

Summary
Tumour‐infiltrating immune cells regulate tumour development and progression either negatively or positively. For example, cytotoxic lymphocytes (CTL) such as CD8+ T and natural killer (NK) cells can recognize and eliminate cancer cells, and thereby restrict the tumour growth and metastasis, if they exert full cytotoxicity. In contrast, tumour‐infiltrating myeloid cells such as tumour‐associated macrophages (TAM) promote the expansion and dissemination of cancer cells depending on their functional states. Given the tumour‐killing ability of CTL, the augmentation of CTL‐induced antitumour immune reactions has been considered as an attractive therapeutic modality for lethal solid tumours and several promising strategies have emerged, which include immune checkpoint inhibitors, cancer vaccines and adoptive CTL transfer. These immunotherapies are now tested in clinical trials and have shown significant antitumour effects in patients with lymphoma and some solid tumours such as melanoma and lung cancer. Despite these encouraging results, these therapies are not efficient in a certain fraction of patients and tumour types with tumour cell‐intrinsic mechanisms such as impaired antigen presentation and/or tumour cell‐extrinsic mechanisms including the accumulation of immunosuppressive cells. Several animal studies suggest that tumour‐infiltrating myeloid cells, especially TAM, are one of the key targets to improve the efficacy of immunotherapies as these cells can suppress the functions of CD8+ T and NK cells. In this review, we will summarize recent animal studies regarding the involvement of TAM in the immune checkpoint, cancer vaccination and adoptive CTL transfer therapies, and discuss the therapeutic potential of TAM targeting to improve the immunotherapies.
Keywords: cancer, immunotherapy, macrophage, tumour immunology
Abbreviations
- ARG1
arginase‐1
- CAR
chimeric antigen receptor
- CCR
CC‐chemokine receptor
- CEA
carcinoembryonic antigen
- CSF1R
colony‐stimulating factor 1 receptor
- CTLA4
cytotoxic T‐lymphocyte‐associated protein 4
- CTL
cytotoxic lymphocyte
- DC
dendritic cell
- FASL
first apoptosis signal ligand
- FcγR
Fc‐γ receptor
- FRβ
folate receptor β
- IL
interleukin
- LLC
Lewis lung carcinoma
- MARCO
macrophage receptor with collagenous structure
- MHC‐I
major histocompatibility complex class I
- NK
natural killer
- PD1
programmed cell death protein 1
- PI3Kγ
phosphoinositide 3‐kinase γ
- PyMT
polyoma middle T oncogene
- TAM
tumour‐associated macrophage
- TGF‐β
transforming growth factor‐β
- TLR
toll‐like receptor
- TNF‐α
tumour necrosis factor‐α
Introduction
Solid tumours are initiated by oncogenic mutations in non‐haematopoietic cells and progress into lethal tumour masses that account for 80% of all mortality of patients with cancer.1 Cytotoxic lymphocytes (CTL) such as CD8+ T and natural killer (NK) cells are critical for suppressing the development and progression of lethal solid tumours, as these cells can eliminate tumour cells once they exert full cytotoxicity.2 Therefore, many efforts have been invested to maximize the tumoricidal abilities of CTLs by understanding CTL regulatory mechanisms and developing CTL engineering technologies, which gives rise to attractive therapeutic modalities – for example, immunotherapies such as cancer vaccination, immune checkpoint therapy and adoptive CTL transfer therapy.
For effective killing of tumour cells, CD8+ T cells require largely three steps. At first, antigen‐presenting cells such as dendritic cells (DC) take up and process immunogenic aberrant proteins produced by genetic mutations in tumour cells (i.e. neoantigens) and present them to naive CD8+ T cells in the lymph node. Second, the primed and activated CD8+ T cells expand clonally and migrate into the tumours. At last, the effector CD8+ T cells recognize the antigenic peptides presented on the surface of tumour cells by major histocompatibility complex class I (MHC‐I) and transmit apoptotic signals into the tumour cells.3 Cancer vaccination and immune checkpoint therapy are developed to amplify this endogenous antitumour response3 and so are suitable for targeting immunogenic tumours that present neoantigens to CD8+ T cells. However, these therapies are not ideal to eliminate tumour cells that do not express tumour antigens or MHC‐I. To eliminate this type of cancer cell, adoptive transfer of CTL such as NK cells or engineered T cells has emerged. Unlike CD8+ T cells, NK cells do not require MHC‐I‐mediated priming or prior activation to kill their target cells. Instead, the cytotoxicity of NK cells is regulated by the balance of signals from inhibitory receptors and activating receptors on their surface. Tumour cells often up‐regulate NK activating receptor ligands and lose NK inhibitory receptor ligands, including MHC‐I molecules,4 and so this type of cancer cells can be eliminated by NK cells, whereas endogenous CD8+ T cells do not recognize them. Adoptive transfer of genetically engineered T cells that directly recognize cell surface proteins on tumour cells has also been developing as another attractive strategy to target such non‐immunogenic tumour cells.5
It is therefore likely that proper selection of therapeutic strategies is important to improve the outcome of immunotherapy, that is monotherapies using cancer vaccine or checkpoint inhibitors for immunogenic tumours and CTL transfer therapies for non‐immunogenic tumours. On the other hand, removal of immune suppressive factors in the tumour microenvironment, along with efficient CTL delivery, has been suggested as key issues to design effective immunotherapies for solid tumours. Most solid tumours include a variety of immune cells such as regulatory T cells, myeloid‐derived suppressor cells and tumour‐associated macrophages (TAM) that can suppress CTL functions.6, 7 Among these cells, TAM is one of the most abundant cell types in solid tumours8 and their infiltration into the tumour associates with poor prognosis in most solid tumours.9, 10, 11, 12, 13, 14 Furthermore, macrophages isolated from mouse and human solid tumours can directly suppress T‐cell responses15, 16 and NK cell cytotoxicity17, 18 in vitro. It is also reported that depletion of TAM enhances CD8+ T‐cell‐mediated antitumour immunity under treatment with chemotherapy in a mouse model of breast cancer.19 In addition, results from other mouse models of breast cancer indicate that physical contact of TAM with tumour‐infiltrating CD8+ T cells suppresses full activation of T cells or their access to the tumour cells.20, 21 Therefore, TAM has been suggested as one of the important therapeutic targets to enhance the efficacy of immunotherapy.22
To prevent the TAM‐mediated immune suppression, there are at least three therapeutic approaches, that is depletion, reprogramming and molecular targeting (Fig. 1). It has been suggested that the continuous accumulation of TAM requires the recruitment of circulating monocytes that differentiate into TAM in the tumour.22 In mouse models of breast cancer, the recruitment of monocytes into the tumour microenvironment is promoted via the CC‐chemokine receptor 2 (CCR2), and hence, blockade of CCR2 can suppress the accumulation of TAM in the tumours.23, 24, 25 It is also well known that colony‐stimulating factor 1 receptor (CSF1R) signal is essential for the recruitment, differentiation and survival of macrophages, and the loss of CSF1/CSF1R dramatically reduces the number of TAM in mouse models of solid tumour.22, 26, 27 Therefore, blockade of signals for monocyte recruitment and/or macrophage survival can reduce the TAM accumulation in tumours and thereby dislodge the immune suppressive tumour microenvironment. Another approach to block immune suppression by TAM is reprogramming of their characteristics. It is now widely accepted that plasticity is a hallmark of macrophages. For example, macrophages cultured with interleukin‐4 (IL‐4) and IL‐13 (called alternatively activated macrophages) produce anti‐inflammatory cytokines such as IL‐10 and transforming growth factor‐β (TGF‐β) that can inhibit CD8+ T‐cell functions, whereas macrophages cultured with lipopolysaccharide and interferon‐γ (called classically activated macrophages) secrete pro‐inflammatory cytokines such as tumour necrosis factor‐α (TNF‐α) and IL‐1β.28 Furthermore, alternatively activated macrophages can change their phenotype to classically activated macrophages when they are exposed to interferon‐γ and lipopolysaccharide.29 As alternatively but not classically activated macrophages suppress in vitro T‐cell proliferation,30 these in vitro studies suggest that targeting macrophage differentiation signals can reprogram TAM from immune suppressive to supportive cells and thereby enhance antitumour immune reactions induced by immunotherapy. Although the precise mechanisms behind TAM‐mediated immune suppression are still unclear, several studies suggest that TAM can suppress T‐cell activities directly via expression of arginase‐1 (ARG1), IL‐10 and TGF‐β, as well as indirectly through recruitment of other immune suppressor cells like regulatory T cells.31 Targeting these molecules can be another attractive approach to eliminate TAM‐mediated restriction of immunotherapy.
Figure 1.
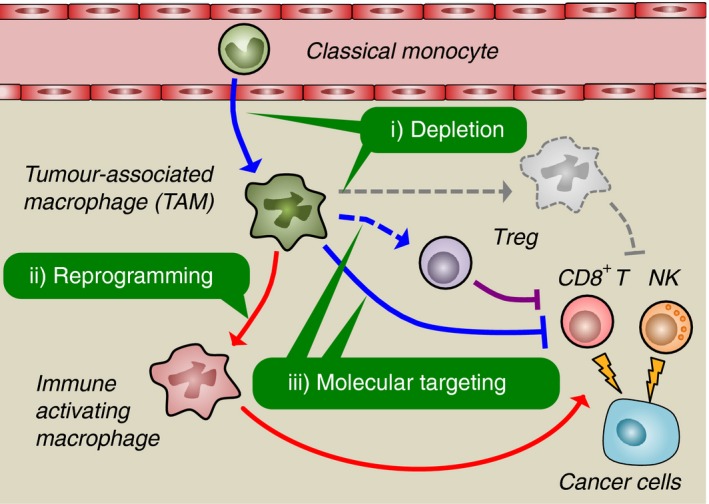
Potential therapeutic approaches to prevent tumour‐associated macrophage (TAM) ‐mediated immune suppression. Classical monocytes in the blood are recruited to the solid tumours where they differentiate to TAM. The TAM suppress cytotoxicity of CD8+ T or natural killer (NK) cells directly via expressing immune suppressive molecules or indirectly via the recruitment of other immune suppressor cells such as regulatory T (Treg) cells. However, TAM can become immune‐activating macrophages in response to certain environmental factors. It is therefore likely that either prevention of TAM accumulation (depletion), alteration of TAM features (reprogramming), or blockade of TAM‐derived immune suppressive molecules (molecular targeting) are attractive approaches to dislodge the TAM‐induced immune suppressive environment in the solid tumours.
TAM targeting for immune checkpoint therapy
To demonstrate strong antitumour responses, effector T cells need to overcome intrinsic negative regulation pathways transmitted via immune checkpoint receptors such as programmed cell death protein 1 (PD1) or cytotoxic T‐lymphocyte‐associated protein 4 (CTLA4). In many cases, cancer cells and stromal cells express ligands for PD1 (PD‐L1, PD‐L2) and/or CTLA4 (CD80, CD86) and thereby restrict the tumoricidal abilities of CD8+ T cells. To overcome this restriction, administration of blocking antibodies against these checkpoint receptors/ligands (checkpoint inhibitors) has been tested in clinical trials and has shown dramatic therapeutic effects in patients with melanoma and lung cancer.32 However, the majority of patients with other types of solid tumours, such as pancreatic and breast cancer, do not fully respond to this type of immunotherapy,33, 34 probably because large numbers of solid tumours including pancreatic and breast cancer (but not melanoma and lung cancer) have a low mutation rate that reduces the opportunity of neoantigen expression to be recognized by CD8+ T cells.35 The excluded immune cell infiltration can be another profile of non‐responding tumours where CD8+ T cells remain outside the tumours (i.e. peritumoral stroma) and fail to reach and eliminate tumour cells.36 Indeed, a recent study demonstrated that urothelial cancers in patients who did not respond to an anti‐PDL1 agent showed a low tumour mutation rate and a preferential accumulation of CD8+ T cells in the fibroblast‐rich peritumoral stroma rather than the tumour parenchyma.37 Another potential reason why checkpoint inhibitors show poor activity is because myeloid cells in the tumour microenvironment, especially TAM, limit the cytotoxicity of the activated CD8+ T cells by unknown mechanisms (Fig. 2a).38
Figure 2.

Effects of tumour‐associated macrophages (TAM) ‐targeting strategies on efficacy of immune checkpoint inhibitors. Immune checkpoint inhibitor (ICI) enhances CD8+ T‐cell cytotoxicity by blocking immune checkpoint pathways via programmed cell death protein 1 (PD1) or cytotoxic T‐lymphocyte‐associated protein 4 (CTLA4) activated by cancer cells and TAM. However, TAM also suppresses CD8+ T‐cell functions via checkpoint pathway‐independent mechanisms and limits the efficacy of ICI (a). Several studies suggest that all potential TAM‐targeting approaches, that is TAM depletion (b), TAM reprogramming (c), and targeting functional molecules of TAM (d), can improve the therapeutic efficacy of ICI. The yellow arrow represents cytotoxicity of CD8+ T cells.
In a mouse model of pancreatic cancer, treatment with a CCR2 antagonist decreases the infiltration of monocyte/macrophage in the tumour. In this model, CCR2 antagonist treatment in combination with anti‐PD1 antibody suppresses tumour growth, whereas single treatment with anti‐PD1 antibody is not effective.39 Therefore, it is likely that blockade of CCR2 signalling prevents TAM accumulation in the tumour and thereby enhances the efficacy of immune checkpoint inhibition. Treatment with CSF1R antagonists (e.g. PLX3397, PLX73086 and BLZ945) also markedly reduces the number of TAM in mouse models of mesothelioma, and of ovarian, cervical, breast and pancreatic cancers.19, 40, 41, 42, 43 In the mouse model of pancreatic cancer, treatment of tumour‐bearing mice with a CSF1R antagonist PLX3397 in combination with anti‐PD1 and anti‐CTLA4 antibodies results in the complete blockade of tumour expansion, although single treatment with PLX3397 or anti‐PD1/anti‐CTLA4 shows only modest suppression of tumour growth.43 A recent study also shows that the depletion of macrophages by PLX3397 in combination with anti‐PD1 therapy increases the accumulation of CD8+ T cells in the tumour and delays tumour progression in a mouse model of breast cancer.21 In mouse models of melanoma, single treatment with anti‐PD1 antibodies can suppress tumour growth, although several mice do not respond to the treatment. In contrast, combined treatment with anti‐PD1 and anti‐CSF1R antibodies induces tumour regression in all mice and prolongs their survival.44 These results indicate that the TAM depletion by targeting CCR2 and/or CSF1R can improve the efficacy of checkpoint inhibitors (Fig. 2b).
Recent studies suggest that TAM reprogramming can also enhance the antitumour effects of checkpoint inhibitors. In mice with melanoma established by subcutaneous injection of B16 cells, administration of neutralizing antibody against macrophage receptor with collagenous structure (MARCO), a scavenger receptor predominantly expressed by TAM, enhances the efficacy of anti‐CTLA4 antibody treatment in suppressing tumour growth.45 As the treatment of anti‐MARCO antibody reduces IL‐10 expression and concomitantly increases IL‐1β expression in TAM, these results suggest that targeting MARCO can switch the TAM phenotype from immunosuppressive (alternatively activated) to immune activating (classically activated) and thereby promote antitumour activities of cytotoxic T cells. Inhibition of phosphoinositide 3‐kinase γ (PI3Kγ) signalling may also change the immune suppressive phenotype of TAM as genetic deletion of PI3Kγ gene (Pik3cg) reduces the expression of ARG1, IL‐10 and TGF‐β in cultured alternatively activated macrophages.46 The loss of Pik3cg also reduces Arg1, Il10 and Tgfb mRNA expression in TAM and enhances the cytotoxicity of T cells in the subcutaneous tumours established by Lewis lung carcinoma (LLC) cells, suggesting that blockade of PI3Kγ signalling promotes the antitumour effects of T‐cell‐based immunotherapies by blocking immune suppressive functions of TAM. In line with this notion, a PI3Kγ inhibitor (TG100‐15) markedly enhances the tumour suppressive effects of anti‐PD1 antibody in a mouse model of head and neck squamous carcinoma.46 In the mammary tumours developed in polyoma middle T oncogene (PyMT) transgenic mice, a selective class IIa histone deacetylase inhibitor (TMP195) alters predominant macrophage populations in the tumour from TAM to highly phagocytic macrophages. In this model, administration of TMP195 combined with anti‐PD1 antibody significantly suppresses tumour development, whereas a single treatment with TMP195 or anti‐PD1 antibody shows modest suppression of the tumour burden.47 Therefore, targeting master regulators of macrophage differentiation (e.g. MARCO, PI3Kγ and histone deacetylase) can be a potential approach to enhance checkpoint therapy by harnessing immune suppressive features and/or drawing antitumour functions in tumour‐infiltrating macrophages (Fig. 2c).
It is well known that alternatively activated macrophages express high levels of ARG1, an l‐arginine processing enzyme that can suppress T‐cell functions by depleting l‐arginine from the environment.31 It is also reported that TAM isolated from the subcutaneous tumours established by C3 fibrosarcoma or LLC cells express high levels of ARG1 and suppress T‐cell proliferation via ARG1‐mediated mechanisms.48, 49 In mice that have received orthotopic injection of 4T1 mammary tumour cells, the treatment with anti‐PD1/anti‐CTLA4 antibodies combined with an ARG1 inhibitor (CB‐1158) significantly suppresses primary tumour growth and lung metastases.50 Likewise, treatment with CB‐1158 enhances the tumour suppressive effect of anti‐PD‐L1 antibody in mice with subcutaneous tumours developed by CT26 colon cancer cells.45 These results highlight the possibility that molecular targeting of TAM‐derived factors can be another approach to prevent TAM‐mediated restriction of checkpoint therapy (Fig. 2d). Although further studies are needed to identify targetable molecules that are expressed by TAM to suppress T‐cell cytotoxicity, a recent study suggests Fcγ receptor (FcγR), a receptor of immunoglobulin, as a candidate. In subcutaneous tumours established by MC38 colon cancer cells, anti‐PD1 antibodies injected into the mice initially bind to T cells but are deprived by TAM via FcγR within 24 hr. Furthermore, injection of anti‐PD1 antibody with FcγR‐blocking antibody completely suppresses tumour growth in all mice whereas the response to a single anti‐PD1 treatment varies among animals,51 suggesting that targeting the interaction of Fc receptors in TAM with the Fc region of checkpoint blocking antibodies can improve the therapeutic efficacy of checkpoint inhibitors. These studies in mouse models confirm that all three TAM‐targeting strategies (depletion, reprogramming and molecular targeting) could potentially be used in combination with checkpoint inhibitors to synergistically improve the response to this kind of immunotherapy.
TAM targeting for cancer vaccination
An additional approach to boost CD8+ T‐cell reactions against tumours is cancer vaccination. Vaccination involves the administration of antigenic materials aimed at the stimulation of the endogenous immune system with the final goal to prevent the onset of infections and tumours (preventive vaccination) or treat the diseases (therapeutic vaccination). Preventive vaccinations, where the antigens are injected before the disease takes place, aim to ‘simulate’ a bacterial or a viral infection so that the immune system, specifically B cells, can generate antibodies against the pathogen and memory for the potential real encounter with the pathogen. On the other side, therapeutic vaccinations aim to educate naive CD8+ T cells to become cytotoxic lymphocytes through antigen presentation by antigen‐presenting cells such as DC and thereby activate the immune system against an ongoing infection or neoplastic lesion that the immune system was not able to detect. Although this therapy is advantageous in cost, ease and safety, its therapeutic response is so far quite limited, probably because each tumour has different mutations, and hence, neoantigens expressed by tumour cells are diverse. However, recent advances in deep‐sequencing technologies enable us to identify the mutations in an individual tumour and thereby predict optimal neoantigens to prime and activate CD8+ T cells.35 It has also been suggested that efficacy of cancer vaccination can be enhanced by co‐injection of immune adjuvants that amplify the host immune responses such as toll‐like receptor (TLR) ligands and DC‐targeted antibodies.52 Another approach for the refinement of this type of therapy is the ex vivo generated DC‐based vaccines where DC cultured with whole tumour cell lysate or antigenic peptide are injected back into patients.53 Advances in all of these components will make therapeutic vaccination more efficient. As in other immunotherapies, however, recent studies have demonstrated that the efficacy of cancer vaccination is strongly linked with the level of accumulation and activation of myeloid cells, especially macrophages.
For example, injection of tumour lysate‐pulsed DC (DC‐based vaccination) prolongs survival of mice that have been orthotopically injected with syngeneic mesothelioma cells, and this therapeutic effect is further enhanced by DC‐based vaccination in combination with injection of PLX3397, a CSF1R inhibitor that depletes macrophages.40 Depletion of TAM also enhances the efficacy of therapeutic vaccination with strong adjuvants. In a murine model of ovarian cancer, immunization with microparticles containing ligands of TLR9 and nucleotide‐binding oligomerization domain 2 leads to the accumulation of T cells in the tumours and prolongs the survival of tumour‐bearing mice. On the other hand, the vaccination also increases accumulation of T‐cell suppressive CD11b+ myeloid cells in the peritoneum.54 In this model, CD11b‐mediated depletion of myeloid cells shows a synergistic effect in combination with the vaccine by further prolonging the survival of tumour‐bearing mice even if no significant reduction in tumour size was observed.54 Similar results are reported in a mouse model of cervical cancer (i.e. subcutaneous injection of TC‐1 cancer cells) in which tumour‐bearing mice are immunized with a di‐palmitoylated peptide, a self‐adjuvanting antigen that stimulates DC maturation and primes CTLs in a TLR2/6‐specific fashion.55 In this model, depletion of myeloid cells by clodronate liposome injection increases the efficacy of the lipopeptide‐based vaccination in reducing tumour size and prolonging survival of tumour‐bearing mice (Fig. 3a). However, macrophage depletion is not always associated with an improved response to vaccination even in the similar cervical tumour mouse model. A recent study shows that injection of a long peptide with incomplete Freund's adjuvant to mice that have established the subcutaneous TC‐1 cervical tumours induces a significant accumulation of CD8+ T cells as well as macrophages in the tumour lesion. Interestingly, vaccination‐induced tumour regression in this case is abrogated rather than enhanced by macrophage depletion before and during the peptide injection using the inhibitor PLX3397, suggesting that the reprogramming of resident macrophages or the recruitment of pro‐inflammatory macrophages by vaccination is responsible for therapeutic efficacy (Fig. 3b).56 These studies suggest that differences in the vaccination protocol affects features of macrophages in the tumours, and hence, TAM‐targeting strategies should be carefully evaluated to combine with cancer vaccination.
Figure 3.
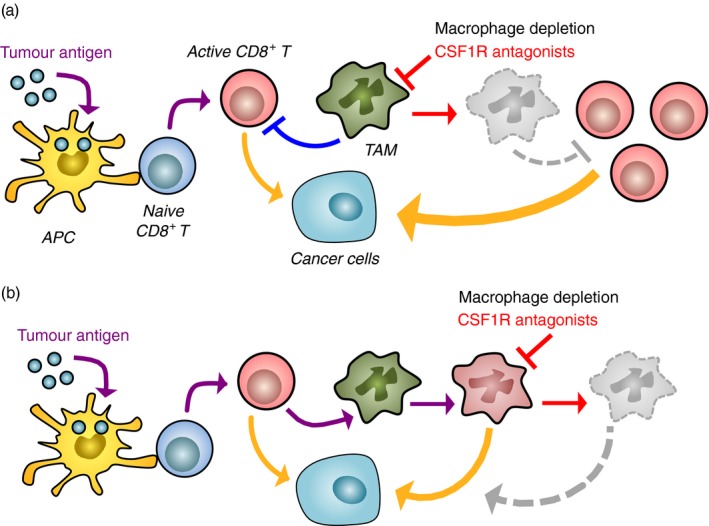
Opposite effects of macrophage depletion on therapeutic cancer vaccination therapy. Administration of tumour antigen with strong adjuvant or dendritic cells pre‐exposed to antigen activates naive CD8+ T cells that exert antitumour ability. However, their cytotoxicity is suppressed by tumour‐associated macrophages (TAM), and hence, pharmacological macrophage depletion, for example treatment with a colony‐stimulating factor 1 receptor (CSF1R) antagonist, enhances efficacy of vaccinations (a). In some cases, however, the activated CD8+ T cells alter the phenotype of TAM from immune suppressive to immune‐activating/tumoricidal. Under such situations, macrophage depletion reduces rather than enhances the efficacy of therapeutic cancer vaccination (b). The yellow arrow represents cytotoxicity of CD8+ T cells or reprogrammed macrophages.
TAM targeting for adoptive CTL transfer therapy
As mentioned above, cancer cells in malignant solid tumours often lose the expression of MHC‐I that is essential for antigen presentation to CD8+ T cells.57 As cancer cells lacking neoantigens or MHC‐I cannot be recognized by pre‐existing tumour‐infiltrating CD8+ T cells, therapeutic strategies aimed at boosting antigen‐dependent T‐cell activation (i.e. vaccination and checkpoint inhibition) may not be suitable for these types of tumours.58 One of the emerging strategies to target the non‐immunogenic cancer cells is an adoptive transfer of CD8+ T cells that are manipulated to express chimeric antigen receptors (i.e. CAR‐T cells).59 Chimeric antigen receptor (CAR) is a genetically engineered T‐cell receptor where the intracellular signalling domain is activated by extracellular single‐chain variable fragments upon direct binding to a specific protein expressed on the surface of tumour cells. Therefore, CAR‐T cells can exert cytotoxicity against tumour cells expressing the target surface molecules without MHC‐I‐restricted antigen presentation.54, 60 The CAR‐T‐cell transfer therapy has been successful for B lymphoma, but its applicability to solid tumours is still under investigation.61 In the solid tumours, the adoptively transferred CAR‐T cells would need to overcome the immune suppressive tumour microenvironment, including TAM in it, to exercise their therapeutic potential (Fig. 4a).
Figure 4.
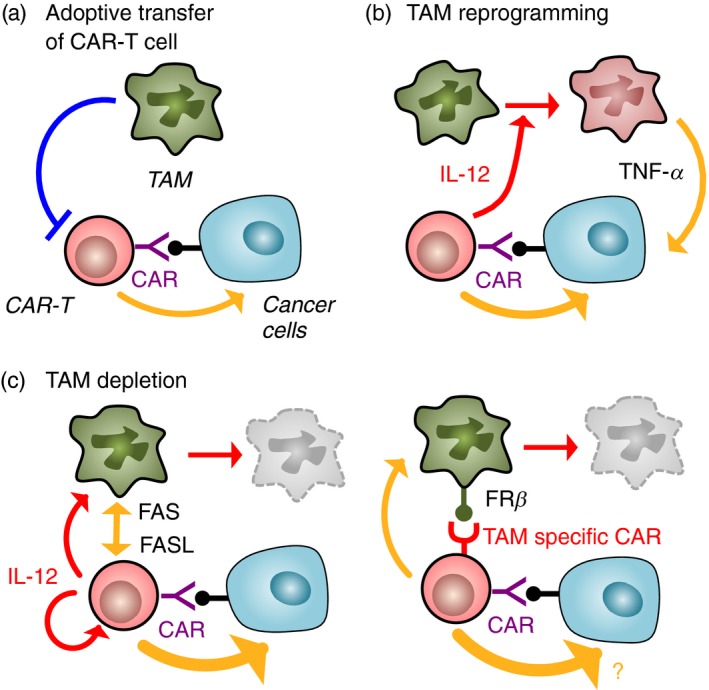
Improvement of chimeric antigen receptor T (CAR‐T) cell transfer therapy via the removal of tumour‐associated macrophages (TAM) ‐mediated immune suppression. T cells manipulated to express CAR‐T recognize surface protein on cancer cells and exert tumour‐killing activity without prior activation, whereas their functions are restricted by TAM (a). It is reported that genetic manipulation of CAR‐T cells to secrete interleukin‐12 (IL‐12) enhances efficacy of the therapy via reprogramming of TAM to tumoricidal macrophages that express tumour necrosis factor‐α (TNF‐α) (b). In another model, expression of IL‐12 in CAR‐T cells enhances therapeutic potential of CAR‐T cells by depletion of TAM as well as augmentation of cytotoxic capacity of CAR‐T cells (c; left). It is also suggested that expression of CAR against macrophage surface protein (e.g. folate receptor β: FRβ) can deplete TAM, whereas its therapeutic impact on CAR‐T cell cytotoxicity against tumour cells is still not known. The yellow arrow represents cytotoxicity of CD8+ T cells or reprogrammed macrophages.
It is suggested that this requirement can be fulfilled by further genetic manipulation of CAR‐T cells to express stimulatory ligands or cytokines. In a subcutaneous tumour model, adoptive transfer of carcinoembryonic antigen (CEA) ‐specific CAR‐T cells modestly suppresses tumour growth of CEA‐expressing C15A3 cancer cells but not their parental CEA‐negative MC38 cells.62 In this model, co‐expression of IL‐12 in the CAR‐T cells dramatically enhances the suppressive effects of transferred CAR‐T cells on the C15A3 tumour. In the C15A3 tumour, the transfer of IL‐12‐producing CAR‐T cells but not the normal CAR‐T cells increases the number of macrophages that express TNF‐α in the tumours. Interestingly, the IL‐12‐producing CAR‐T cells can also inhibit tumour growth of CEA‐negative MC38 cells when these cancer cells are co‐injected with CEA‐positive C15A3 cells, and such suppressive effects disappear with anti‐TNF‐α‐neutralizing antibody treatment. Furthermore, cultured macrophages stimulated with IL‐12 directly kill MC38 cancer cells via production of TNF‐α.62 It has been reported that systemic injection of IL‐12 into the LLC tumour‐bearing mice significantly reduces IL‐10 and TGF‐β expression in TAM and concomitantly increases expression IL‐6 and TNF‐α.63 These results therefore collectively suggest that IL‐12 from CAR‐T cells reprogram macrophages recruited into the tumours to be tumoricidal cells that co‐operate with CAR‐T cells for elimination of cancer cells (Fig. 4b).
On the other hand, IL‐12 has been shown to stimulate antitumour responses through activation of CD8+ T cells in solid tumour models.64 In mice with abdominal cancer developed by ID8 ovarian cancer cells that express ectodomain of mucin‐16 (Muc16ecto), intraperitoneal injection of Muc16ecto‐specific CAR‐T cells modified to secrete IL‐12 prolongs the survival of tumour‐bearing mice.65 As this therapeutic effect is not observed in mice treated with IL‐12‐expressing CAR‐T cells developed from IL‐12 ‘receptor’ knockout mice, IL‐12 secreted from modified CAR‐T cells seems to act in an autocrine manner. Indeed, the Muc16ecto‐specific CAR‐T cells increase cytokine secretion, proliferation, cytotoxicity and survival in vitro when they are modified to secrete IL‐12. In this ovarian cancer model, IL‐12‐expressing CAR‐T cells increase their expression of first apoptosis signal ligand (FASL) and up‐regulate FAS expression on TAM and thereby eliminate TAM in the ascites via the FAS/FASL apoptosis pathway. As macrophage depletion by clodronate treatment significantly prolongs overall survival of tumour‐bearing mice treated with the IL‐12‐expressing CAR‐T cells, these results suggest that co‐expression of IL‐12 intensifies the therapeutic potential of CAR‐T cells by depletion of TAM in addition to the augmentation of CAR‐T‐cell cytotoxic capacity (Fig. 4c).65 Although IL‐12 armoured CAR‐T cells deplete TAM via an indirect mechanism,65 it is suggested that genetic manipulation of T cells to express TAM‐specific CAR can directly eliminate TAM in the tumours. In the ID8 ovarian cancer model, TAM in peritoneal tumour ascites expresses a high level of folate receptor β (FRβ) and adoptive transfer of FRβ‐specific CAR‐T cells into the tumour‐bearing mice results in destruction of TAMs and delayed tumour growth (Fig. 4c).66 However, further studies are needed to determine whether co‐expression of two different CARs, that is one for TAM and another for cancer cells, enhances therapeutic effects of CAR‐T‐cell transfer therapy. All of these studies suggest that future CAR‐T‐cell engineering should focus not only to target cancer cells but also to modulate the phenotype of TAM by their reprogramming or by their selective depletion in the tumour microenvironment.
Although therapeutic effects of TAM targeting on NK cell infusion therapy are not clear, suppressive effects of TAM on NK cell functions have been reported. For example, high infiltration of TAM in the tumours correlates with low number of interferon‐γ‐expressing active NK cells in human hepatocellular carcinoma and TAM from the tumour reduce activation and survival of NK cells.17 It is also reported that TAM isolated from the PyMT mouse mammary tumours significantly reduces cytotoxicity of NK cells in vitro.18 These results suggest that TAM depletion by blocking TAM recruitment or survival signals (e.g. treatment with CCR2 or CSF1R antagonist) can improve the efficacy of NK cell infusion therapy. After the encouraging results of CAR‐T‐cell transfer therapy, genetic engineering of NK cells has been tested at preclinical stage.67 Further development of this technology may enable us to manipulate NK cells to express CAR that binds to TAM (e.g. FRβ) or TAM regulatory cytokines (e.g. IL‐12), which would enhance NK cell cytotoxicity in the solid tumours by depletion or reprogramming of TAM.
Perspective
Cancer immunotherapy is currently being tested in several clinical trials in combination with standard therapies, and preclinical research is also active to optimize better ways to stimulate the immune system in combination with pre‐existing or novel therapies.68, 69 Unfortunately, however, not all the strategies work for all tumours and not all patients seem to respond even in those cancer subtypes where the therapy works. This is caused by different aspects such as the immunogenic properties of the tumour but also by the tumour microenvironment composition. It is becoming clear that boosting the antitumour abilities of CTL is not sufficient to exert significant tumour depletion and that a certain level of intervention in the immune suppressive tumour microenvironment must accompany the CTL activation. As summarized in this review, different studies have indicated that TAM targeting is able to synergistically enhance the response to almost all immunotherapies. Although most TAM‐targeting therapies are still at the preclinical stage, antagonists or blocking antibodies that can be used for TAM depletion (e.g. CCR2 or CSF1R antagonists) have already been tested in clinical trials for malignant solid tumours.70, 71 Further investigation of synergistic effects of these agents with immunotherapies will lead to the improvement of ongoing immunotherapeutic strategies. On the other hand, data suggest that macrophages in the solid tumours can engage a robust antitumour immune response in some cases and so TAM depletion is not necessarily associated with good outcome in some immunotherapies such as cancer vaccination. Therefore, deciphering the exact molecular mechanisms responsible for macrophage polarization during treatment is necessary to determine effective TAM‐targeting approaches to improve immunotherapies. It is also necessary to identify specific markers and immune suppressive molecules of TAM in different cancers and cancer subtypes for the targeting of only the tumour‐promoting macrophage subpopulations. Results from these basic studies will be helpful to choose the appropriate TAM‐targeting strategy (depletion, reprogramming and molecular targeting) and tailor it based on the type of administered immunotherapy.
Disclosures
The authors declare no conflict of interests.
Acknowledgements
This work was supported by grants from the Wellcome Trust (109657/Z/15/Z, 615KIT/J22738, 101067/Z/13/Z, UK), and the MRC (MR/N022556/1, UK). This work was also kindly backed by the COST Action BM1404 Mye‐EUNITER (http://www.mye-euniter.eu). COST is supported by the EU Framework Programme Horizon 2020.
References
- 1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin 2017; 67:7–30. [DOI] [PubMed] [Google Scholar]
- 2. Dunn GP, Old LJ, Schreiber RD. The immunobiology of cancer immunosurveillance and immunoediting. Immunity 2004; 21:137–48. [DOI] [PubMed] [Google Scholar]
- 3. Durgeau A, Virk Y, Corgnac S, Mami‐Chouaib F. Recent advances in targeting cd8 t‐cell immunity for more effective cancer immunotherapy. Front Immunol 2018; 9:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ljunggren HG, Malmberg KJ. Prospects for the use of NK cells in immunotherapy of human cancer. Nat Rev Immunol 2007; 7:329–39. [DOI] [PubMed] [Google Scholar]
- 5. Kershaw MH, Westwood JA, Darcy PK. Gene‐engineered T cells for cancer therapy. Nat Rev Cancer 2013; 13:525–41. [DOI] [PubMed] [Google Scholar]
- 6. Smyth MJ, Ngiow SF, Ribas A, Teng MW. Combination cancer immunotherapies tailored to the tumour microenvironment. Nat Rev Clin Oncol 2016; 13:143–58. [DOI] [PubMed] [Google Scholar]
- 7. Sharma P, Hu‐Lieskovan S, Wargo JA, Ribas A. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell 2017; 168:707–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Qian B‐Z, Pollard JW. Macrophage diversity enhances tumour progression and metastasis. Cell 2010; 141:39–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zhang QW, Liu L, Gong CY, Shi HS, Zeng YH, Wang XZ et al Prognostic significance of tumour‐associated macrophages in solid tumour: a meta‐analysis of the literature. PLoS ONE 2012; 7:e50946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gentles AJ, Newman AM, Liu CL, Bratman SV, Feng W, Kim D et al The prognostic landscape of genes and infiltrating immune cells across human cancers. Nat Med 2015; 21:938–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Guo B, Cen H, Tan X, Ke Q. Meta‐analysis of the prognostic and clinical value of tumour‐associated macrophages in adult classical Hodgkin lymphoma. BMC Med 2016; 14:159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mei J, Xiao Z, Guo C, Pu Q, Ma L, Liu C et al Prognostic impact of tumour‐associated macrophage infiltration in non‐small cell lung cancer: a systemic review and meta‐analysis. Oncotarget 2016; 7:34217–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yin S, Huang J, Li Z, Zhang J, Luo J, Lu C et al The prognostic and clinicopathological significance of tumour‐associated macrophages in patients with gastric cancer: a meta‐analysis. PLoS ONE 2017; 12:e0170042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhao X, Qu J, Sun Y, Wang J, Liu X, Wang F et al Prognostic significance of tumour‐associated macrophages in breast cancer: a meta‐analysis of the literature. Oncotarget 2017; 8:30576–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ruffell B, Coussens LM. Macrophages and therapeutic resistance in cancer. Cancer Cell 2015; 27:462–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kitamura T, Qian BZ, Pollard JW. Immune cell promotion of metastasis. Nat Rev Immunol 2015; 15:73–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wu Y, Kuang DM, Pan WD, Wan YL, Lao XM, Wang D et al Monocyte/macrophage‐elicited natural killer cell dysfunction in hepatocellular carcinoma is mediated by CD48/2B4 interactions. Hepatology 2013; 57:1107–16. [DOI] [PubMed] [Google Scholar]
- 18. Krneta T, Gillgrass A, Chew M, Ashkar AA. The breast tumour microenvironment alters the phenotype and function of natural killer cells. Cell Mol Immunol 2016; 13:628–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. DeNardo DG, Brennan DJ, Rexhepaj E, Ruffell B, Shiao SL, Madden SF et al Leukocyte complexity predicts breast cancer survival and functionally regulates response to chemotherapy. Cancer Discov 2011; 1:54–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Engelhardt JJ, Boldajipour B, Beemiller P, Pandurangi P, Sorensen C, Werb Z et al Marginating dendritic cells of the tumor microenvironment cross‐present tumor antigens and stably engage tumor‐specific T cells. Cancer Cell 2012; 21:402–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Peranzoni E, Lemoine J, Vimeux L, Feuillet V, Barrin S, Kantari‐Mimoun C et al Macrophages impede CD8 T cells from reaching tumor cells and limit the efficacy of anti‐PD‐1 treatment. Proc Natl Acad Sci USA 2018; 115:E4041–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mantovani A, Marchesi F, Malesci A, Laghi L, Allavena P. Tumour‐associated macrophages as treatment targets in oncology. Nat Rev Clin Oncol 2017; 14:399–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lu X, Kang Y. Chemokine (C‐C motif) ligand 2 engages CCR2+ stromal cells of monocytic origin to promote breast cancer metastasis to lung and bone. J Biol Chem 2009; 284:29087–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Qian B‐Z, Li J, Zhang H, Kitamura T, Zhang J, Campion LR et al CCL2 recruits inflammatory monocytes to facilitate breast‐tumour metastasis. Nature 2011; 475:222–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kitamura T, Qian B‐Z, Soong D, Cassetta L, Noy R, Sugano G et al CCL2‐induced chemokine cascade promotes breast cancer metastasis by enhancing retention of metastasis‐associated macrophages. J Exp Med 2015; 212:1043–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lin EY, Nguyen AV, Russell RG, Pollard JW. Colony‐stimulating factor 1 promotes progression of mammary tumours to malignancy. J Exp Med 2001; 193:727–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ries CH, Cannarile MA, Hoves S, Benz J, Wartha K, Runza V et al Targeting tumour‐associated macrophages with anti‐CSF‐1R antibody reveals a strategy for cancer therapy. Cancer Cell 2014; 25:846–59. [DOI] [PubMed] [Google Scholar]
- 28. Zhang YH, He M, Wang Y, Liao AH. Modulators of the balance between m1 and m2 macrophages during pregnancy. Front Immunol 2017; 8:120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Smith TD, Tse MJ, Read EL, Liu WF. Regulation of macrophage polarization and plasticity by complex activation signals. Integr Biol (Camb) 2016; 8:946–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Oishi S, Takano R, Tamura S, Tani S, Iwaizumi M, Hamaya Y et al M2 polarization of murine peritoneal macrophages induces regulatory cytokine production and suppresses T‐cell proliferation. Immunology 2016; 149:320–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Noy R, Pollard JW. Tumour‐associated macrophages: from mechanisms to therapy. Immunity 2014; 41:49–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sharma P, Allison JP. Immune checkpoint targeting in cancer therapy: toward combination strategies with curative potential. Cell 2015; 161:205–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Foley K, Kim V, Jaffee E, Zheng L. Current progress in immunotherapy for pancreatic cancer. Cancer Lett 2016; 381:244–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Polk A, Svane IM, Andersson M, Nielsen D. Checkpoint inhibitors in breast cancer – current status. Cancer Treat Rev 2018; 63:122–34. [DOI] [PubMed] [Google Scholar]
- 35. Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science 2015; 348:69–74. [DOI] [PubMed] [Google Scholar]
- 36. Chen DS, Mellman I. Elements of cancer immunity and the cancer‐immune set point. Nature 2017; 541:321–30. [DOI] [PubMed] [Google Scholar]
- 37. Mariathasan S, Turley SJ, Nickles D, Castiglioni A, Yuen K, Wang Y et al TGFβ attenuates tumour response to PD‐L1 blockade by contributing to exclusion of T cells. Nature 2018; 554:544–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Cassetta L, Kitamura T. Targeting tumour‐associated macrophages as a potential strategy to enhance the response to immune checkpoint inhibitors. Front Cell Dev Biol 2018; 4:38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Janson C, Jung H, Ertl L, Liu S, Dang T, Zeng Y et al Abstract 5655: inhibition of CCR2 potentiates checkpoint inhibitor immunotherapy in murine model of pancreatic cancer. Cancer Res 2017; 77(Supplement):5655. [Google Scholar]
- 40. Dammeijer F, Lievense LA, Kaijen‐Lambers ME, van Nimwegen M, Bezemer K, Hegmans JP et al Depletion of tumour‐associated macrophages with a CSF‐1R kinase inhibitor enhances antitumour immunity and survival induced by DC immunotherapy. Cancer Immunol Res 2017; 5:535–46. [DOI] [PubMed] [Google Scholar]
- 41. Lyons YA, Pradeep S, Wu SY, Haemmerle M, Hansen JM, Wagner MJ et al Macrophage depletion through colony stimulating factor 1 receptor pathway blockade overcomes adaptive resistance to anti‐VEGF therapy. Oncotarget 2017; 8:96496–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Strachan DC, Ruffell B, Oei Y, Bissell MJ, Coussens LM, Pryer N et al CSF1R inhibition delays cervical and mammary tumour growth in murine models by attenuating the turnover of tumour‐associated macrophages and enhancing infiltration by CD8+ T cells. Oncoimmunology 2013; 2:e26968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zhu Y, Knolhoff BL, Meyer MA, Nywening TM, West BL, Luo J et al CSF1/CSF1R blockade reprograms tumour‐infiltrating macrophages and improves response to T‐cell checkpoint immunotherapy in pancreatic cancer models. Cancer Res 2014; 74:5057–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Neubert NJ, Schmittnaegel M, Bordry N, Nassiri S, Wald N, Martignier C et al T cell‐induced CSF1 promotes melanoma resistance to PD1 blockade. Sci Transl Med 2018; 10:eaan3311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Georgoudaki AM, Prokopec KE, Boura VF, Hellqvist E, Sohn S, Östling J et al Reprogramming tumour‐associated macrophages by antibody targeting inhibits cancer progression and metastasis. Cell Rep 2016; 15:2000–11. [DOI] [PubMed] [Google Scholar]
- 46. Kaneda MM, Messer KS, Ralainirina N, Li H, Leem CJ, Gorjestani S et al PI3Kγ is a molecular switch that controls immune suppression. Nature 2016; 539:437–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Guerriero JL, Sotayo A, Ponichtera HE, Castrillon JA, Pourzia AL, Schad S et al Class IIa HDAC inhibition reduces breast tumours and metastases through anti‐tumour macrophages. Nature 2017; 543:428–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Rodriguez PC, Quiceno DG, Zabaleta J, Ortiz B, Zea AH, Piazuelo MB et al Arginase I production in the tumour microenvironment by mature myeloid cells inhibits T‐cell receptor expression and antigen‐specific T‐cell responses. Cancer Res 2004; 64:5839–49. [DOI] [PubMed] [Google Scholar]
- 49. Kusmartsev S, Gabrilovich DI. STAT1 signaling regulates tumour‐associated macrophage‐mediated T cell deletion. J Immunol 2005; 174:4880–91. [DOI] [PubMed] [Google Scholar]
- 50. Steggerda SM, Bennett MK, Chen J, Emberley E, Huang T, Janes JR et al Inhibition of arginase by CB‐1158 blocks myeloid cell‐mediated immune suppression in the tumour microenvironment. J Immunother Cancer 2017; 5:101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Arlauckas SP, Garris CS, Kohler RH, Kitaoka M, Cuccarese MF, Yang KS et al In vivo imaging reveals a tumour‐associated macrophage‐mediated resistance pathway in anti‐PD‐1 therapy. Sci Transl Med 2017; 9:eaal3604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hu Z, Ott PA, Wu CJ. Towards personalized, tumour‐specific, therapeutic vaccines for cancer. Nat Rev Immunol 2018; 18:168–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Pizzurro GA, Barrio MM. Dendritic cell‐based vaccine efficacy: aiming for hot spots. Front Immunol 2015; 6:91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Khan AN, Kolomeyevskaya N, Singel KL, Grimm MJ, Moysich KB, Daudi S et al Targeting myeloid cells in the tumour microenvironment enhances vaccine efficacy in murine epithelial ovarian cancer. Oncotarget 2015; 6:11310–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Shen KY, Song YC, Chen IH, Chong P, Liu SJ. Depletion of tumour‐associated macrophages enhances the anti‐tumour immunity induced by a Toll‐like receptor agonist‐conjugated peptide. Hum Vaccin Immunother 2014; 10:3241–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. van der Sluis TC, Sluijter M, van Duikeren S, West BL, Melief CJ, Arens R et al Therapeutic peptide vaccine‐induced CD8 T cells strongly modulate intratumoural macrophages required for tumour regression. Cancer Immunol Res 2015; 3:1042–51. [DOI] [PubMed] [Google Scholar]
- 57. Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity's roles in cancer suppression and promotion. Science 2011; 331:1565–70. [DOI] [PubMed] [Google Scholar]
- 58. Ribas A, Wolchok JD. Cancer immunotherapy using checkpoint blockade. Science 2018; 359:1350–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. June CH, O'Connor RS, Kawalekar OU, Ghassemi S, Milone MC. CAR T cell immunotherapy for human cancer. Science 2018; 359:1361–5. [DOI] [PubMed] [Google Scholar]
- 60. Irving M, Vuillefroy de Silly R, Scholten K, Dilek N, Coukos G. Engineering chimeric antigen receptor t‐cells for racing in solid tumours: don't forget the fuel. Front Immunol 2017; 8:267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Li J, Li W, Huang K, Zhang Y, Kupfer G, Zhao Q. Chimeric antigen receptor T cell (CAR‐T) immunotherapy for solid tumours: lessons learned and strategies for moving forward. J Hematol Oncol 2018; 11:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Chmielewski M, Kopecky C, Hombach AA, Abken H. IL‐12 release by engineered T cells expressing chimeric antigen receptors can effectively Muster an antigen‐independent macrophage response on tumour cells that have shut down tumour antigen expression. Cancer Res 2011; 71:5697–706. [DOI] [PubMed] [Google Scholar]
- 63. Watkins SK, Egilmez NK, Suttles J, Stout RD. IL‐12 rapidly alters the functional profile of tumour‐associated and tumour‐infiltrating macrophages in vitro and in vivo . J Immunol 2007; 178:1357–62. [DOI] [PubMed] [Google Scholar]
- 64. Portielje JE, Gratama JW, van Ojik HH, Stoter G, Kruit WH. IL‐12: a promising adjuvant for cancer vaccination. Cancer Immunol Immunother 2003; 52:133–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Yeku OO, Purdon TJ, Koneru M, Spriggs D, Brentjens RJ. Armored CAR T cells enhance antitumour efficacy and overcome the tumour microenvironment. Sci Rep 2017; 7:10541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Lynn RC, Matshuyama T, Powell DJ. Targeting FRb+ tumour associated macrophages with CAR T cells in ovarian cancer. J Immunother Cancer 2015; 3(Suppl 2):32.26199728 [Google Scholar]
- 67. Guillerey C, Huntington ND, Smyth MJ. Targeting natural killer cells in cancer immunotherapy. Nat Immunol 2016; 17:1025–36. [DOI] [PubMed] [Google Scholar]
- 68. Wraith DC. The future of immunotherapy: a 20‐year perspective. Front Immunol 2017; 8:1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Bookstaver ML, Tsai SJ, Bromberg JS, Jewell CM. Improving vaccine and immunotherapy design using biomaterials. Trends Immunol 2018; 39:135–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Nywening TM, Wang‐Gillam A, Sanford DE, Belt BA, Panni RZ, Cusworth BM et al Targeting tumour‐associated macrophages with CCR2 inhibition in combination with FOLFIRINOX in patients with borderline resectable and locally advanced pancreatic cancer: a single‐centre, open‐label, dose‐finding, non‐randomised, phase 1b trial. Lancet Oncol 2016; 17:651–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Cannarile MA, Weisser M, Jacob W, Jegg AM, Ries CH, Rüttinger D. Colony‐stimulating factor 1 receptor (CSF1R) inhibitors in cancer therapy. J Immunother Cancer 2017; 5:53. [DOI] [PMC free article] [PubMed] [Google Scholar]