

Summary
Although low‐molecular‐mass hyaluronan (LMMHA) has been implicated in pulmonary inflammatory diseases, the signalling pathway of LMMHA (200 000 molecular weight) that initiates the inflammatory response in lung is still unknown. In this study, we evaluate the role of phosphoinositide 3‐kinase (PI3K) and its downstream signalling pathway in LMMHA‐induced lung inflammatory responses. Our results indicate that pharmacological inhibition of PI3K or genetic deletion of Akt1 enhances neutrophil apoptosis, attenuates neutrophil influx into the lungs of mice and diminishes the expression of pro‐inflammatory factors such as interleukin‐6, keratinocyte cell‐derived chemokine and pro‐matrix metalloproteinase‐9 in bronchoalveolar lavage fluid after intratracheal administration of LMMHA. More importantly, we found that PI3K/Akt1 participates in LMMHA‐induced inflammatory responses, which are mainly mediated by the myeloid leukaemia cell differentiation protein (Mcl‐1). Our study suggests that LMMHA induced significantly increased levels of inflammatory factors in bronchoalveolar lavage fluid and activation of the PI3K/Akt1 pathway, which up‐regulates the expression of the anti‐apoptotic protein Mcl‐1 and inhibits the activation of caspase‐3, thereby suppressing neutrophil apoptosis to trigger lung inflammation. These findings reveal a novel molecular mechanism underlying sterile inflammation and provides a new potential target for the treatment of pulmonary disease.
Keywords: low‐molecular‐weight hyaluronan (200 000 MW), lung inflammation, Mcl‐1, neutrophil, phosphoinositide 3‐kinase/Akt1
Abbreviations
- BALF
bronchoalveolar lavage fluid
- ECM
extracellular matrix
- ERK 1/2
extracellular signal‐regulated kinase 1/2
- IFN‐β
interferon‐β
- IL‐1β
interleukin‐1β
- IRF‐3
interferon regulatory factor 3
- KC
keratinocyte cell‐derived chemokine
- LMMHA
low‐molecular‐mass hyaluronan
- Mcl‐1
myeloid leukaemia cell differentiation protein
- MMP‐9
matrix metalloproteinase‐9
- MPO
myeloperoxidase
- PBS
phosphate‐buffered saline
- PI3K
phosphoinositide 3‐kinase
- TLR
Toll‐like receptor
- WT
wild‐type
Introduction
Hyaluronan (or hyaluronic acid, HA), a major extracellular matrix (ECM) component, is a non‐sulphated glycosaminoglycan composed of repeating polymeric disaccharides d‐glucuronic acid and N‐acetyl‐d‐glucosamine linked by a glucuronidic bond.1 Hyaluronan regulates many aspects of molecular mechanisms of tissue repair, such as activation of inflammatory cells to mount an immunological response.2 Low‐molecular‐mass HA (LMMHA), a product of rapid turnover and degradation of HA, can serve as an intracellular signalling molecule in inflammation and has been found to be pro‐inflammatory.3 LMMHA is dramatically increased in patients with idiopathic pulmonary fibrosis, chronic obstructive pulmonary disease, asthma and acute respiratory distress syndrome.4, 5, 6, 7 A previous study on the role of LMMHA in lung injury revealed that blocking the effect of LMMHA with specific peptides decreases neutrophil infiltration and fibrosis activity and alleviates lung tissue damage in a bleomycin‐mediated LMMHA‐induced mouse lung injury model.8 These studies establish the importance of LMMHA in respiratory disorders and encourage further study of the pro‐inflammatory properties of LMMHA.
In previous studies on the response to LMMHA stimulation, murine macrophages were identified as the principle effector cells in the production of pro‐inflammatory cytokines and chemokines.9 Furthermore, LMMHA has the potential to induce the production of pro‐inflammatory cytokines in airway epithelial cells, as well as the ability to up‐regulate adhesion molecules in endothelial cells.10, 11 Many pro‐inflammatory mediators also play a key role in LMMHA‐mediated lung inflammatory responses including keratinocyte cell‐derived chemokine (KC), interleukin‐1β (IL‐1β), IL‐6, macrophage inflammatory protein 2 and tumour nerosis factor‐α.12, 13 It is known that IL‐6 is an important inflammatory factor that has the ability to enhance the adhesion and aggregation of neutrophils and delay spontaneous apoptosis of neutrophils.14 Matrix metalloproteinase 9 (MMP9) stored in the tertiary granules of neutrophils is a protease with the potential to catalyse the ECM component and produce active forms of pro‐inflammatory cytokines and chemokines, thereby contributing to tissue remodelling and inflammatory responses.15, 16
In large quantities, LMMHA may promote inflammatory responses by endowing structural and immune cells with pro‐inflammatory phenotypes. Although there are limited and contradictory reports on neutrophil migration in the context of LMMHA‐mediated responses,17 the critical role of neutrophils in inflammatory pulmonary diseases is well studied.18 Direct study of the LMMHA effect on the airway system is scarce, and despite the evidence that supports the pro‐inflammatory effects of LMMHA, the molecular mechanism by which LMMHA drives or regulates the inflammatory reaction has been incompletely investigated.
In this study, an in vivo system was used to mimic the stressed environment by intratracheal administration of LMMHA. Our studies demonstrate for the first time that phosphoinositide 3‐kinase (PI3K)/Akt1 signalling plays a critical role in LMMHA (200 000 MW) ‐induced lung inflammation. More importantly, the PI3K/Akt1 pathway is shown to be responsible for delaying lung neutrophil apoptosis, and the anti‐apoptotic molecule myeloid leukaemia cell differentiation protein (Mcl‐1) was identified as a downstream molecule in LMMHA‐inhibited neutrophil apoptosis. These new findings will surely offer a new perspective to our understanding of molecular mechanisms involved in pulmonary diseases.
Materials and methods
Mice and LMMHA administration
Wild‐type control mice (C57BL/6J) were purchased from the Jackson Laboratory (Bar Harbor, ME). Akt1−/− mice were provided by Taconic (Rensselaer, NY). The LMMHA (200 000 MW) intratracheal mouse model was generated as described previously.13 The mice were given 50 μl of wortmannin (40 μg/kg), LY294002 (2 mg/kg), or 0·1% dimethyl sulphoxide (DMSO) vehicle by intranasal administration before LMMHA treatment. All procedures involving mice were approved and monitored by Zhejiang University of Technology.
Bronchoalveolar lavage fluid
The trachea was exposed through a midline incision and cannulated with a sterile 22‐gauge needle. Bronchoalveolar lavage fluid (BALF) was obtained by instilling and collecting four 0·5‐ml volumes of cold phosphate‐buffered saline (PBS) through the incised trachea. BALF was performed three times and 1·4 ml of lavage fluid was retrieved per mouse. Each BALF sample was centrifuged, and the supernatants were stored at −80° until use. Total cell numbers in BALF were counted from each sample in a haemocytometer. BALF neutrophil counts were determined on/in cytospin preparations stained with a Diff‐Quickstaining kit (IMEB, San Marcos, CA).12, 13
Enzyme‐linked immunosorbent assay
Interleukin‐6, KC and pro‐MMP9 were measured in the BALF using mouse enzyme‐linked immunosorbent assay (R&D Systems, Minneapolis, MN) according to the manufacturer's protocol.
Neutrophil detection and activity identification in lung tissues
To detect the infiltration of neutrophils in the lung, tissue sections were fixed in formalin for 24 hr, followed by paraffin‐embedding. Neutrophils were measured with a histochemical Leder stain for naphthol‐ASD‐chloroacetate esterase. For the analysis of lung neutrophil activity, flushed lungs were excised, digested in RPMI‐1640 medium containing 5% fetal bovine serum, 20 U/ml collagens, and 1 μg/ml DNase I, and incubated at 37° for 60 min. The remaining lung tissue was disrupted by passage through a 21‐gauge needle. Cell pellets were resuspended for further analysis. Lung neutrophils were positively selected using an anti‐Ly‐6G monoclonal antibody (Miltenyi Biotec, Bergisch Gladbach, Germany) as described previously.13
Myeloperoxidase assay
For myeloperoxidase (MPO) activity analysis, lung tissues were perfused to remove all blood, frozen in liquid nitrogen and stored at −80° for use. The homogenates were prepared in lysis buffer that consisted of 200 mm NaCl, 5 mm EDTA, 10 mm Tris–HCl, 10% glycerine, 1 mm PMSF, 1 μg/ml leupeptin and 28 μg/ml aprotinin (pH 7·4). This mixture was centrifuged at 4° at 1500 g for 15 min. Supernatants were diluted at a ratio of 1 : 4 and subjected to MPO analysis following the manufacturer's protocol (Hycult Biotechnology, Uden, the Netherlands). MPO activity was determined by absorbance at 450 nm.
Examination of protein expression and kinase phosphorylation
Neutrophil extracts were prepared using the neutrophil isolation kit (Miltenyi Biotec). The protein concentration was assayed using the BCA protein assay (Pierce, Rockford, IL). For the immunoblot analysis, 500 μg of total protein was separated by 10% SDS–PAGE and transferred to polyvinylidene difluoride membranes (Millipore). After being blocked, the membrane was incubated overnight at 4° with antibody to phos‐ and total‐Akt1, p38, extracellular signal‐regulated kinase 1/2 (ERK1/2), β‐actin or Mcl‐1 using a dilution of 1 : 1000, followed by horseradish peroxidase‐coupled secondary antibody at a dilution of 1 : 2000. All antibodies were purchased from Cell Signaling Technology (Danvers, MA). The signals were detected using chemiluminescence Western blot detection reagents (ECL Detection System, Amersham Pharmacia Biotech, Piscat‐away, NJ).
Real‐time PCR
RNA was extracted from neutrophils using TRIzol (Invitrogen, Carlsbad, CA). cDNA was synthesized using the superscript™ first‐strand synthesis system (Invitrogen). Real‐time PCR analysis was performed on the GeneAmp 5700 Sequence Detection System (Applied Biosystems, Foster City, CA) using the Quantitect SYBR Green PCR kit, as recommended by the manufacturer (Qiagen, Hilden, Germany). Specific primers were as follows: Mcl‐1 (forward, 5′‐GGGCAGGATTGTGACTCTCATT‐3′; reverse, 5′‐GATGCAGCTTTCTTGGTTTATGG‐3′);and β‐actin (forward, 5′‐AAGGATTCCTATGTGGGCGAC‐3′; reverse, 5′‐CGCGGTTGGCCTTGGGGTTCA‐3′). The relative value of target gene expression was calculated from the standardized target gene and β‐actin amplified curves.
Assessment of neutrophil apoptosis and caspase‐3 activity
Cells from BALF (1 × 106 cells/ml) were incubated with Annexin V‐FITC and propidium iodide (BD Biosciences, San Jose, CA) for 15 min in the dark at room temperature. Cells were analysed with FACS Calibur (Becton Dickinson, Franklin Lakes, NJ). Neutrophils were identified by forward‐scatter versus side‐scatter characteristics and by Ly6G staining.19, 20 Data analysis was performed with cell quest software (Becton Dickinson, Franklin Lakes, NJ). To analyse caspase‐3 activity in neutrophils, a caspase‐3 colorimetric activity assay kit (BioVision Research Products, Milpitas, CA) was applied in accordance with the manufacturer's instructions. Cell lysates, prepared from 5 × 106 neutrophils, were incubated with a labelled DEVD‐pNA substrate at 37° for 1·5 hr. The pNA light emission was quantified using a microtitre plate reader at 405 nm.
Statistical analysis
All data were expressed as mean ± SEM. The Tukey–Kramer multiple comparisons test and a one‐way analysis of variance were used for multiple groups, followed by Student's t‐test for comparisons between two groups. Values of P < 0·05 were considered significant.
Results
LMMHA induces lung inflammation following intratracheal administration
Hyaluronan fragments, representing the molecules frequently found at inflammatory sites in the lung,21 with a molecular mass of 200 000 are known to induce lung inflammation;12, 13 however, the effect of LMMHA (200 000 MW) on neutrophil activity during the very early stages of lung inflammation in vivo is still unknown. A single intratracheal dose of LMMHA (200 000 MW, 65 mg/kg) resulted in a remarkable increase in total cell and neutrophil counts in BALF (Fig. 1a). To further quantify the sequestration of neutrophils, MPO activity in the lung tissue was measured. The results demonstrate a substantial increase in MPO activity in LMMHA‐treated mice 6 hr after LMMHA administration (Fig. 1b). Esterase staining of lung tissue consistently indicates that the neutrophils infiltrate the lung in response to LMMHA administration (Fig. 1c). To further characterize the inflammatory response triggered by LMMHA in the airway, levels of pro‐inflammatory cytokines and chemokines in BALF were examined. Production of IL‐6 and KC was rapidly induced when the mice were given LMMHA (Fig. 1d). Importantly, we found that pro‐MMP9, a protease involved in the degradation of ECM and the processing of cytokines and chemokines, was also released into the BALF at this early stage (Fig. 1e). Hence, intratracheal administration of LMMHA causes lung inflammation and results in neutrophils infiltration into the lungs of the mice.
Figure 1.
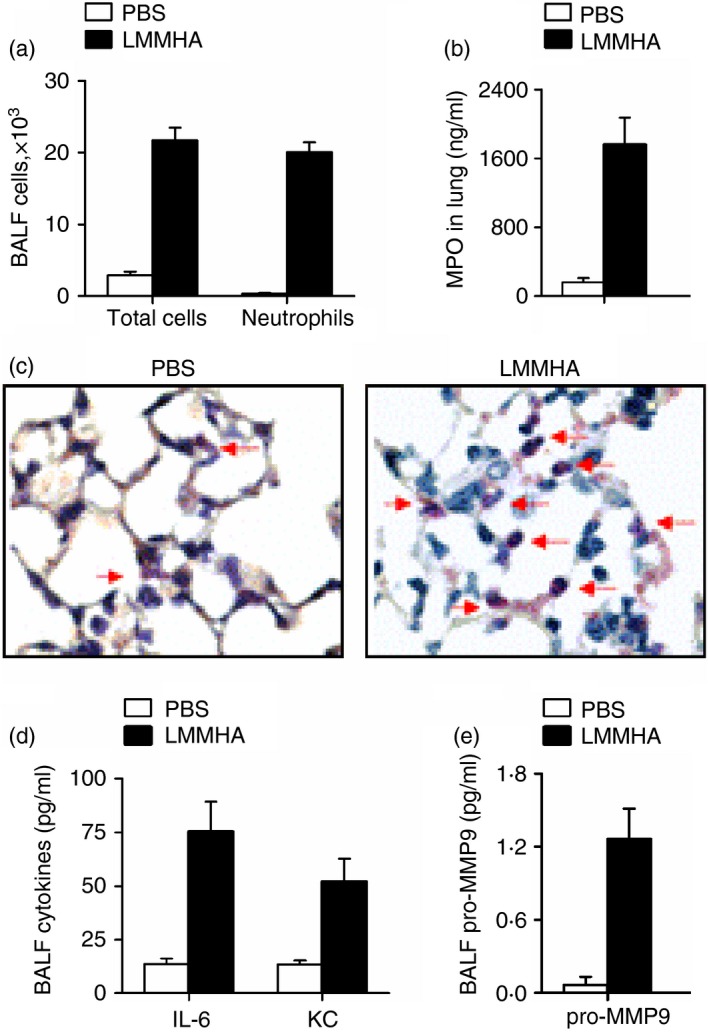
Low‐molecular‐mass hyaluronan (LMMHA) (200 000 MW) induces an early lung inflammatory response. Mice were intratracheally administered with LMMHA (200 000 MW, 65 mg/kg) and killed after 6 hr. Total cell and neutrophil counts were measured in bronchoalveolar lavage fluid (BALF) (a). The myeloperoxidase (MPO) level of whole lung homogenates was measured (b). Leder staining for neutrophils that infiltrated the lung (c). Arrows indicate the esterase‐positive neutrophils, ×1000. The levels of interleukin‐6 (IL‐6), keratinocyte cell‐derived chemokine (KC) and pro‐matrix metalloproteinase 9 (MMP9) in BALF were measured by ELISA (d and e).
Akt1 signalling is critical in LMMHA‐induced neutrophilic inflammation
Because the suppression of neutrophil apoptosis in acute lung injury is associated with the Akt‐signalling pathway,22 we investigated the molecular mechanism by which LMMHA (200 000 MW) drives the neutrophilic response in the airway system. Western blot analysis of lung homogenates revealed a more potent and sustained activation of Akt1 in the LMMHA‐challenged lungs than in that of mice treated with PBS (Fig. 2a). The phosphorylation of Akt1 in lung neutrophils was also remarkably induced at 6 hr following LMMHA administration (Fig. 2b). Moreover, the phosphorylation of p38 and ERK1/2 was significantly decreased in Akt1−/− mouse neutrophils compared with those of wild‐type (WT) mice after intratracheal administration of LMMHA (Fig. 2c). This indicates that Akt1 plays a pivotal role in regulating LMMHA‐induced neutrophil activity.
Figure 2.
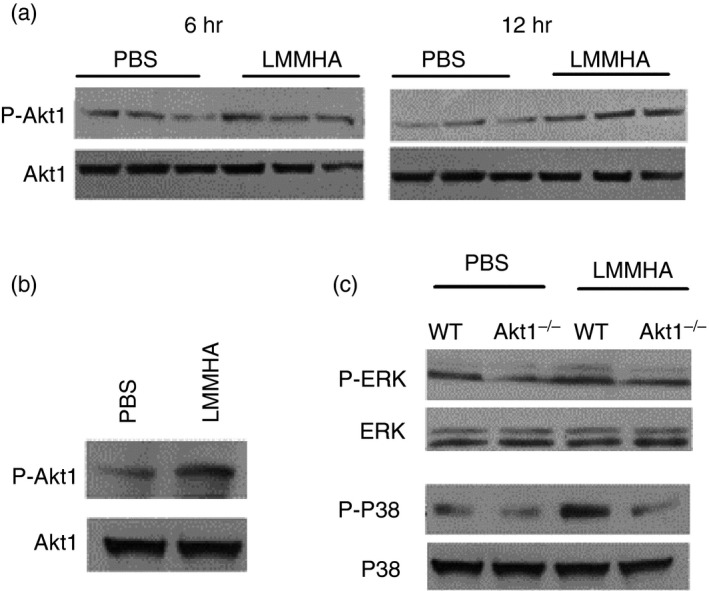
Low‐molecular‐mass hyaluronan (LMMHA) (200 000 MW) stimulates the activation of Akt1 kinase. Phosphorylation of Akt1 in lung homogenates was assessed by Western blot analysis 6 hr or 12 hr after intratracheal administration of LMMHA (200 000 MW, 65 mg/kg) (a). Phosphorylation of Akt1 in whole lung neutrophils was assessed 6 hr after LMMHA administration (b). Phosphorylation of p38 and ERK1/2 in whole lung neutrophils 6 hr after LMMHA (c). Mouse whole lung neutrophils were isolated as described in the Materials and methods.
To further demonstrate the role of Akt1 in LMMHA‐mediated airway inflammatory responses, WT and Akt1−/− mice were exposed to LMMHA. Neutrophil recruitment into the lung was remarkably decreased by a deficiency in Akt1, demonstrated by BALF neutrophil counts (Fig. 3a). Since decreased neutrophil accumulation is the result of either impaired sequestration of neutrophils into lungs or the defective migration of neutrophils in the tissue, an MPO assay was conducted. Our results show a significant reduction in MPO activity in Akt1−/− mice (Fig. 3b), which suggests the need for Akt1 signalling in neutrophil extravasation. Furthermore, the substantial elevations of IL‐6, KC and pro‐MMP9 in BALF induced by LMMHA stimulation have been compromised in Akt1−/− mice (Fig. 3c). This suggests that Akt1 signalling selectively influences the generation of inflammatory mediators.
Figure 3.
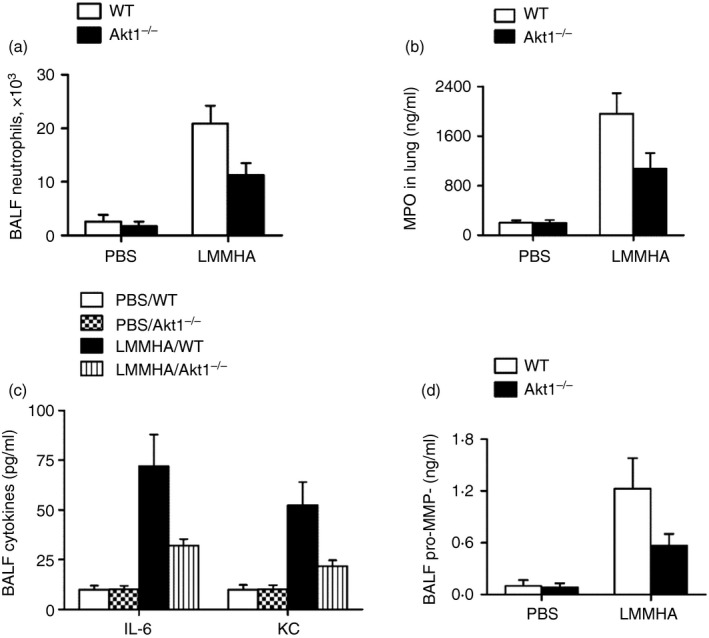
Akt1 signalling plays a key role in low‐molecular‐mass hyaluronan (LMMHA) (200 000 MW)‐induced lung inflammation. Numbers of neutrophils in bronchoalveolar lavage fluid (BALF) (a). Myeloperoxidase MPO activity in lung tissue (b). BALF concentrations of interleukin 6 (IL‐6), keratinocyte cell‐derived chemokine (KC) and pro‐matrix metalloproteinase 9 (MMP9) (c, d). The data were evaluated 6 hr after intratracheal administration of PBS or LMMHA. Each bar represents mean ± SEM from eight to ten mice.
PI3K participates in LMMHA‐induced inflammatory response
Given that Akt1 is involved in LMMHA‐induced lung inflammation, we sought to extend the understanding of this pathway by identifying the upstream and downstream molecule(s) in this cascade. It is known that PI3K is the most primary upstream molecule of a large number of pathways including the Akt1 pathway,23 so the involvement of PI3K in LMMHA‐induced lung inflammation was examined. Through the application of the PI3K‐specific inhibitor wortmannin before LMMHA administration, we found that LMMHA‐triggered Akt1 phosphorylation in neutrophils was significantly reduced upon PI3K inhibition (Fig. 4a), which indicates the dependency of Akt1 activation on PI3K. Further demonstrating the functional relevance of PI3K, a greater reduction in BALF neutrophils was observed in mice pretreated with wortmannin than in those treated only with PBS (Fig. 4b). Furthermore, blockade of PI3K with wortmannin induced a marked reduction in MPO activity (Fig. 4c), which is associated with a decrease in the levels of IL‐6, KC and pro‐MMP9 in BALF (Fig. 4d,e). Therefore, PI3K plays an essential role in LMMHA‐induced airway responses, and Akt1 is a downstream target activated by PI3K in the inflammatory signalling pathway.
Figure 4.
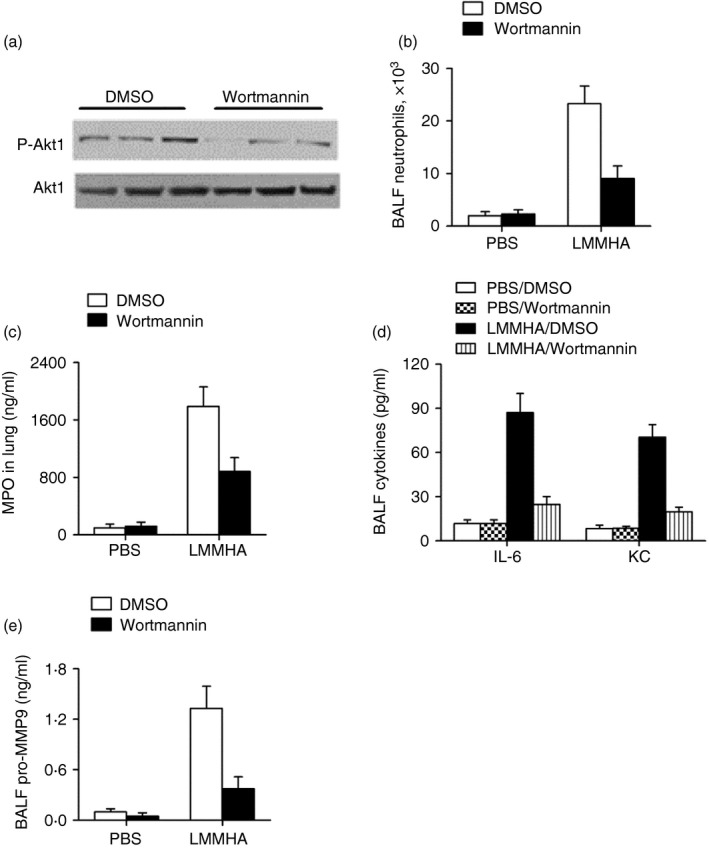
The phosphoinositide 3‐kinase (PI3K) involves in low‐molecular‐mass hyaluronan (LMMHA) (200 000 MW) ‐induced lung inflammation. Mice were pretreated with or without the PI3K chemical inhibitor, wortmannin (40 μg/kg) intranasally for 30 min and then challenged with LMMHA. Mice were killed at 6 hr after LMMHA administration for functional analysis. Akt1 activation in bronchoalveolar lavage fluid (BALF) neutrophils (a), BALF neutrophil counts (b), myeloperoxidase (MPO) activity in lung homogenates after BALF (c). The levels of interleukin 6 (IL‐6), keratinocyte cell‐derived chemokine (KC) and pro‐matrix metalloproteinase 9 (MMP9) in BALF (d, e). Each bar represents mean ± SEM from eight to ten mice.
PI3K/Akt1 regulates LMMHA‐induced neutrophil apoptosis through Mcl‐1
Because of a profound decrease in BALF neutrophil numbers induced by either pharmacological inhibition of PI3K or genetic deletion of Akt1, PI3K/Akt1 signalling was found to be responsible for the LMMHA‐induced neutrophil counts in the lungs (Figs 3a and 4b). After considering the well‐recognized role of Akt1 in mediating survival signals in diverse cells,24, 25, 26 it was hypothesized that Akt1 may have a regulatory role in sustaining neutrophil lifespan. To address this possibility, apoptosis of BALF neutrophils from WT and Akt1−/− mice was evaluated by flow cytometry. The analysis revealed that the number of apoptotic neutrophils in BALF was significantly higher in Akt1−/− mice than in WT mice at 18 hr after LMMHA administration (Fig. 5a). Next, we determined the role of PI3K in neutrophil apoptosis and observed a similar increase in neutrophil apoptosis in mice treated with wortmannin or LY2940002 (Fig. 5a). Apoptosis of neutrophils was further corroborated by increased caspase‐3 activities when PI3K/Akt1 signalling was blocked (Fig. 5b). Hence, PI3K/Akt1 signalling promotes neutrophil survival.
Figure 5.
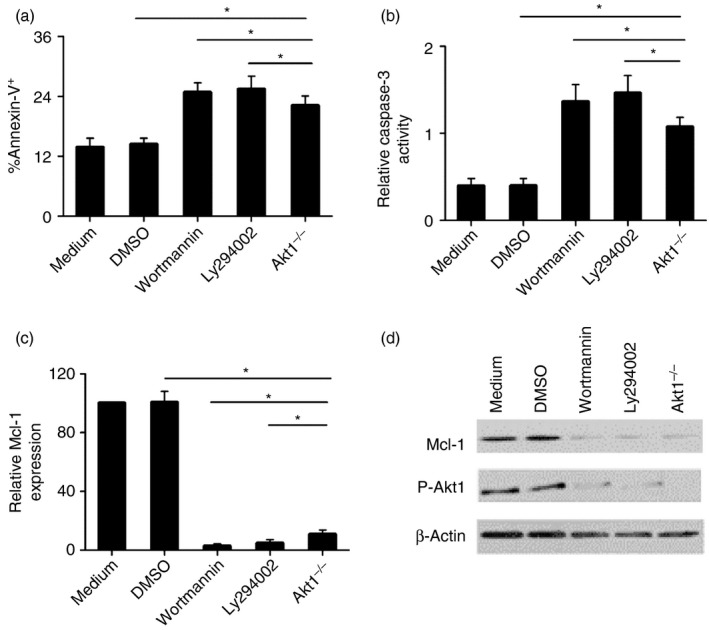
Mcl‐1 participates in phosphoinositide 3‐kinase (PI3K)/Akt1‐mediated bronchoalveolar lavage fluid (BALF) neutrophil apoptosis. Percentage of BALF neutrophils undergoing apoptosis (Annexin V+/PI –) (a), The levels of caspase‐3 activities (b), Mcl‐1 mRNA (c) and Mcl‐1 protein in BALF neutrophils (d). BALF neutrophils were recovered 18 hr after low‐molecular‐mass hyaluronan (LMMHA) (200 000 MW) administration. The results are representative of three separate experiments. *P < 0·05.
To investigate the mechanisms behind the neutrophil‐protecting effects of LMMHA, potential downstream components were examined. As Bcl‐2 family members constitute a network of anti‐apoptotic regulators, and some of these regulators have been proved to be closely associated with Akt signalling,27 our initial targets were these Bcl‐2 family members. Bcl‐2 and Bcl‐X existed at almost undetectable levels in the accumulated lung neutrophils (data not shown), as is consistent with the previous findings.28 When anti‐apoptotic protein Mcl‐1 was analysed in BALF neutrophils after LMMHA stimulation, its mRNA and protein levels were significantly increased. Nevertheless, Mcl‐1 levels were markedly decreased in Akt1−/− neutrophils and in neutrophils from mice treated with wortmannin or LY2940002 (Fig. 5c,d). These results indicate that PI3K/Akt1 signalling plays a regulatory role of in Mcl‐1 production. More importantly, the decreased expression of Mcl‐1 upon Akt1 deletion or PI3K inhibition was inversely related to neutrophil apoptosis and to caspase‐3 activities (Fig. 5b,c,d). Therefore, we conclude that Mcl‐1 functions as the downstream target of the PI3K/Akt1 pathway in inhibiting neutrophil apoptosis in this setting (Fig. 6).
Figure 6.
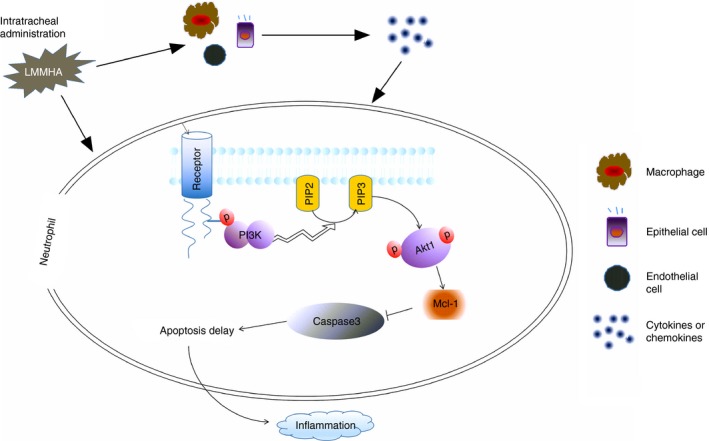
The pathological mechanism of low‐molecular‐mass hyaluronan (LMMHA) (200 000 MW) induces pulmonary inflammation in bronchoalveolar lavage fluid (BALF) neutrophils. Intratracheal administration of LMMHA induced significantly increased levels of inflammatory factors, resulted in activation of phosphoinositide 3‐kinase (PI3K) signalling pathway and markedly augmented phosphorylation of Akt1 in neutrophils of BALF. Further, the expression of the anti‐apoptotic protein Mcl‐1 was significantly increased in BALF neutrophils, adversely, the caspase‐3 activity was inhibited, which delayed the apoptosis of neutrophils during LMMHA‐induced pulmonary inflammation.
Discussion
In the present study, our data demonstrate that intratracheal administration of LMMHA triggers lung inflammation and induces a dramatic infiltration of neutrophils into the interstitial and intra‐alveolar compartments in mice. This is consistent with previous studies that show IL‐6 and KC are significantly elevated in LMMHA‐induced lung injury.12, 13 Notably, we found that the level of pro‐MMP9, a pro‐inflammatory cytokine MMP9 precursor, was remarkably increased in BALF. MMP9 has been shown to be related to several inflammatory lung diseases, such as chronic obstructive pulmonary disease, asthma and acute respiratory distress syndrome.29, 30, 31 Hence, the participation of pro‐MMP9 highlights the importance of this protease in LMMHA‐induced inflammation response. The catalytic property of MMP9 enables it to degrade extracellular structures and facilitate the extravasation and migration of neutrophils, so promoting neutrophilic inflammation.
Despite evidence that supports the critical role of LMMHA in airway inflammatory disorders, its signalling pathway in vivo is not clearly understood. Akt1, a member of the three serine/threonine protein kinase families (Akt1, Akt2, Akt3), is expressed profusely and ubiquitously in the cytoplasm. The other two isoforms are localized to certain tissues and subcellular loci. Several studies have suggested that Akt1 plays an extensive role in regulating cell survival, differentiation and proliferation.32, 33 Akt1 was recently identified as a crucial regulator in inflammatory responses.34 Although previous studies have shown that Akt1 activation in murine macrophages enhances lipopolysaccharide‐induced inflammatory responses,35 the importance of Akt1 in LMMHA‐challenged inflammatory neutrophils had not been elucidated. Here, we report that phosphorylation of Akt1 in pulmonary neutrophils was significantly elevated following administration of LMMHA. Both p38 and ERK1/2 phosphorylation in lung neutrophils were significantly reduced in Akt1−/− mice by LMMHA induction compared with WT mice. Additionally, we found that the severity of inflammatory responses in Akt1−/− mice was remarkably weaker than that in WT mice. These data demonstrate that Akt1‐mediated signalling contributes to LMMHA‐induced neutrophil activity. We propose that Akt1 influences neutrophil activity consistently. This is in accordance with the sustained activation of Akt1 during the response.
Activation of PI3K leads to generation of the 3′ hydroxyl group of phosphatidylinositols, which function as secondary messengers in the recruitment and activation of a number of kinase enzymes, including phosphoinositide‐dependent kinase‐1, protein kinase B/Akt, and several protein kinase C isoforms.36 PI3K/Akt signalling pathways negatively regulate lipopolysaccharide‐induced acute inflammatory responses in vitro.37 Moreover, inhibition of PI3K/Akt signalling pathways have been shown to block tumour necrosis factor‐stimulated gene 6‐mediated acute airway inflammation and hyper‐responsiveness in response to ozone or short‐fragment HA.38 Here we confirm that the inclusion of PI3K as an upstream activator of Akt1 was confirmed by using specific inhibitors, wortmannin and LY2940002, which consistently suppressed Akt1 phosphorylation and neutrophil counts. Akt1 is regulated by PI3K in LMMHA‐induced neutrophil responses. These results suggest that the PI3K/Akt1 pathway is activated in LMMHA‐induced lung inflammation and wortmannin or LY2940002 pre‐treatment markedly reduced the LMMHA‐induced phosphorylation of Akt1. This is consistent with the previous studies that show that inhibition of the PI3K/Akt signalling pathway exerts a protective role in lung inflammatory responses.39 Given the importance of the PI3K/Akt1 pathway in transmitting anti‐apoptotic signalling in neutrophils, we hypothesize that in addition to their direct inflammation‐promoting properties, these molecules may have an effect on the neutrophil lifespan. Indeed, when apoptosis of BALF neutrophils from LMMHA‐treated mice was examined in detail, an increase in apoptotic neutrophils was observed in Akt1−/− mice compared with WT mice. This phenomenon was reproduced in the mice with PI3K inhibition, which supports the notion that LMMHA‐activated PI3K/Akt1 signalling is capable of sustaining the lifespan of neutrophils.
Recently, studies have reported that Mcl‐1 plays an inhibitory role in apoptosis.40 In our experiments, we show that LMMHA‐induced up‐regulation of Mcl‐1 was markedly decreased by the abrogation of PI3K/Akt1, at either the mRNA or the protein level. This suggests that PI3K/Akt1 participates in the transcription of Mcl‐1. Our identification of Mcl‐1 as a major component of the LMMHA‐induced neutrophil survival pathway, operating mostly downstream of PI3K/Akt1, uncovers a previously unknown signalling molecule in LMMHA‐mediated neutrophil apoptosis. Caspase‐3 is known to be a key performer downstream of cell apoptosis. We observed that caspase‐3 activity is significantly elevated in the absence of Akt1, or PI3K inhibition is inversely related to Mcl‐1 levels. It was shown that LMMHA‐induced neutrophil apoptosis is regulated by downstream signalling molecules Mcl‐1 and caspase‐3 of the PI3K/Akt1 signalling pathway. This suggests that the high expression of Mcl‐1 and inhibition of caspase‐3 activity are important mechanisms of LMMHA‐induced delayed apoptosis of pulmonary neutrophils. Based on the above findings, we propose that LMMHA‐induced PI3K/Akt1 signalling, in cooperation with Mcl‐1, not only induces neutrophil activation and recruitment, but also prevents neutrophils from constitutive apoptosis, thereby amplifying and prolonging the acute inflammatory response.
PI3K/Akt1‐regulated pro‐inflammatory responses additionally correlate with the Toll‐like receptor (TLR) signalling pathway.41 Several studies have shown that Akt1 expression has a significant inhibitory effect on the TLR4 pathway and impairs interferon‐β (IFN‐β) promoter activity by suppressing IFN regulatory factor 3 (IRF3) activity in the Toll/IL‐1 receptor domain‐containing adapter‐inducing IFN‐β‐dependent pathway in vitro.42 Our previous studies revealed severe inflammatory infiltration in the alveolar space of IRF3−/−, TLR4−/− and IFN‐β−/− mice within 24 hr after LMMHA (200 000 MW) intratracheal administration. Levels of IFN‐β were also decreased in TLR4‐deficient neutrophils compared with those of WT neutrophils, leading to impaired apoptosis of neutrophils and abnormal accumulation of neutrophils in the lungs of LMMHA‐treated mice.12, 13 Consequently, we hypothesized that Akt1 inhibits neutrophil apoptosis in LMMHA‐induced pulmonary inflammation associated with the TLR4 pathway, which acts as a negative regulator of TLR4, and distinctively suppresses IRF3 activity and decrease the levels of IFN‐β in triggering inflammation. Although the exact relationship and mechanisms of action require further experiments to verify, these findings help to extend our understanding of the biological function of LMMHA in the airway system.
In conclusion, our results demonstrate that LMMHA (200 000 MW) is a critical signal in neutrophil activation and attraction, leading to lung inflammation. Moreover, LMMHA‐challenged PI3K/Akt1 signalling through the up‐regulation of the expression of the anti‐apoptotic protein Mcl‐1 delays lung neutrophil apoptosis. Therefore, a novel PI3K/Akt1‐dependent pathway may contribute to the LMMHA‐triggered inflammatory response in the airway system. Hence, inhibition of the PI3K/Akt1 pathway exerts a protective role in LMMHA‐induced lung inflammation. Our study sheds a light on the functions and mechanisms of specific ECM fragments in the airway response and helps to improve our understanding of the pathogenesis of pulmonary inflammatory diseases, so providing new information on novel target molecules for treatment.
Disclosures
The authors have no financial conflict of interest.
Acknowledgements
We gratefully acknowledge the National Natural Science Foundation of China (81470004) and Zhejiang Provincial Natural Science Foundation of China under Grant No.LY18H010009 for their financial support.
References
- 1. Weissmann B, Meyer K, Sampson P, Linker A. Isolation of oligosaccharides enzymatically produced from hyaluronic acid. J Biol Chem 1954; 208:417–29. [PubMed] [Google Scholar]
- 2. Jiang D, Liang J, Noble PW. Hyaluronan as an immune regulator in human diseases. Physiol Rev 2011; 91:221–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lauer ME, Dweik RA, Garantziotis S, Aronica MA. The rise and fall of hyaluronan in respiratory diseases. Int J Cell Biol 2015; 2015:712507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Papakonstantinou E, Roth M, Klagas I, Karakiulakis G, Tamm M, Stolz D. COPD exacerbations are associated with proinflammatory degradation of hyaluronic acid. Chest 2015; 148:1497. [DOI] [PubMed] [Google Scholar]
- 5. Li Y, Jiang D, Liang J, Meltzer EB, Gray A, Miura R et al Severe lung fibrosis requires an invasive fibroblast phenotype regulated by hyaluronan and CD44. J Exp Med 2011; 208:1459–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Nakamura Y, Suzuki R, Mizuno T, Abe K, Chiba S, Horii Y et al Therapeutic implication of genetic variants of IL13 and STAT4 in airway remodelling with bronchial asthma. Clin Exp Allergy 2016; 46:1152–61. [DOI] [PubMed] [Google Scholar]
- 7. Kredel M, Muellenbach RM, Brock RW, Wilckens HH, Brederlau J, Roewer N et al Liver dysfunction after lung recruitment manoeuvres during pressure‐controlled ventilation in experimental acute respiratory distress. Crit Care 2007; 11:R13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Jiang D, Liang J, Fan J, Yu S, Chen S, Luo Y et al Regulation of lung injury and repair by Toll‐like receptors and hyaluronan. Nat Med 2005; 11:1173–9. [DOI] [PubMed] [Google Scholar]
- 9. Jiang D, Liang J, Noble PW. Hyaluronan in tissue injury and repair. Annu Rev Cell Dev Biol 2006; 23:435–61. [DOI] [PubMed] [Google Scholar]
- 10. Taylor KR, Trowbridge JM, Rudisill JA, Termeer CC, Simon JC, Gallo RL. Hyaluronan fragments stimulate endothelial recognition of injury through TLR4. J Biol Chem 2004; 279:17079–84. [DOI] [PubMed] [Google Scholar]
- 11. Boodoo S, Spannhake EW, Powell JD, Horton MR. Differential regulation of hyaluronan‐induced IL‐8 and IP‐10 in airway epithelial cells. Am J Physiol Lung Cell Mol Physiol 2006; 291:L479. [DOI] [PubMed] [Google Scholar]
- 12. Zhao H, Leu SW, Shi L, Dedaj R, Zhao G, Garg HG et al TLR4 is a negative regulator in noninfectious lung inflammation. J Immunol 2010; 184:5308–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Leu SW, Shi L, Xu C, Zhao Y, Liu B, Li Y et al TLR4 through IFN‐β promotes low molecular mass hyaluronan‐induced neutrophil apoptosis. J Immunol 2011; 186:556–62. [DOI] [PubMed] [Google Scholar]
- 14. Ottonello L, Frumento G, Arduino N, Bertolotto M, Dapino P, Mancini M et al Differential regulation of spontaneous and immune complex‐induced neutrophil apoptosis by proinflammatory cytokines. Role of oxidants, Bax and caspase‐3. J Leukocyte Biol 2002; 72:125. [PubMed] [Google Scholar]
- 15. Chakrabarti S, Zee JM, Patel KD. Regulation of matrix metalloproteinase‐9 (MMP‐9) in TNF‐stimulated neutrophils: novel pathways for tertiary granule release. J Leukoc Biol 2006; 79:214–22. [DOI] [PubMed] [Google Scholar]
- 16. Lindsey M, Wedin K, Brown MD, Qi H, Wu ZY, Shu HQ et al Matrix‐dependent mechanism of neutrophil‐mediated release and activation of matrix metalloproteinase 9 in myocardial ischemia/reperfusion. Circulation 2001; 103:2181–7. [DOI] [PubMed] [Google Scholar]
- 17. Khan AI, Kerfoot SM, Heit B, Liu L, Andonegui G, Ruffell B et al Role of CD44 and hyaluronan in neutrophil recruitment. J Immunol 2004; 173:7594–601. [DOI] [PubMed] [Google Scholar]
- 18. Yan B, Chen F, Xu L, Xing J, Wang X. HMGB1‐TLR4‐IL23‐IL17A axis promotes paraquat‐induced acute lung injury by mediating neutrophil infiltration in mice. Sci Rep‐UK 2017; 7:597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Conus S, Perozzo R, Reinheckel T, Peters C, Scapozza L, Yousefi S et al Caspase‐8 is activated by cathepsin D initiating neutrophil apoptosis during the resolution of inflammation. J Exp Med 2008; 205:685–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Turino GM, Cantor JO. Hyaluronan in respiratory injury and repair. Am J Respir Crit Care Med 2003; 167:1169–75. [DOI] [PubMed] [Google Scholar]
- 21. Liang J, Jiang D, Noble PW. Hyaluronan as a therapeutic target in human diseases. Adv Drug Deliver Rev 2016; 97:186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gong J, Liu H, Wu J, Qi H, Wu ZY, Shu HQ et al Maresin 1 prevents lipopolysaccharide‐induced neutrophil survival and accelerates resolution of acute lung injury. Shock 2015; 44:371. [DOI] [PubMed] [Google Scholar]
- 23. Lu S, Li H, Gao R, Gao X, Xu F, Wang Q et al IL‐17A, but not IL‐17F, is indispensable for airway vascular remodeling induced by exaggerated Th17 cell responses in prolonged ovalbumin‐challenged mice. J Immunol 2015; 194:3557–66. [DOI] [PubMed] [Google Scholar]
- 24. Wang Q, Yu WN, Chen X, Peng XD, Jeon SM, Birnbaum MJ et al Spontaneous hepatocellular carcinoma after the combined deletion of Akt isoforms. Cancer Cell 2016; 29:523–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Larson‐Casey JL, Deshane JS, Ryan AJ, Thannickal VJ, Carter AB. Macrophage Akt1 kinase‐mediated mitophagy modulates apoptosis resistance and pulmonary fibrosis. Immunity 2016; 44:582–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chen ML, Xu PZ, Peng XD, Chen WS, Guzman G, Yang X et al The deficiency of Akt1 is sufficient to suppress tumor development in Pten+/– mice. Gene Dev 2006; 20:1569–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. François S, El BJ, Dang PM, Pedruzzi E, Gougerotpocidalo MA, Elbim C. Inhibition of neutrophil apoptosis by TLR agonists in whole blood: involvement of the phosphoinositide 3‐kinase/Akt and NF‐κB signaling pathways, leading to increased levels of Mcl‐1, A1, and phosphorylated Bad. J Immunol 2005; 174:3633–42. [DOI] [PubMed] [Google Scholar]
- 28. Epling‐Burnette PK, Zhong B, Bai F, Jiang K, Bailey RD, Garcia R et al Cooperative regulation of Mcl‐1 by Janus kinase/stat and phosphatidylinositol 3‐kinase contribute to granulocyte–macrophage colony‐stimulating factor‐delayed apoptosis in human neutrophils. J Immunol 2001; 166:7486–95. [DOI] [PubMed] [Google Scholar]
- 29. Zhou H, Wu Y, Jin Y, Zhou J, Zhang C, Che L et al Genetic polymorphism of matrix metalloproteinase family and chronic obstructive pulmonary disease susceptibility: a meta‐analysis. Sci Rep‐UK 2013; 3:2818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chaudhuri R, Mcsharry C, Brady J, Grierson C, Messow CM, Spears M et al Low sputum MMP‐9/TIMP ratio is associated with airway narrowing in smokers with asthma. Eur Respir J 2014; 44:895–904. [DOI] [PubMed] [Google Scholar]
- 31. Takashima K, Matsushima M, Hashimoto K, Nose H, Sato M, Hashimoto N et al Protective effects of intratracheally administered quercetin on lipopolysaccharide‐induced acute lung injury. Resp Res 2014; 15:150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Fernández‐Hernando C, Ackah E, Yu J, Suárez Y, Murata T, Iwakiri Y et al Loss of Akt1 leads to severe atherosclerosis and occlusive coronary artery disease. Cell Metab 2007; 6:446–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Franke TF. PI3K/Akt: getting it right matters. Oncogene 2008; 27:6473–88. [DOI] [PubMed] [Google Scholar]
- 34. Lorenzo AD, Fernandez‐Hernando C, Cirino G, Sessa WC, Ignarro LJ. Akt1 is critical for acute inflammation and histamine‐mediated vascular leakage. Proc Natl Acad Sci U S A 2009; 106:14552–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tang B, Li X, Ren Y, Wang J, Xu D, Hang Y et al MicroRNA‐29a regulates lipopolysaccharide (LPS)‐induced inflammatory responses in murine macrophages through the Akt1/NF‐κB pathway. Exp Cell Res 2017; 360:74. [DOI] [PubMed] [Google Scholar]
- 36. Le GJ, Ziegler WH, Parekh DB, Alessi DR, Cohen P, Parker PJ. Protein kinase C isotypes controlled by phosphoinositide 3‐kinase through the protein kinase PDK1. Science 1998; 281:2042–5. [DOI] [PubMed] [Google Scholar]
- 37. Guha M, Mackman N. The phosphatidylinositol 3‐kinase‐Akt pathway limits lipopolysaccharide activation of signaling pathways and expression of inflammatory mediators in human monocytic cells. J Biol Chem 2002; 277:32124–32. [DOI] [PubMed] [Google Scholar]
- 38. Stober VP, Johnson CG, Majors A, Lauer ME, Cali V, Midura RJ et al TNF‐stimulated gene 6 promotes formation of hyaluronan‐inter‐α‐inhibitor heavy chain complexes necessary for ozone‐induced airway hyperresponsiveness. J Biol Chem 2017; 292:M116–756627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zhao M, Li C, Shen F, Wang M, Jia N, Wang C. Naringenin ameliorates LPS‐induced acute lung injury through its anti‐oxidative and anti‐inflammatory activity and by inhibition of the PI3K/AKT pathway. Exp Ther Med 2017; 14:2228–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kodama T, Hikita H, Kawaguchi T, Shigekawa M, Shimizu S, Hayashi Y et al Mcl‐1 and Bcl‐xL regulate Bak|[sol]|Bax‐dependent apoptosis of the megakaryocytic lineage at multistages. Cell Death Differ 2012; 19:1856–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Fukao T, Koyasu S. PI3K and negative regulation of TLR signaling. Trends Immunol 2003; 24:358–63. [DOI] [PubMed] [Google Scholar]
- 42. Zenke K, Muroi M, Tanamoto K. AKT1 distinctively suppresses MyD88‐dependent and TRIF‐dependent Toll‐like receptor signaling in a kinase activity‐independent manner. Cell Signal 2018; 43:32–9. [DOI] [PubMed] [Google Scholar]