

Summary
Regulatory T (Treg) cells play an essential role in the maintenance of intestinal homeostasis. In Peyer's patches (PPs), which comprise the most important IgA induction site in the gut‐associated lymphoid tissue, Treg cells promote IgA isotype switching. However, the mechanisms underlying their entry into PPs and isotype switching facilitation in activated B cells remain unknown. This study, based on the dextran sulphate sodium (DSS)‐induced colitis model, revealed that Treg cells are significantly increased in PPs, along with CD11b+ B‐cell induction. Immunofluorescence staining showed that infiltrated Treg cells were located around CD11b+ B cells and produced transforming growth factor‐β, thereby inducing IgA+ B cells. Furthermore, in vivo and in vitro studies revealed that CD11b+ B cells in PPs had the capacity to recruit Treg cells into PPs rather than promoting their proliferation. Finally, we found that Treg cell recruitment was mediated by the chemokine CXCL9 derived from CD11b+ B cells in PPs. These findings demonstrate that CD11b+ B cells induced in PPs during colitis actively recruit Treg cells to accomplish IgA isotype switch in a CXCL9‐dependent manner.
Keywords: CD11b+ B cells, colitis, regulatory T cells
Abbreviations
- APC
allophycocyanin
- CCL
CC chemokine ligand
- CCR
CC chemokine receptor
- DSS
dextran sulphate sodium
- FBS
fetal bovine serum
- MLN
mesentery lymph nodes
- PBS
phosphate‐buffered saline
- PE
phycoerythrin
- PP
Peyer's patch
- TGF‐β
transforming growth factor‐β
- Th17
T helper type 17
- Treg
regulatory T
- WT
wild‐type
Introduction
B cells have been widely reported to exert regulatory functions that maintain immune homeostasis.1, 2 Regulatory B cells inhibit excessive inflammatory responses via multiple mechanisms, including antigen presentation, cytokine production and cellular interactions.3 We have previously demonstrated that B cells attenuate the severity of acute dextran sulphate sodium (DSS) ‐induced colitis and maintain gut homeostasis through an increase in regulatory T (Treg) cells.4 Moreover, Treg cells are considered important facilitators of the induction and maintenance of intestinal IgA+ B‐cell responses.5 Cong and colleagues showed that Treg cells can regulate intestinal IgA expression of B cells via transforming growth factor β (TGF‐β).6 Therefore, the interaction between Treg and B cells may play a role in the maintenance of intestinal immune homeostasis. However, how Treg cells enter the B zone, in particular Peyer's patches (PPs), to interact with B cells, remains unknown.
CD11b, also known as macrophage‐1 antigen (Mac‐1), is widely expressed on various immune cells, including B cells, and multiple studies indicate that it exerts a suppressive function. For example, CD11b expressed in antigen‐presenting cells negatively regulates immune responses via Toll‐like receptor pathways.7, 8 Moreover, CD11b expression can inhibit T‐cell activation and T helper type 17 (Th17) cell differentiation,9, 10 and can maintain autoreactive B‐cell tolerance through negative regulation of B‐cell receptor signalling.11 The suppressive function of CD11b has also been reported in autoimmune diseases, including DSS‐induced colitis.12 In our previous study, CD11b‐expressed B cells acted as regulatory cells in experimental autoimmune hepatitis induced by self‐liver antigens.13 However, little is known about the role of induced CD11b+ B cells in colitis.
Highly organized follicular structures termed PPs constitute the most important IgA‐inducing sites in the gut‐associated lymphoid tissue.14, 15 The development of IgA+ B cells in PPs depends on the antigenic stimulation and generation of germinal centres.16, 17 The PPs are equipped with a follicle‐associated epithelium that contains microfold cells (M cells), able to recognize and capture invaded antigens.18 Following antigen‐specific stimulation, PPs form germinal centres while B cells undergo proliferation, class switch recombination and somatic hypermutation to induce high‐affinity IgA.19, 20 Induction of protective IgA+ B cells depends on the cognate interaction between B cells and CD4+ T cells. Distinguishable from other effector helper T cells, CXCR5+ PD‐1+ Bcl‐6+ T follicular helper cells are considered essential for B‐cell survival and activation in PPs.21 Moreover, Th17 was reported to mediate gut‐IgA induction for specific members of the commensal microbiota, such as segmented filamentous bacteria colonization.22, 23, 24 Recent studies have shown that Treg cells also play a critical role in the differentiation of IgA+ B cells in PPs.5 Our previous study revealed that B cells increase Treg cells in the lamina propria.4 Whether this also occurs in PPs remains unclear.
Using a DSS‐induced colitis mouse model, we found that the CD11b+ B cells were inductively up‐regulated in PPs, and that their adoptive transfer could increase Treg cells in PPs. Moreover, we found that the induced CD11b+ B cells in PPs actively secrete CXCL9 to recruit Treg cells into PPs and help them differentiate into IgA+ cells.
Materials and methods
Mice
Wild‐type (WT) C57BL/6 mice were purchased from the Shanghai SLAC Laboratory Animal Co., Ltd (Shanghai, China). CD11b−/− mice and μMT mice on a C57BL/6 background were obtained from the Institute Pasteur of the Chinese Academy of Science (Shanghai, China). All mice were 6–12 weeks of age and were housed in the animal facility of Fudan University (Shanghai, China) under specific pathogen‐free barrier conditions. Animal care and use were in compliance with the guidelines outlined in the Guide for the Care and Use of Laboratory Animals.
DSS‐induced colitis
According to a previously established protocol, experimental colitis was established by administering 2·5% DSS (w/v; MP Biomedicals, Santa Ana, CA) in the mice's drinking water for 7 days.4, 25
Immunofluorescence analysis
The PPs were removed and fixed in 4% paraformaldehyde for 24 h, washed with phosphate‐buffered saline (PBS) and sequentially treated with 20% sucrose for 12 hr at 4°. The tissues were embedded in OCT compound (Sakura Finetechnical, Tokyo, Japan). Cryostat sections (6 μm) were pre‐blocked with 5% (w/v) fetal bovine serum (FBS) for 15 min at room temperature, stained with purified antibodies for mCD19 and mCD11b (Abcam, Cambridge, MA) for 15 hr at 4° and then stained with Alexa Fluor® 647 anti‐rat IgG monoclonal antibody (Cell Signaling Technology, Danvers, MA) and DyLight 405 anti‐rabbit IgG monoclonal antibody (Jackson ImmunoResearch, West Grove, PA) and fluorescein isothiocyanate Foxp3 monoclonal antibody (BD Biosciences, San Diego, CA) for 1 hr at room temperature. The specimens were analysed using fluorescence microscopy.
Flow cytometry
Mononuclear cells were first incubated with anti‐CD16/32 antibody (BD Biosciences) and then reacted with the following anti‐mouse antibodies: allophycocyanin (APC) ‐Cy7‐CD11b (M1/70); fluorescein isothiocyanate‐CD19 (1D3); phycoerythrin (PE) ‐CD25 (PC61.5); PE‐IgA (Ma‐6E1) (all from eBioscience, San Diego, CA) and PE‐Cy7‐CD45 (30‐F11); Pacific blue‐CD4 (RM4‐5) (from BD Biosciences). To stain intracellular (CXCL9, TGF‐β) and intranuclear (Foxp3, Ki67) markers, cells were fixed and permeabilized using a Staining Buffer set (eBioscience) and stained with PE‐Ki67 (SolA15; eBioscience); APC‐Foxp3 (MF23; BD Biosciences); APC‐CXCL9 (MIG‐2F5.5; BioLegend, San Diego, CA); and Pacific blue‐TGF‐β (TW7‐16B4; BD Biosciences).
Concentration‐matched isotype antibodies were used as negative controls. Flow‐cytometric analysis was performed with CyAn ADP Analyzer (Beckman Coulter, Carlsbad, CA), and data were analysed using the flowjo software (Tree Star, Ashland, OR). Absolute numbers of multiple cell populations were calculated by flow cytometry, according to their respective percentage.
Cell isolation and adoptive transfer assays
CD19+ cells from CD11b−/− mice and CD11b+ B cells from PPs in WT mice were sorted with a MoFlo flow cytometer (Beckman Coulter). CD19+ CD11b+/CD11b− B cells from PPs in mice with colitis were similarly purified by flow cytometry. The purity of both cell populations was at least 90%. The role of CD11b+ B cells in colitis was determined through adoptive transfer of approximately 5 × 106 purified single‐cell suspensions into μMT mice through the inferior ophthalmic vein, followed by DSS administration 48 hr later. The purified CD19+ CD11b+/CD11b− B cells (5 × 106 cells, from DSS‐induced mice) were adoptively transferred into healthy WT mice, respectively. Control groups received PBS instead.
Enzyme‐linked immunosorbent assay
To evaluate CXCL9‐producing CD11b+ B cells in PPs, cells were purified from DSS‐treated mice on days 0, 4, 7 and 10 and cultured with RPMI‐1640 medium in 96‐well plates (2 × 105 cells/well) for 24 hr. The supernatants were collected and coated into the plates (100 μl/well) overnight, at 4°, after which they were blocked with 250 μl PBS containing 5% (w/v) fat‐free milk at 37° for 2 hr. After washing the plates twice with PBS containing 0·05% Tween‐20, anti‐CXCL9 antibodies (AF‐492‐NA; R&D Systems, Minneapolis, MN) were diluted as 1 μg/ml and added for incubation at 37° for 1·5 hr. Horseradish peroxidase‐conjugated donkey anti‐goat antibodies (Santa Cruz Biotechnology, Heidelberg, Germany) were added, after which the plates were incubated at 37° for 1 hr. After the final washing, the colour reaction was developed with 4,4′‐Bi‐2,6‐xylidine (eBioscience) at room temperature for 15 min and terminated with 2 m H2SO4. The optical density value was measured at a 450 nm wavelength (OD450).
Real‐time polymerase chain reaction analysis
Total RNA of CD11b+ B cells was prepared with an RNeasy kit (Qiagen, Crawley, UK) followed by DNase I treatment and reverse transcribed into cDNA. Real‐time polymerase chain reaction (PCR) was performed in SYBR Green Master Mix (TaKaRa Biotechnology Co., Ltd, Dalian, China) plus optimal volumes of cDNA and primers on the ABI 7500 Thermocycler (Applied Biosystems, Foster City, CA). The amplification parameter was used for 40 cycles of PCR, and all PCRs were set up at least in triplicate. The relative gene expression compared with β‐actin was calculated by the ∆∆Ct method. The chemokine‐expression profile was made by hemi software. The sense CXCL9 primer was 5′‐GGCATCATCTTCCTGGAGCAGTGTGGAGTT‐3′ and the antisence CXCL9 primer was 5′‐TTGTAGTGGATCGTGCCTCGGCTGGTG‐3′. The sense β‐actin primer was 5′‐CCAGCCTTCCTTCTTGGGTATG‐3′, and the antisense primer was 5′‐TGTGTTGGCATAGAGGTCTTTACG‐3′.
Treg cell migration and proliferation assays
To investigate the effect of CD11b+ and CD11b− B cells on Treg cell migration, we sorted B cells from PPs on days 4, 7 and 10 following DSS treatment and cultured them into RPMI‐1640 medium (without FBS) in 48‐well plates (3 × 105 cells/well) for 24 hr. Either 600 μl cell supernatant containing recombinant mouse CXCL9 (rmCXCL9, 50 ng/ml; Peprotech Inc.) or anti‐CXCL9 antibody (1 μg/ml) was placed in the lower chamber of 6·5‐mm diameter, 5·0‐μm pore‐size polycarbonate membrane filter Transwell plates (Costar Corning, Cambridge, MA). Total lymphocyte suspension (1 × 107 cells/ml) from PPs (on days 4, 7 and 10) was added to the upper filters (200 μl/well) and incubated for 4 hr. Migrated cells in the lower chamber were collected and stained with PE‐CD4 (GK1.5; eBioscience) and APC‐Foxp3 (MF23; BD Bioscience) followed by flow cytometry analysis.
CD4+ CD25+ Treg cells were purified from WT mice with MicroBead (Miltenyi Biotec, Bergisch‐Gladbach, Germany). The combinations of fluorescence‐activated cell sorted (FACS) CD11b+ B cells (4 × 105 cells/well) and CD4+ CD25+ Treg cells (2 × 105 cells/well) were co‐cultured in 48‐well plates for 72 hr in complete RPMI‐1640 containing 10% FBS, 2 mm l‐glutamine, 0·05 mm 2‐mercaptoethanol and 100 U/ml penicillin/streptomycin. The control group was given the same dose of CD4+ CD25+ Treg cells alone. Proliferation of Treg cells was measured with PE‐Ki67 expression by flow cytometry.
Statistical analysis
Multiple group comparisons were performed using one‐way analysis of variance, followed by either Bonferroni correction or Mann–Whitney U‐test to compare two individual groups. Statistical analysis was performed using graphpad prism (GraphPad Software, San Diego, CA). The P‐value of < 0·05 was considered statistically significant.
Results
Treg cells increased following CD11b+ B‐cell induction in PPs during colitis
Our previous study showed that Treg cells were indispensable helper cells to induce and maintain IgA in gut‐associated lymphoid tissue during DSS‐induced colitis.4 As IgA isotype switch was reported to occur in PPs,26 we first determined the distribution of Treg cells in PPs during DSS‐induced colitis. Cytometric analysis of Treg cells on days 4, 7 and 10 after DSS induction showed that 10% of CD4+ T cells in control mice‐derived PPs are Foxp3+ Treg cells (Fig. 1a). No significant change was observed on day 4 regarding the frequency of Treg cells, whereas on days 7 and 10, they increased to ~15% of CD4+ T cells. The absolute number consistently increased from 1 × 105 up to nearly 3 × 105 cells in PPs, with up‐regulated production of TGF‐β and IgA (Fig. 1a; see Supplementary material, Figs S1 and S2). We have shown that CD11b+ B cells are up‐regulated and play an important suppressive role in experimental autoimmune hepatitis.13 We next determined whether CD11b+ B cells can also be induced in colitis. The results showed that, before colitis induction, PPs comprised 1–2% of CD11b+ B cells. From day 4 to day 10, they had a twofold to fivefold increase in the frequency range and reached the normal level around day 20 (Fig. 1b; see Supplementary material, Fig. S3a). Moreover, the absolute number of CD11b+ B cells also increased on day 4 and thereafter (Fig. 1b). The increase in CD11b+ B cells in PPs was much earlier than that of Treg cells (see Supplementary material, Fig. S3b).
Figure 1.
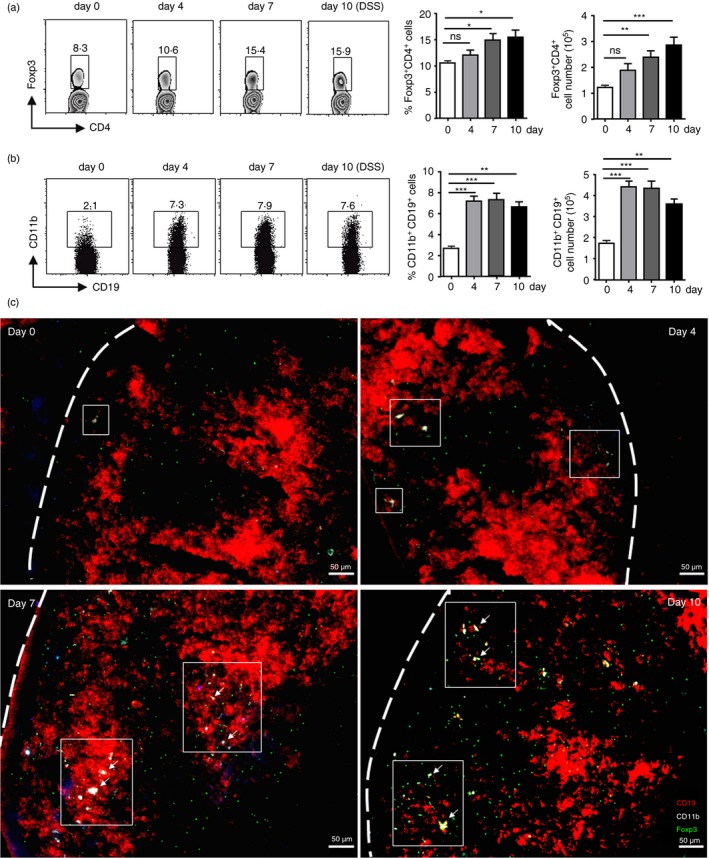
Both regulatory T (Treg) cells and CD11b+ B cells increased in Peyer's patches (PPs) during dextran sulphate sodium (DSS) ‐induced colitis. (a, b) Flow cytometry was used to determine the percentage and absolute number of Treg cells and CD11b+ B cells in PPs of wild‐type (WT) mice on days 0, 4, 7 and 10 after colitis induction. Treg cell expressions (CD4+ Foxp3+) within the CD4‐gated population is shown in (a). CD11b+ B‐cell expressions within the CD19‐gated population are shown in (b). *P < 0·05; ***P < 0·001. Data shown are the mean ± SEM from one experiment with eight mice, which was repeated at least three times with similar results. (c) Immunofluorescence microscopic analysis was performed to examine the distribution of cells expressing CD19 (red), CD11b (cyan) (CD19 and CD11b double‐positive, yellow) and Foxp3 (green) in the representative PP sections obtained from WT mice on days 0, 4, 7 and 10 after colitis induction. Bars indicate 50 μm.
To further confirm the results, we performed an immunofluorescence assay using frozen sections obtained from DSS‐induced mice. The stained PP sections derived from untreated mice showed large quantities of CD19+ B cells, but few CD11b+ B cells (showed CD19+ CD11b+) or Treg cells (showed Foxp3+) (Figs 1c; see Supplementary material, Fig. S4). Moreover, few Foxp3+ Treg cells were observed in B‐cell zones where activated B cells differentiate to IgA+ B cells. However, CD11b+ B cells increased in PPs along with the progression of colitis. Consistent with flow cytometric analysis, the Treg cells, which were still unobservable on day 4, infiltrated after the appearance of CD11b+ B cells. On days 7 and 10, the quantity of Foxp3+ cells observed in the PPs increased. Moreover, Treg cells entered the B zones along with their increase, some of which, interestingly, were located around the CD11b+ B‐cell area (Fig. 1c). Taken together, these results reveal that CD11b+ B cells were induced before the increase of Treg cells, and that a large proportion of Treg cells was located in B‐cell zones.
Increase in Treg cells was promoted by induced CD11b+ B cells during colitis
Given that CD11b+ B cells expanded into PPs much earlier than Treg cells, we questioned whether CD11b+ B‐cell induction facilitated the increase in Treg cells. Both the B cells of PPs obtained from WT (CD11b+/+ B cells) and CD11b‐deficient mice (CD11b−/− B cells) were sorted by FACS. Equal numbers of purified cells were adoptively transferred into B‐lymphocyte‐deficient μMT mice, 2 days before DSS administration, and Treg cells in PPs were detected 7 days later. The frequency of Treg cells within both CD4+ and CD45+ T cells increased about threefold following adoptive CD11b+/+ B‐cell transfer, compared with other groups (Fig. 2a). To further confirm that CD11b+ B cells play a positive role in in vivo Treg cell expansion, purified CD19+ CD11b+ B cells or CD19+ CD11b− B cells in PPs from 7 days DSS‐induced mice were adoptively transferred into WT mice. On post‐transfer day 4, about 10% of transferring B cells were found in the recipients' PPs (see Supplementary material, Fig. S5). As shown in Fig. 2(b), mice that received CD11b+ B cells showed a significant increase compared with naive WT mice. These results demonstrate that induced CD11b+ B cells in PPs promote the expansion of Treg cells in vivo.
Figure 2.
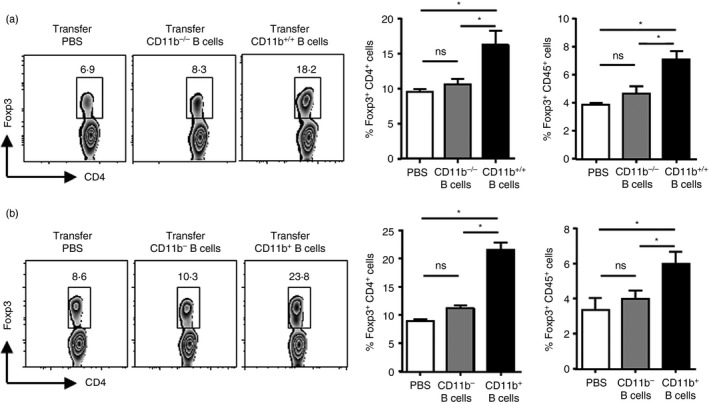
Induced CD11b+ B cells may increase regulatory T (Treg) cells in Peyer's patches (PPs) in vivo. (a) μMT mice were intravenously (i.v.) administered with PP‐derived B cells from wild‐type (WT) mice and CD11b‐deficient mice, respectively, and administered with dextran sulphate sodium (DSS) 2 days later after adoptive transfer. Treg cell percentages (Foxp3+) in PPs within the CD4‐ or CD45‐gated population were detected on day 7 after colitis induction. (b) Purified CD19+ CD11b+ B cells and CD19+ CD11b− B cells in PPs were sorted from colitis mice on day 7 after DSS administration and were adoptively transferred into WT mice. Treg cell percentages (Foxp3+) in PPs within the CD4‐ or CD45‐gated population were detected 4 days later. The number of transferred cells were at 5 × 106 cells suspended in 200 μl sterile phosphate‐buffered saline (PBS). The control group mice receive PBS instead. *P < 0·05; data are representative of six independent experiments.
Treg cells are recruited by induced CD11b+ B cells rather than proliferated in situ during colitis
We have previously found that splenic B cells promote lamina propria Treg cell proliferation both in vivo and in vitro.4 Hence, we hypothesized that induced CD11b+ B cells in PPs contribute to Treg cell expansion by influencing their proliferation. To investigate this hypothesis, CD19+ CD11b+ B cells were sorted from PPs on day 7 after colitis induction, and co‐cultured with purified Treg cells at the ratio of 2 : 1 in vitro. Unexpectedly, CD11b+ B cells had no effect on Treg cell proliferation (Fig. 3a). We tested whether the Treg cell increase was mediated by the recruitment of CD11b+ B cells using a transwell assay. We observed that Treg cells migrated towards either CD11b+ B cells or CD11b− B cells obtained from different stages of colitis. However, Treg cells exhibited a significantly increased chemotaxis towards CD11b+ B cells compared with CD11b− B cells at different stages. These results indicate that induced CD11b+ B cells contribute to the increase in Treg cells by recruitment.
Figure 3.
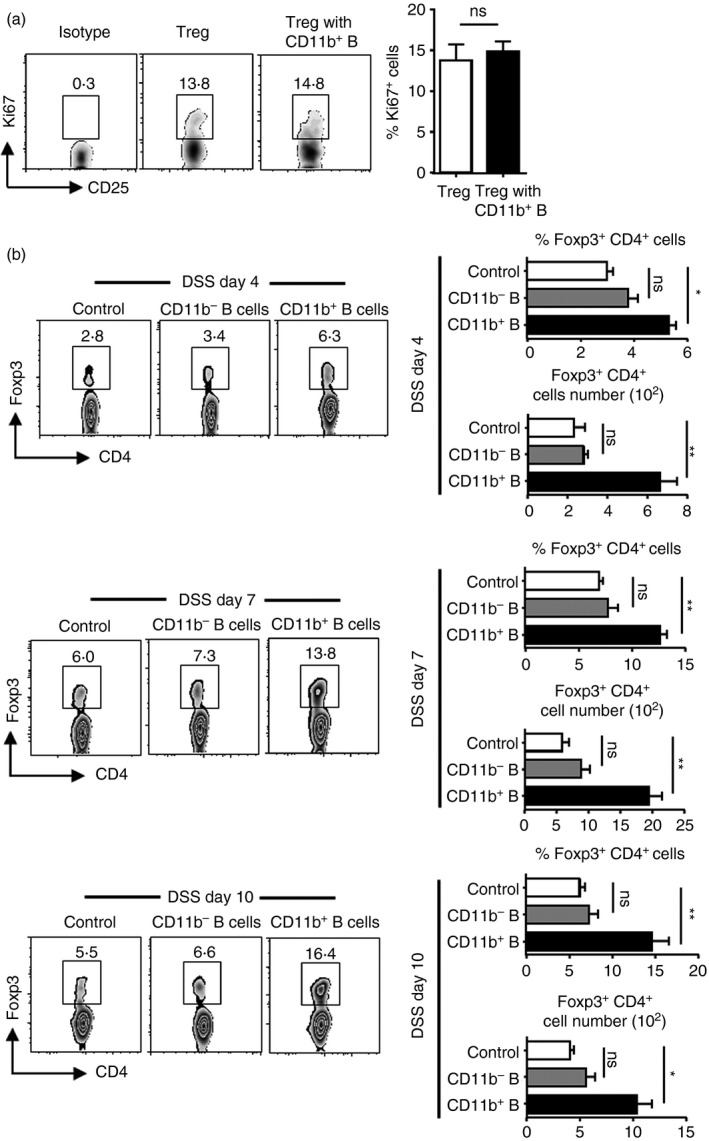
Induced CD11b+ B cells, but not CD11b− B cells, could increase regulatory T (Treg) cells though recruiting route rather than proliferation in vitro. (a) CD11b+ B cells of Peyer's patches (PPs) from day 7 in mice with colitis were sorted with a MoFlo high‐speed cell sorter (Beckman Coulter, Pasadena, CA). Treg cells (CD4+ CD25+; 2 × 105 cells/well) were purified from naive wild‐type (WT) mice with MicroBeads (Miltenyi Biotec) and were cultured alone or with CD11b+ B cells (4 × 105 cells/well) in 48‐well plates for 72 hr in complete RPMI‐1640. The percentage of Ki‐67+ Treg cells was determined by FACS. CD11b+ B cells and Treg cells were prepared from four mice. (b) The supernatant samples were obtained either from purified CD11b+ B cells or CD11b− B cells (3 × 105 cells/well) sorted from PPs on days 4, 7 and 10, from mice with colitis and then cultured in RPMI‐1640 (without fetal bovine serum) for 24 hr. Total PP lymphocyte suspensions (2 × 106 cells/well) were prepared from the respective period (days 4, 7 and 10) of mice with colitis. Supernatants and cell suspensions were separated by Transwell in a 24‐well plate for 4 hr. Percentage and absolute number of Treg cells were determined by FACS. *P < 0·05; **P < 0·01. Results presented as the means of four independent experiments.
Treg cell migration into PPs during colitis is mediated by CD11b+ B‐cell‐derived CXCL9
We have previously reviewed the function of activated B cells as chemokine producers.27 Hence, we next investigated whether CD11b+ B cells functioned as chemokine‐producing cells in Treg cell recruitment in PPs. We obtained CD11b+ B cells from mouse PPs on days 0, 1, 4, 7 and 10 after colitis induction and analysed their expression of Treg cell recruitment‐related chemokines by quantitative PCR. We found that CD11b+ B cells from naive mice had a limited chemokine secretion ability. However, on day 1 after DSS administration, various chemokines were up‐regulated, including Mig(CXCL9); SLC(CCL21); MIP‐α(CCL20); MIP‐3β(CCL19); MCP‐5(CCL12); and SDF‐1(CXCL12). On days 1 and 4, CXCL9 was the only chemokine that remained up‐regulated (Fig. 4a).
Figure 4.
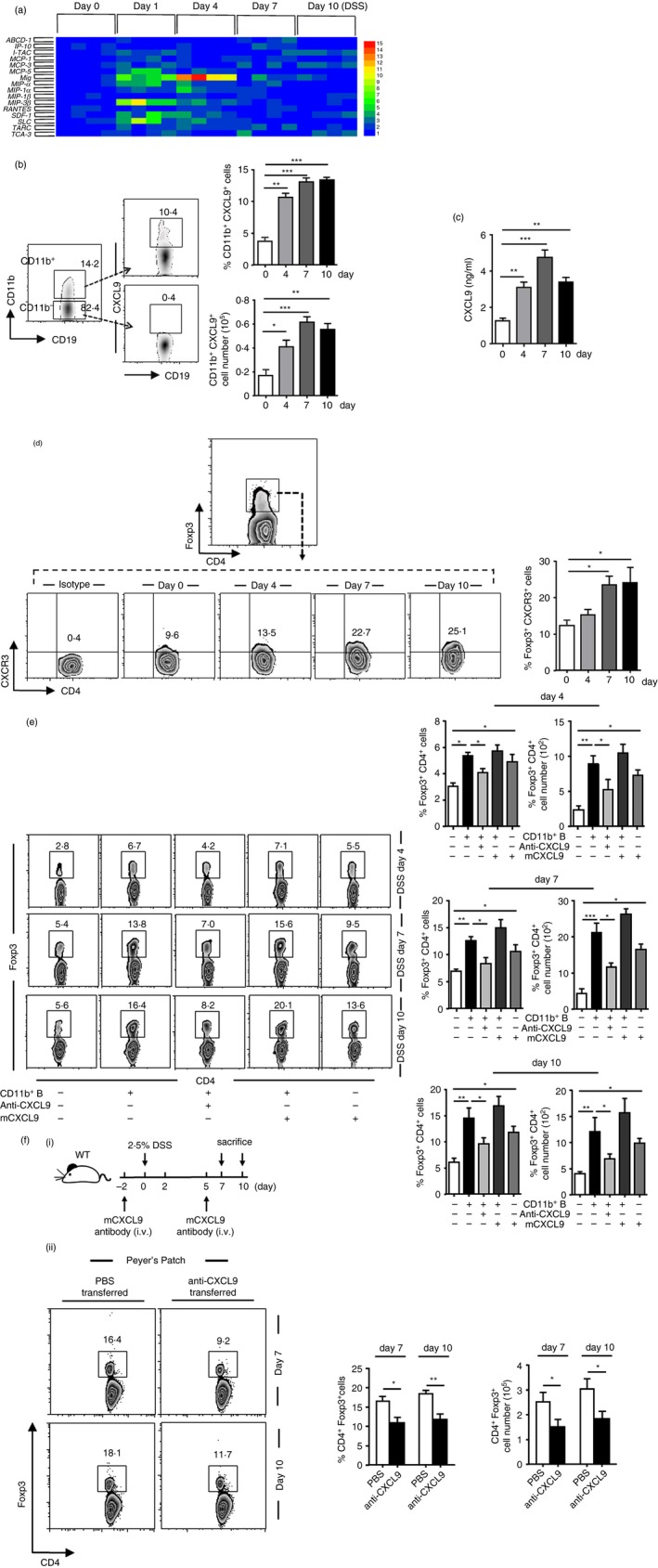
The induced CD11b+ B cells initially produce CXCL9 to increase regulatory T (Treg) cells. (a) Real‐time PCR analysis was performed to examine the chemokine expressions of Peyer's patches (PPs) CD11b+ B cells purified from dextran sulphate sodium (DSS) ‐treated mice. The chemokine profile was developed using the hemi software. Samples of every phage in colitis contain four mice. (b) CXCL9+ expression within the CD11b+ or CD11b−‐gated population was obtained in PPs on days 0, 4, 7 and 10 after colitis induction. (c) PP‐derived CD11b+ B cells from mice with colitis were sorted and cultured in complete RPMI‐1640 for 24 hr. The cell supernatant was harvested, and the level of chemokine CXCL9 was determined by enzyme‐linked immunosorbent assay. (d) CXCR3 expressions gated on Foxp3+ T cells from PPs during the process of colitis were detected using flow cytometry. (e) Total PP‐derived lymphocyte suspension (described as in Fig. 3b, 2 × 106 cells/well) was either cultured alone or with CXCL9 (50 ng/ml), CD11b+ B‐cell supernatant (described as in Fig. 3b) CD11b+ B cells supernatant and CXCL9 or CD11b+ B‐cell supernatant and anti‐CXCL9 antibody (1 μg/ml), which were separated in a 24‐well Transwell plate for 4 hr. Percentage and absolute number of Treg cells were determined by FACS. (f; i) Illustration of the protocol of anti‐CXCL9 antibody (4·5 μg/g) administration in DSS‐induced mice. (f; ii) Frequencies and absolute numbers of PP‐derived Treg cells on days 7 and 10 after CXCL9 neutralization were analysed by flow cytometry. *P < 0·05; *P < 0·05; **P < 0·01. Results presented as the mean ± SEM of one experiment, which was repeated at least three times yielding similar results.
To confirm the results, the chemokine increase was analysed by flow cytometry, which revealed that only CXCL9 increased at protein level (data not shown). Moreover, the expression of CXCL9 was significantly higher in induced CD11b+ B cells than in CD11b− B cells (Fig. 4b). Supernatant of cultured CD11b+ B cells also had a high CXCL9 concentration (Fig. 4c). Accordingly, CXCR3, the receptor of CXCL9, was highly expressed on Treg cells at both day 7 and day 10 in DSS‐treated mice (Fig. 4d). Taken together, these results show that CXCL9+ CD11b+ B cells were significantly up‐regulated during colitis compared with controls. Intriguingly, the expression of CXCL9 was undetected in CD11b− B cells at any stage of colitis.
Further, a migration assay was performed using CXCL9 neutralization antibody (anti‐CXCL9) and recombinant mouse CXCL9 (rmCXCL9). We found that anti‐CXCL9 blocked the ability of PP CD11b+ B cells to attract Treg cells (Fig. 4e). The number of Treg cells was significantly reduced after addition of anti‐CXCL9 antibody. In contrast, rmCXCL9 addition promoted CD11b+ B‐cell recruitment of Treg cells. Moreover, Treg cells were also recruited when rmCXCL9 was added alone. To investigate whether the anti‐CXCL9 antibody could block the effect of CXCL9 and decrease Treg cell accumulation in PPs, CXCL9 was neutralized with anti‐CXCL9 antibody to DSS‐induced mice in vivo. Results revealed that the populations of Treg cells were significantly reduced on both day 7 and day 10, after anti‐CXCL9 antibody administration (Fig. 4f). Taken together, these results suggest that induced CD11b+ B cells recruit Treg cells into PPs in a process mediated by CXCL9.
Discussion
The present study provides evidence that CD11b+ B cells are induced in PPs, in a DSS‐induced colitis model. On the course of the disease, induced CD11b+ B cells can attract Treg cells into PPs. Moreover, we determined that chemokine CXCL9 mediates the recruitment of Treg cells into PPs by CD11b+ B cells during disease.
Although our previous study indicated that CD11b+ B cells play an important suppressive role in experimental hepatitis,13 the present study, in line with previous ones, demonstrates that Treg cells also play an essential role in the maintenance of intestinal homeostasis.4 There is no evidence for the presence of Th1 and Th17 cells in PPs, although they play an important role for the induction and perpetuation of intestinal inflammation.28, 29 The present study shows that DSS‐induced mice are characterized by few Th1 and Th17 cells in PPs over the course of disease (data not shown). The present study shows that B cells promote the recruitment of Treg cells into PPs, which in turn triggers the IgA isotype switch of B cells. We found that, although CD11b+ B are induced by day 3, a significant increase in Treg cells in PPs occurs only at the peak of disease (day 7). Despite the obviously increased accumulation of induced CD11b+ B cells in PPs from day 3 after DSS induction, no obvious increase in Treg cell population was detected until day 6 (see Supplementary material, Fig. S3b). Given that IgA class switching is dependent on TGF‐β and T‐cell‐derived TGF‐β plays an important role in colitis,30, 31, 32, 33, 34 we determined the Treg‐derived TGF‐β in our system. Consistently, we detected that Foxp3+ T cells in PPs produced higher levels of TGF‐β expression during disease, accompanied by significantly increased IgA expression (see Supplementary material, Figs S1 and S2). Consistent with previous studies, which demonstrated that T‐cell‐derived TGF‐β plays an important role in colitis.33, 34
In a previous study, we reported that, rather than recruiting splenic and mesentery lymph node (MLN) Treg cells, B cells promote their proliferation.4 Hence, we first investigated the proliferation of Treg cells in PPs during disease, which, surprisingly, remained unaltered (Fig. 3a), indicating that increase in PP Treg cells during colitis was not due to proliferation. In the present study, we found that CD11b+ B cells increased much earlier than Treg cells and that transferring CD11b+ B cells induced the increase in Treg cells (Figs 1 and 2). We hypothesized that CD11b+ B cells may increase Treg cells via chemotactic attraction. In our previous study, we found that part of CD11b− B cells could differentiate into CD11b+ B cells in PPs when they were adoptively transferred into mice a week later. To obtain purified B cells without induced CD11b expression, B cells must be sorted from CD11b knockout mice (CD11b−/− B cells) as control group in the transfer assay. It is noteworthy that CD11b‐deficient B cells failed to increase additional Treg cells in PPs (Fig. 2a), indicating that these intestinal B cells play a positive role in the recruitment of Treg cells in a CD11b‐dependent manner. In addition, the increase in Treg cells and CD11b+ B cells was not observed in MLN (data not shown). It remains to be clarified whether this is due to a deficiency of induced CD11b+ B cells in MLN or to MLN not comprising an IgA‐producing site.
As we summarized before, B cells can act as chemokine producers, which is important to understand the additional functions of B cells apart from their roles via immunoglobulin secretion or antigen presentation.27 We found that the transcriptional levels of various chemokines in CD11b+ B cells were up‐regulated during disease, possibly induced by the pro‐inflammatory immune environment (Fig. 4a). We detected MIG(CXCL9), MIP‐3β(CCL19) and SLC(CCL21) expression using flow cytometry to confirm the results; however, only the increase in CXCL9 was observed during colitis.
CXCL9 is an interferon‐γ‐inducible CXC chemotactic factor, known to mediate T‐cell migration, particularly following T‐cell activation.35, 36, 37 However, mice lacking CXCL9 showed a significant decrease in the antibodies against the intracellular bacterium,38 indicating that this chemokine may serve as a link between B‐cell and T‐cell responses. In the present study, we identified that B cells in PPs could produce CXCL9 to recruit Treg cells during colitis. Furthermore, CXCL9 is present in induced CD11b+ B cells but not in CD11b− B cells. These findings indicate that not all B cells have the ability to produce CXCL9 during colitis. CXCR3 is the only known receptor for CXCL9 and is reportedly expressed by various immune cells, including Treg cells.39, 40, 41 We observed that, on days 7 and 10, Treg cells up‐regulated CXCR3, which is consistent with previous studies. In summary, our findings demonstrate that induced CD11b+ B cells in PPs actively recruit Treg cells through the expression of CXCL9, assisting IgA isotype switch during DSS‐induced colitis. Our work may provide new insights into the regulatory mechanisms underlying gut‐associated lymphoid tissue and the development of new therapies against inflammatory bowel disease.
Authorship
W.Z. and W.L. designed experiments, performed experiments, analysed data and wrote the manuscript. C.Y. designed experiments and wrote the manuscript. H.E. improved the manuscript. Z.H., L.R., Q.T., L.J. C.Z and L.Z. performed experiments and analysed data.
Disclosures
The authors declare no conflict of interests.
Supporting information
Figure S1. Expression of transforming growth factor‐β (TGF‐β) by regulatory T (Treg) cells from Peyer's patches (PPs) in dextran sulphate sodium (DSS) ‐treated mice.
Figure S2. Expression of IgA on Peyer's patch (PP) ‐derived B cells in mice with colitis.
Figure S3. Regulatory T (Treg) cells and/or CD11b+ B‐cell populations in Peyer's patches (PPs) from mice with colitis.
Figure S4. Regulatory T (Treg) cells and CD11b+B cells co‐localize in (PPs) during dextran sulphate sodium (DSS) ‐induced colitis.
Figure S5. The transfer efficiency of GFP‐tagged CD11b+ or CD11b− B cells in recipients.
Acknowledgements
This work was supported by the Major Programme of the National Natural Science Foundation of China (81730045), the General Programme of National Natural Science Foundation of China (31770992, 81771736), the National Natural Science Funds for Distinguished Young Scholar (81501394) and the Young Elite Scientists Sponsorship Programme by CAST (YESS20160094). The authors have no other relevant affiliations nor financial involvement with any organization or entity with a financial interest, or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Contributor Information
Luman Wang, Email: lumanwang@fudan.edu.cn.
Yiwei Chu, Email: yiweichu@fudan.edu.cn.
References
- 1. Mauri C, Bosma A. Immune regulatory function of B cells. Annu Rev Immunol 2012; 30:221–41. [DOI] [PubMed] [Google Scholar]
- 2. Bouaziz JD, Yanaba K, Tedder TF. Regulatory B cells as inhibitors of immune responses and inflammation. Immunol Rev 2008; 224:201–14. [DOI] [PubMed] [Google Scholar]
- 3. Lund FE, Randall TD. Effector and regulatory B cells: modulators of CD4+ T cell immunity. Nat Rev Immunol 2010; 10:236–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wang L, Ray A, Jiang X, Wang JY, Basu S, Liu X et al T regulatory cells and B cells cooperate to form a regulatory loop that maintains gut homeostasis and suppresses dextran sulfate sodium‐induced colitis. Mucosal Immunol 2015; 8:1297–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kawamoto S, Maruya M, Kato LM, Suda W, Atarashi K, Doi Y et al Foxp3+ T cells regulate immunoglobulin a selection and facilitate diversification of bacterial species responsible for immune homeostasis. Immunity 2014; 41:152–65. [DOI] [PubMed] [Google Scholar]
- 6. Cong Y, Feng T, Fujihashi K, Schoeb TR, Elson CO. A dominant, coordinated T regulatory cell‐IgA response to the intestinal microbiota. Proc Natl Acad Sci U S A 2009; 106:19256–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bai Y, Qian C, Qian L, Ma F, Hou J, Chen Y et al Integrin CD11b negatively regulates TLR9‐triggered dendritic cell cross‐priming by upregulating microRNA‐146a. J Immunol 2012; 188:5293–302. [DOI] [PubMed] [Google Scholar]
- 8. Han C, Jin J, Xu S, Liu H, Li N, Cao X. Integrin CD11b negatively regulates TLR‐triggered inflammatory responses by activating Syk and promoting degradation of MyD88 and TRIF via Cbl‐b. Nat Immunol 2010; 11:734–42. [DOI] [PubMed] [Google Scholar]
- 9. Varga G, Balkow S, Wild MK, Stadtbaeumer A, Krummen M, Rothoeft T et al Active MAC‐1 (CD11b/CD18) on DCs inhibits full T‐cell activation. Blood 2007; 109:661–9. [DOI] [PubMed] [Google Scholar]
- 10. Ehirchiou D, Xiong Y, Xu G, Chen W, Shi Y, Zhang L. CD11b facilitates the development of peripheral tolerance by suppressing Th17 differentiation. J Exp Med 2007; 204:1519–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ding C, Ma Y, Chen X, Liu M, Cai Y, Hu X et al Integrin CD11b negatively regulates BCR signalling to maintain autoreactive B cell tolerance. Nat Commun 2013; 4:2813. [DOI] [PubMed] [Google Scholar]
- 12. Abdelbaqi M, Chidlow JH, Matthews KM, Pavlick KP, Barlow SC, Linscott AJ et al Regulation of dextran sodium sulfate induced colitis by leukocyte β2 integrins. Lab Invest 2006; 86:380–90. [DOI] [PubMed] [Google Scholar]
- 13. Liu X, Jiang X, Liu R, Wang L, Qian T, Zheng Y et al B cells expressing CD11b effectively inhibit CD4+ T‐cell responses and ameliorate experimental autoimmune hepatitis in mice. Hepatology 2015; 62:1563–75. [DOI] [PubMed] [Google Scholar]
- 14. Tseng J. Transfer of lymphocytes of Peyer's patches between immunoglobulin allotype congenic mice: repopulation of the IgA plasma cells in the gut lamina propria. J Immunol 1981; 127:2039–43. [PubMed] [Google Scholar]
- 15. Tseng J. A population of resting IgM‐IgD double‐bearing lymphocytes in Peyer's patches: the major precursor cells for IgA plasma cells in the gut lamina propria. J Immunol 1984; 132:2730–5. [PubMed] [Google Scholar]
- 16. Butcher EC, Rouse RV, Coffman RL, Nottenburg CN, Hardy RR, Weissman IL. Surface phenotype of Peyer's patch germinal center cells: implications for the role of germinal centers in B cell differentiation. J Immunol 1982; 129:2698–707. [PubMed] [Google Scholar]
- 17. Weinstein PD, Cebra JJ. The preference for switching to IgA expression by Peyer's patch germinal center B cells is likely due to the intrinsic influence of their microenvironment. J Immunol 1991; 147:4126–35. [PubMed] [Google Scholar]
- 18. Kraehenbuhl JP, Neutra MR. Epithelial M cells: differentiation and function. Annu Rev Cell Dev Biol 2000; 16:301–32. [DOI] [PubMed] [Google Scholar]
- 19. Macpherson AJ, McCoy KD, Johansen FE, Brandtzaeg P. The immune geography of IgA induction and function. Mucosal Immunol 2008; 1:11–22. [DOI] [PubMed] [Google Scholar]
- 20. Fagarasan S, Kawamoto S, Kanagawa O, Suzuki K. Adaptive immune regulation in the gut: T cell‐dependent and T cell‐independent IgA synthesis. Annu Rev Immunol 2010; 28:243–73. [DOI] [PubMed] [Google Scholar]
- 21. Vinuesa CG, Tangye SG, Moser B, Mackay CR. Follicular B helper T cells in antibody responses and autoimmunity. Nat Rev Immunol 2005; 5:853–65. [DOI] [PubMed] [Google Scholar]
- 22. Lecuyer E, Rakotobe S, Lengliné‐Garnier H, Lebreton C, Picard M, Juste C et al Segmented filamentous bacterium uses secondary and tertiary lymphoid tissues to induce gut IgA and specific T helper 17 cell responses. Immunity 2014; 40:608–20. [DOI] [PubMed] [Google Scholar]
- 23. Ivanov II, Atarashi K, Manel N, Brodie EL, Shima T, Karaoz U et al Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell 2009; 139:485–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Talham GL, Jiang HQ, Bos NA, Cebra JJ. Segmented filamentous bacteria are potent stimuli of a physiologically normal state of the murine gut mucosal immune system. Infect Immun 1999; 67:1992–2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wirtz S, Neufert C, Weigmann B, Neurath MF. Chemically induced mouse models of intestinal inflammation. Nat Protoc 2007; 2:541–6. [DOI] [PubMed] [Google Scholar]
- 26. Craig SW, Cebra JJ. Peyer's patches: an enriched source of precursors for IgA‐producing immunocytes in the rabbit. J Exp Med 1971; 134:188–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhiming W, Luman W, Tingting Q, Yiwei C. Chemokines and receptors in intestinal B lymphocytes. J Leukoc Biol 2018; 103:807–19. [DOI] [PubMed] [Google Scholar]
- 28. Roberts‐Thomson IC, Fon J, Uylaki W, Cummins AG, Barry S. Cells, cytokines and inflammatory bowel disease: a clinical perspective. Expert Rev Gastroenterol Hepatol 2011; 5:703–16. [DOI] [PubMed] [Google Scholar]
- 29. Hundorfean G, Neurath MF, Mudter J. Functional relevance of T helper 17 (Th17) cells and the IL‐17 cytokine family in inflammatory bowel disease. Inflamm Bowel Dis 2012; 18:180–6. [DOI] [PubMed] [Google Scholar]
- 30. Cerutti A. The regulation of IgA class switching. Nat Rev Immunol 2008; 8:421–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Borsutzky S, Cazac BB, Roes J, Guzmán CA. TGF‐β receptor signaling is critical for mucosal IgA responses. J Immunol 2004; 173:3305–9. [DOI] [PubMed] [Google Scholar]
- 32. Cazac BB, Roes J. TGF‐β receptor controls B cell responsiveness and induction of IgA in vivo . Immunity 2000; 13:443–51. [DOI] [PubMed] [Google Scholar]
- 33. Becker C, Fantini MC, Neurath MF. TGF‐β as a T cell regulator in colitis and colon cancer. Cytokine Growth Factor Rev 2006; 17:97–106. [DOI] [PubMed] [Google Scholar]
- 34. Feagins LA. Role of transforming growth factor‐β in inflammatory bowel disease and colitis‐associated colon cancer. Inflamm Bowel Dis 2010; 16:1963–8. [DOI] [PubMed] [Google Scholar]
- 35. Farber JM. A macrophage mRNA selectively induced by γ‐interferon encodes a member of the platelet factor 4 family of cytokines. Proc Natl Acad Sci U S A 1990; 87:5238–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Farber JM. HuMig: a new human member of the chemokine family of cytokines. Biochem Biophys Res Commun 1993; 192:223–30. [DOI] [PubMed] [Google Scholar]
- 37. Rabin RL, Park MK, Liao F, Swofford R, Stephany D, Farber JM. Chemokine receptor responses on T cells are achieved through regulation of both receptor expression and signaling. J Immunol 1999; 162:3840–50. [PubMed] [Google Scholar]
- 38. Park MK, Amichay D, Love P, Wick E, Liao F, Grinberg A et al The CXC chemokine murine monokine induced by IFN‐ (CXC Chemokine Ligand 9) is made by APCs, targets lymphocytes including activated B cells, and supports antibody responses to a bacterial pathogen in vivo . J Immunol 2002; 169:1433–43. [DOI] [PubMed] [Google Scholar]
- 39. Lacotte S, Brun S, Muller S, Dumortier H. CXCR3, inflammation, and autoimmune diseases. Ann N Y Acad Sci 2009; 1173:310–7. [DOI] [PubMed] [Google Scholar]
- 40. Qin S, Rottman JB, Myers P, Kassam N, Weinblatt M, Loetscher M et al The chemokine receptors CXCR3 and CCR5 mark subsets of T cells associated with certain inflammatory reactions. J Clin Invest 1998; 101:746–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Koch MA, Tucker‐Heard G, Perdue NR, Killebrew JR, Urdahl KB, Campbell DJ. The transcription factor T‐bet controls regulatory T cell homeostasis and function during type 1 inflammation. Nat Immunol 2009; 10:595–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Expression of transforming growth factor‐β (TGF‐β) by regulatory T (Treg) cells from Peyer's patches (PPs) in dextran sulphate sodium (DSS) ‐treated mice.
Figure S2. Expression of IgA on Peyer's patch (PP) ‐derived B cells in mice with colitis.
Figure S3. Regulatory T (Treg) cells and/or CD11b+ B‐cell populations in Peyer's patches (PPs) from mice with colitis.
Figure S4. Regulatory T (Treg) cells and CD11b+B cells co‐localize in (PPs) during dextran sulphate sodium (DSS) ‐induced colitis.
Figure S5. The transfer efficiency of GFP‐tagged CD11b+ or CD11b− B cells in recipients.