature20805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang X, Sun Q. TP53 mutations, expression and interaction networks in human cancers. Oncotarget. 2017;8:624–643. doi: 10.18632/oncotarget.13483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Testa U, Castelli G, Pelosi E. Esophageal cancer: genomic and molecular characterization, stem cell compartment and clonal evolution. Medicines (Basel) 2017;4 doi: 10.3390/medicines4030067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Comprehensive molecular characterization of gastric adenocarcinomaNature. 2014;513:202–209. doi: 10.1038/nature13480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nones K., Waddell N., Wayte N., Patch A.M., Bailey P., Newell F., Holmes O., Fink J.L., Quinn M.C.J., Tang Y.H. Genomic catastrophes frequently arise in esophageal adenocarcinoma and drive tumorigenesis. Nat Commun. 2014;5:5224. doi: 10.1038/ncomms6224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Song Y., Li L., Ou Y., Gao Z., Li E., Li X., Zhang W., Wang J., Xu L., Zhou Y. Identification of genomic alterations in oesophageal squamous cell cancer. Nature. 2014;509:91–95. doi: 10.1038/nature13176. [DOI] [PubMed] [Google Scholar]
- 7.Alexandrov L.B., Ju Y.S., Haase K., Van Loo P., Martincorena I., Nik-Zainal S., Totoki Y., Fujimoto A., Nakagawa H., Shibata T. Mutational signatures associated with tobacco smoking in human cancer. Science. 2016;354:618–622. doi: 10.1126/science.aag0299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Olshen AB, Venkatraman ES, Lucito R, Wigler M. Circular binary segmentation for the analysis of array-based DNA copy number data. Biostatistics. 2004;5:557–572. doi: 10.1093/biostatistics/kxh008. [DOI] [PubMed] [Google Scholar]
- 9.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Krempel R., Kulkarni P., Yim A., Lang U., Habermann B., Frommolt P. Integrative analysis and machine learning on cancer genomics data using the Cancer Systems Biology Database (CancerSysDB) BMC Bioinformatics. 2018;19:156. doi: 10.1186/s12859-018-2157-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mermel C.H., Schumacher S.E., Hill B., Meyerson M.L., Beroukhim R., Getz G. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol. 2011;12:R41. doi: 10.1186/gb-2011-12-4-r41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Holscher AH, Schneider PM, Gutschow C, Schroder W. Laparoscopic ischemic conditioning of the stomach for esophageal replacement. Ann Surg. 2007;245:241–246. doi: 10.1097/01.sla.0000245847.40779.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schneider P.M., Metzger R., Schaefer H., Baumgarten F., Vallbohmer D., Brabender J., Wolfgarten E., Bollschweiler E., Baldus S.E., Dienes H.P. Response evaluation by endoscopy, rebiopsy, and endoscopic ultrasound does not accurately predict histopathologic regression after neoadjuvant chemoradiation for esophageal cancer. Ann Surg. 2008;248:902–908. doi: 10.1097/SLA.0b013e31818f3afb. [DOI] [PubMed] [Google Scholar]
- 14.Simon R, Mirlacher M, Sauter G. Tissue microarrays. Methods Mol Med. 2005;114:257–268. doi: 10.1385/1-59259-923-0:257. [DOI] [PubMed] [Google Scholar]
- 15.Helbig D., Ihle M.A., Putz K., Tantcheva-Poor I., Mauch C., Buettner R., Quaas A. Oncogene and therapeutic target analyses in atypical fibroxanthomas and pleomorphic dermal sarcomas. Oncotarget. 2016;7:21763–21774. doi: 10.18632/oncotarget.7845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thway K., Wang J., Swansbury J., Min T., Fisher C. Fluorescence in situ hybridization for MDM2 amplification as a routine ancillary diagnostic tool for suspected well-differentiated and dedifferentiated liposarcomas: experience at a tertiary center. Sarcoma. 2015;2015:812089. doi: 10.1155/2015/812089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Peifer M., Fernandez-Cuesta L., Sos M.L., George J., Seidel D., Kasper L.H., Plenker D., Leenders F., Sun R., Zander T. Integrative genome analyses identify key somatic driver mutations of small-cell lung cancer. Nat Genet. 2012;44:1104–1110. doi: 10.1038/ng.2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Moniz S, Jordan P. Emerging roles for WNK kinases in cancer. Cell Mol Life Sci. 2010;67:1265–1276. doi: 10.1007/s00018-010-0261-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gallolu Kankanamalage S., Lee A.Y., Wichaidit C., Lorente-Rodriguez A., Shah A.M., Stippec S., Whitehurst A.W., Cobb M.H. WNK1 is an unexpected autophagy inhibitor. Autophagy. 2017;13:969–970. doi: 10.1080/15548627.2017.1286431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yamauchi T., Kamon J., Ito Y., Tsuchida A., Yokomizo T., Kita S., Sugiyama T., Miyagishi M., Hara K., Tsunoda M. Cloning of adiponectin receptors that mediate antidiabetic metabolic effects. Nature. 2003;423:762–769. doi: 10.1038/nature01705. [DOI] [PubMed] [Google Scholar]
- 21.Huang B., Cheng X., Wang D., Peng M., Xue Z., Da Y., Zhang N., Yao Z., Li M., Xu A. Adiponectin promotes pancreatic cancer progression by inhibiting apoptosis via the activation of AMPK/Sirt1/PGC-1alpha signaling. Oncotarget. 2014;5:4732–4745. doi: 10.18632/oncotarget.1963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lin G, Aranda V, Muthuswamy SK, Tonks NK. Identification of PTPN23 as a novel regulator of cell invasion in mammary epithelial cells from a loss-of-function screen of the 'PTP-ome'. Genes Dev. 2011;25:1412–1425. doi: 10.1101/gad.2018911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Menigatti M., Cattaneo E., Sabates-Bellver J., Ilinsky V.V., Went P., Buffoli F., Marquez V.E., Jiricny J., Marra G. The protein tyrosine phosphatase receptor type R gene is an early and frequent target of silencing in human colorectal tumorigenesis. Mol Cancer. 2009;8:124. doi: 10.1186/1476-4598-8-124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Su P.H., Lin Y.W., Huang R.L., Liao Y.P., Lee H.Y., Wang H.C., Chao T.K., Chen C.K., Chan M.W., Chu T.Y. Epigenetic silencing of PTPRR activates MAPK signaling, promotes metastasis and serves as a biomarker of invasive cervical cancer. Oncogene. 2013;32:15–26. doi: 10.1038/onc.2012.29. [DOI] [PubMed] [Google Scholar]
- 25.Munkley J., Lafferty N.P., Kalna G., Robson C.N., Leung H.Y., Rajan P., Elliott D.J. Androgen-regulation of the protein tyrosine phosphatase PTPRR activates ERK1/2 signalling in prostate cancer cells. BMC Cancer. 2015;15:9. doi: 10.1186/s12885-015-1012-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Motaghed M, Al-Hassan FM, Hamid SS. Thymoquinone regulates gene expression levels in the estrogen metabolic and interferon pathways in MCF7 breast cancer cells. Int J Mol Med. 2014;33:8–16. doi: 10.3892/ijmm.2013.1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Landau D.A., Tausch E., Taylor-Weiner A.N., Stewart C., Reiter J.G., Bahlo J., Kluth S., Bozic I., Lawrence M., Bottcher S. Mutations driving CLL and their evolution in progression and relapse. Nature. 2015;526:525–530. doi: 10.1038/nature15395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sasaki S., Kitagawa Y., Sekido Y., Minna J.D., Kuwano H., Yokota J., Kohno T. Molecular processes of chromosome 9p21 deletions in human cancers. Oncogene. 2003;22:3792–3798. doi: 10.1038/sj.onc.1206589. [DOI] [PubMed] [Google Scholar]
- 29.Zhang H, Chen ZH, Savarese TM. Codeletion of the genes for p16INK4, methylthioadenosine phosphorylase, interferon-alpha1, interferon-beta1, and other 9p21 markers in human malignant cell lines. Cancer Genet Cytogenet. 1996;86:22–28. doi: 10.1016/0165-4608(95)00157-3. [DOI] [PubMed] [Google Scholar]
- 30.Linsley P.S., Speake C., Whalen E., Chaussabel D. Copy number loss of the interferon gene cluster in melanomas is linked to reduced T cell infiltrate and poor patient prognosis. PLoS One. 2014;9 doi: 10.1371/journal.pone.0109760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rayess H., Wang M.B., Srivatsan E.S. Cellular senescence and tumor suppressor gene p16. Int J Cancer. 2012;130:1715–1725. doi: 10.1002/ijc.27316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ye Z., Fang J., Dai S. MicroRNA-34a induces a senescence-like change via the down-regulation of SIRT1 and up-regulation of p53 protein in human esophageal squamous cancer cells with a wild-type p53 gene background. Cancer Lett. 2016;370:216–221. doi: 10.1016/j.canlet.2015.10.023. [DOI] [PubMed] [Google Scholar]
- 33.Ye Z., Fang J., Dai S., Wang Y., Fu Z., Feng W., Wei Q., Huang P. MicroRNA control of p53. J Cell Biochem. 2017;118:7–14. doi: 10.1002/jcb.25609. [DOI] [PubMed] [Google Scholar]
- 34.Corney D.C., Flesken-Nikitin A., Godwin A.K., Wang W., Nikitin A.Y. MicroRNA-34b and MicroRNA-34c are targets of p53 and cooperate in control of cell proliferation and adhesion-independent growth. Cancer Res. 2007;67:8433–8438. doi: 10.1158/0008-5472.CAN-07-1585. [DOI] [PubMed] [Google Scholar]
- 35.Ji Q., Hao X., Meng Y., Zhang M., Desano J., Fan D., Xu L. Restoration of tumor suppressor miR-34 inhibits human p53-mutant gastric cancer tumorspheres. BMC Cancer. 2008;8:266. doi: 10.1186/1471-2407-8-266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Perdomo C., Campbell J.D., Gerrein J., Tellez C.S., Garrison C.B., Walser T.C., Drizik E., Si H., Gower A.C., Vick J. MicroRNA 4423 is a primate-specific regulator of airway epithelial cell differentiation and lung carcinogenesis. Proc Natl Acad Sci U S A. 2013;110:18946–18951. doi: 10.1073/pnas.1220319110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ghandadi M., Sahebkar A. MicroRNA-34a and its target genes: key factors in cancer multidrug resistance. Curr Pharm Des. 2016;22:933–939. doi: 10.2174/1381612822666151209153729. [DOI] [PubMed] [Google Scholar]
- 38.Fang C., Chen Y.X., Wu N.Y., Yin J.Y., Li X.P., Huang H.S., Zhang W., Zhou H.H., Liu Z.Q. MiR-488 inhibits proliferation and cisplatin sensibility in non-small-cell lung cancer (NSCLC) cells by activating the eIF3a-mediated NER signaling pathway. Sci Rep. 2017;7 doi: 10.1038/srep40384. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Genomic Alterations in the TP53 WT Group According to TCGA.
Fold-Change of miRNAs and Coding Genes.