

Summary
As a result of its strategic location, the epithelium is constantly exposed to a wide variety of pathogen and danger signals. Traditionally, the epithelium has been perceived as a defensive but passive barrier; however, it has now become evident that the epithelium senses and actively responds to these signals in order to maintain barrier homeostasis and contributes to the inflammatory response. One way it does this is by producing pro‐inflammatory cytokines including interleukin‐1β (IL‐1β) and IL‐18. These two cytokines are synthesized as inactive precursors, the maturation of which is mediated by pro‐inflammatory caspases after the activation and assembly of macromolecular complexes called inflammasomes. Epithelial cells express a large panel of inflammasome components, and although the molecular mechanisms underlying the activation of these complexes in haematopoietic cells are well understood, how epithelial cells react to danger signals to activate the inflammasome remains unclear. We review and discuss how different inflammasomes contribute to barrier homeostasis and inflammation at several barrier sites, their mechanisms and how their aberrant regulation contributes to disease at the different epithelia.
Keywords: epithelial cell, inflammasome, inflammation
Introduction
As a result of its essential role as a physical barrier between our body and the external environment, epithelial cells are some of the most specialized cells of the human body. The epithelium is often targeted by a myriad of pathogens, including bacteria, fungi and viruses, but also by particulate matter such as tobacco smoke and air pollution.1 When it comes to respond to these threats, resident macrophages play an essential role.2 Macrophages express numerous membrane and cytosolic receptors to detect threats, and the study of those (including the activation of inflammasome) has been widely studied. However, in recent years and breaking the paradigm of epithelial cells as a passive barrier, epithelial cells have also been shown to play an important and active role in these immune responses.
Epithelial cells can sense threats through membrane‐bound receptors such as Toll‐like receptors (TLRs) TLR4 and TLR5,3 but also through intracellular receptors such as TLR7, TLR9, absent in melanoma 2 (AIM2; a receptor that recognizes cytosolic foreign DNA) and nucleotide oligomerization‐domain‐like receptors (NLRs) recognizing sterile and pathogen‐associated signals.4, 5 Upon sensing damage‐ and pathogen‐associated molecular patterns (DAMPs and PAMPs), the epithelium secretes a plethora of immune mediators, constituting the first layer of defence that alerts the immune system to the presence of a threat. Among these immune mediators, there are mucins, defensins and lysozymes; cytokines such as granulocyte–macrophage colony‐stimulating factor, tumour necrosis factor‐α, interleukin‐6 (IL‐6) and also the two unique cytokines IL‐1β and IL‐18.6 Unlike other cytokines, IL‐1β and IL‐18 are synthesized as pro‐forms that need to be cleaved by caspase‐1 after the activation of a multiprotein complex called inflammasome.
The inflammasome: a well‐regulated immune platform also present in non‐myeloid cells
The canonical assembly of the inflammasome is a well‐regulated process that starts after the recognition of PAMPs or DAMPs by cytosolic receptors. The cytosolic receptor acts as a sensor protein and typically belongs to the NLR family but the activation of other receptors such as AIM2 also leads to the formation of an inflammasome5 (Table 1). The most studied inflammasome is NLRP3 because of its ability to respond not only to pathogens but also to sterile stimuli.7 Activation of the cytosolic receptor leads to the recruitment of the effector enzyme capase‐1. Depending on which cytosolic receptor is activated, the recruitment of the effector enzyme, caspase‐1, will require an adaptor molecule known as apoptosis‐associated speck‐like protein containing a caspase activation and recruitment domain (ASC) or not (Table 1). Upon oligomerization of the inflammasome, the effector enzyme caspase‐1 is activated and leads to the cleavage, maturation and release of IL‐1β and IL‐18, and to a form of cell death known as pyroptosis8 (Fig. 1). It is important to mention that, although most of the inflammasome‐forming receptors need ASC for their function, NLRP1b and NLRC4 have been shown to have both ASC‐dependent and ASC‐independent functions. Although ASC is dispensable (but significantly increases) NLRP1b function, NLRC4‐mediated pyroptosis (but not cytokine release) is ASC‐independent.9, 10, 11
Table 1.
Main inflammasome‐forming receptors in epithelial cells
| Subfamily | Receptor | Requirement for ASC | Main activating signal | Main epithelia involved |
|---|---|---|---|---|
| NLRP | NLRP1 | Yesa | Anthrax toxin and ATP | Oral, airway22, 23, 24 |
| NLRP3 | Yes | Ionophores, crystals, ATP, bacterial toxins | Oral, airway, intestinal, skin22, 23, 25, 26 | |
| NLRP6b | Yes | Microbiota‐modulated metabolites (taurine) | Intestinal27 | |
| NLRP9b | Yes | Rotavirus dsRNA | Intestinal28 | |
| NLRC | NLRC4/IPAF | Yesa | Flagellin (in mouse)/Type III secretion system (in human) | Intestinal29 |
| Other inflammasome‐forming sensors | AIM2 | Yes | dsDNA | Oral, skin23, 30 |
Not all the inflammasome‐forming receptor proteins require the adaptor protein apoptosis‐associated speck‐like protein containing a caspase activation and recruitment domain (ASC) to activate caspases‐1, however, when present, ASC potentiates inflammasome activity. Also shown the main activating signals and epithelial distribution.
Both nucleotide NACHT, LRR and PYD domains‐containing protein 1 (NLRP1) and NLRC4 protein have a caspase recruitment domain and have shown activity in the presence or absence of ASC.9, 31
The ability of NLRP6 to form inflammasomes has only been suggested but not directly shown.
Figure 1.
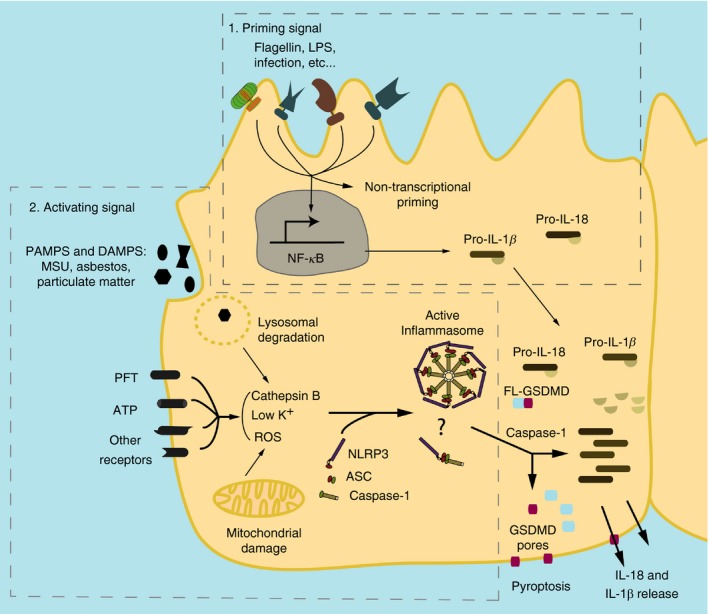
Canonical activation of NACHT, LRR and PYD domains‐containing protein 3 (NLRP3) inflammasome. Inflammasome needs a priming signal1 such as lipopolysaccharide (LPS) that triggers the nuclear factor‐κB (NF‐κB) pathway leading to an increase in the production of pro‐interleukin‐1β (pro‐IL‐1β). The priming signal also has a non‐transcriptional function, deubiquitinating NLRP3.12 A second signal, named activation signal,2 is triggered after the recognition of pathogen‐associated molecular patterns (PAMPs) and damage‐associated molecular patterns (DAMPs) through specific receptors, membrane disruption or lysosomal uptake. This activating signal will trigger a disruption of cell homeostasis. This disruption will be sensed in different ways, including the release of cathepsin B by damaged lysosomes, increased production of mitochondrial reactive oxygen species (ROS) and/or potassium efflux. This potassium efflux will be detected by cytosolic receptors such as NLRP3. The recognition of the signal will foster NLRP3 oligomerization and the formation of the active complex of the inflammasome after recruitment of apoptosis‐associated speck‐like protein containing a caspase activation and recruitment domain (ASC) and caspase‐1. The active inflammasome will cleave procaspase‐1 generating active caspase‐1, which will lead to the production and release of the pro‐inflammatory cytokines IL‐1β and IL‐18. Caspase‐1 will also cleave full‐length gasdermin‐D (FL‐GSDMD), which will create pores in the membrane. These pores are suggested to be one mechanism of release for IL‐18 and IL‐1β but not the only one. The gasdermin‐D pore will also produce a form of pro‐inflammatory cell death known as pyroptosis. ?, it is unclear whether epithelial cells can form inflammasome multimeric specks or present only filament oligomerization; MSU, monosodium urate; PFT, pore‐forming toxins.
Although canonical inflammasomes rely on the activation of caspase‐1, cells can also form ‘non‐canonical’ inflammasomes, which lead to the activation of caspase‐11 (caspase‐4 and caspase‐5 in humans) or caspase‐8.13, 14 Caspase‐11 acts as an intracellular lipopolysaccharide (LPS) sensor by direct binding using its caspase activation and recruitment domain (CARD). Interestingly, this process is widely extended into myeloid and non‐myeloid cells.15 Therefore, caspase‐11 could be a widespread mechanism to detect intracellular Gram‐negative bacteria in cells, including infected epithelial cells. Upon LPS recognition, caspase‐11 is activated and causes K+ efflux, which induces NLRP3 canonical inflammasome formation and IL‐1β and IL‐18 release.16
Both IL‐1β and IL‐18 are potent pro‐inflammatory cytokines with a pivotal role during the first steps of inflammation and their deregulation is extremely detrimental to health. Hence, these are tightly regulated proteins, not only at transcriptional level, but also post‐transcriptionally by their activation within the inflammasome. Although IL‐1β and IL‐18 are activated by the inflammasome in a similar manner, their contribution to the inflammatory response is very different. Interleukin‐1β drives inflammation by controlling the recruitment of neutrophils to the site of infection, the induction of IL‐8 by epithelial cells and the release of IL‐17 from T cells.17 On the other hand, the main role of IL‐18 is to foster the activation of natural killer and T cells and the release of interferon‐γ.18, 19, 20 Although most studies focus on the release of IL‐1β and IL‐18 by immune cells such as macrophages, epithelial cells are also able to release these pro‐inflammatory cytokines.19, 21 However, the mechanistic insights by which this occurs remain unclear.
As a result of their essential role as triggers of innate immunity, and in order to tightly control their release, IL‐1β and IL‐18 are synthesized as pro‐forms lacking a signal peptide. The maturation and release of these pro‐inflammatory cytokines are regulated by the assembly of a multiprotein complex known as the inflammasome, also present in epithelial cells (Table 1; Fig. 2).22
Figure 2.
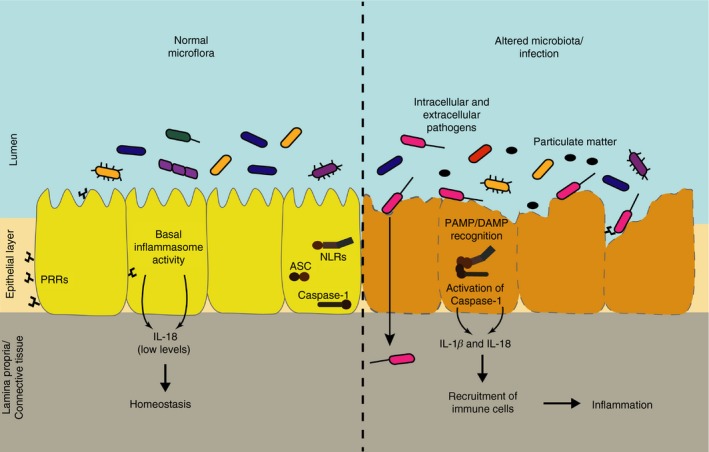
During healthy conditions epithelial cells do not respond (or have a weak response) to commensal bacteria or non‐invasive microorganisms. However, epithelial cells still release basal levels of interleukin‐18 (IL‐18).19, 45, 46 The lack of response from epithelial cells to commensal bacteria is partially due to differential distribution of pattern recognition receptors (PRRs) in the membrane, predicted to foster recognition of only invasive pathogens. After invasion and disruption of the epithelial layer by bacterial pathogens, viruses or exposure to sterile agents, epithelial cells recognize pathogen‐ or danger‐associated molecular patterns (PAMPS and DAMPS) and activate inflammasome. Inflammasome activation in epithelial cells leads to the release of IL‐1β and IL‐18, recruiting immune cells to the site of infection and inflammation.
Despite caspase‐1 being the main caspase involved in this process, others such as caspase‐4/5 (human orthologues of murine caspase‐11) and caspase‐8 can also play an important role.13, 32 Recently, a new protein‐mediating pyroptosis has been described: gasdermin‐D.33, 34 Gasdermin‐D is activated by caspase‐1 and forms pores in the membrane, which leads to cell death.35, 36, 37, 38 It has been recently proposed that IL‐1β and IL‐18 release happens through gasdermin‐D pores;39 even so, further research is needed. Gasdermin‐D is also expressed in epithelial cells, and its involvement on inflammasome activity has been reported. Although no direct cleavage of gasdermin‐D has been shown in epithelial cells, the pore‐forming protein has been shown to be necessary for NLRC4‐mediated and NLRP9b‐mediated pyroptosis.28, 40, 41 This would show that gasdermin‐D is functional and important for epithelial cell inflammasome function. Interestingly, in recent years, it has been reported that under certain circumstances the release of pro‐inflammatory cytokines and cell death are uncoupled upon inflammasome activation in several cell types such as neutrophils, monocytes and macrophages.39, 42, 43 This is particularly important for epithelial cells where the conservation of the integrity of the epithelial layer is key to maintain barrier homeostasis44 (Fig. 2). Whether the release of IL‐1β and IL‐18 is uncoupled from cell death in epithelial cells remains unknown.
Despite increasing research in this area, there are still unanswered questions regarding inflammasome activation in the epithelium (Fig. 2). The epithelium is exposed to mechanical and chemical damage associated with the exterior milieu as well as to a wide range of microorganisms colonizing our body. Hence, the inflammasome activity has to be tightly regulated in epithelial cells, not only to prevent an inflammasome response to healthy microbiota but also to potentiate the response to an altered microbiome. In this review, we will summarize the current knowledge on inflammasome activation at different barrier sites, and how these molecular complexes contribute not only to maintenance of barrier homeostasis but also to disease.
Gut epithelial cells
The most studied inflammasome‐forming epithelium is the gut epithelium. In this barrier, the heavy colonization of commensal microbiota makes the tight regulation of the inflammatory cascade crucial. How gut epithelial cells respond to invasive pathogens and not to commensal bacteria remains only partially understood. It has been shown that differential distribution of TLRs on the membrane of gut epithelial cells allows the response to invasive microorganisms not just after apical contact but after the disruption of the epithelium47 (Fig. 2). The requirement of invasion or damage of epithelium integrity to activate the immune function of epithelial cells guarantees that repair mechanisms and inflammatory processes are triggered only when needed. In addition to TLRs, the cytosolic localization of the inflammasome‐forming sensors NLRs and AIM2, and their ability to respond to damage signals as well as to pathogens, places them as ideal platforms to detect pathogen invasion.
The role of inflammasomes at this barrier became apparent after early reports showed that intestinal epithelial cells (IEC) were able to up‐regulate IL‐1β in response to infection48 as well as being the principal contributors to the total levels of IL‐18 in the gut under physiological conditions.49 Since then, an increasing amount of evidence has shown an involvement of the inflammasome in maintaining homeostasis in this barrier as well as responding to pathogens.
The gut epithelium has been shown to rely on the NLRC4 inflammasome to respond to Citrobacter rodentium, a mouse model of enteropathogenic Escherichia coli infection.29, 50 Interestingly, epithelial NLRP3 is also involved in C. rodentium clearance as Nlrp3 −/− mice show higher bacterial burden and higher epithelium invasion.51 This effect is likely to be mediated by both IL‐18 and IL‐1β as Il‐18 −/− and Il‐1β −/− mice present a similar phenotype to the Nlrp3 −/− mouse when challenged with C. rodentium.50 These findings have also been confirmed in human cells after E. coli infection of the epithelial cell line Caco‐2 induced increased levels of IL‐18 release.52 Apart from this role in infection, caspase‐1 and NLRC4 have also been reported as essential in preventing colonic inflammation‐induced tumorigenesis by regulating the response of epithelial cells to injury, highlighting the role of NLRC4 in epithelial cells.53, 54
In another model of infection, inflammasome‐dependent release of IL‐18 has been shown to mediate natural killer cell recruitment after Salmonella typhimurium infection.55 Although authors do not investigate the origin of gut IL‐18 in this paper, the above‐mentioned studies suggest that this IL‐18 is likely to be of epithelial cell origin. Using the same infection model, it has been shown that inflammasome‐forming sensors are also involved in the extrusion and expulsion of intestinal epithelial cells. During S. typhimurium infection, caspase‐4 mediates epithelial cell expulsion through a process dependent on pyroptosis.56 This process is epithelial cell intrinsic and independent of cytokine signalling, showing a specific role for pyroptosis and for non‐canonical inflammasomes.57 However, this is not the only inflammasome‐dependent mechanisms for epithelial cell expulsion. In another study exploring the role of NLRC4 in gut epithelial cells, authors showed that caspase‐1 and gasdermin‐D are dispensable for IEC expulsion from the epithelial layer upon treatment with the NLRC4 activator FlaTox (Legionella pneumophila flagellin fused to Bacillus anthracis lethal factor), as cells were still expelled from the epithelium in the absence of these mediators. In contrast, the expelled cells were not dead, implying that both caspase‐1 and gasdermin‐D are required for IEC pyroptosis upon NLRC4 activation.41 How the two mechanisms can act together is still not understood. Although this is an important mechanism necessary for the maintenance of normal microbiota and pathogen clearance, all these studies highlight the danger of excessive inflammasome response and gasdermin‐D‐mediated pyroptosis in gut epithelial cells. An excessive activation of pyroptosis could eventually lead to loss of membrane integrity and systemic infection.
Another NLR involved in the relationship between gut epithelium and bacteria is NLRP6. Recent findings emphasize the importance of having proper controls to elucidate the role of different inflammasomes in immunity, especially in maintaining commensal bacteria. Although the role of NLRP6 in gut epithelial cells is still not totally clear, NLRP6 is highly expressed and functional in gut epithelial cells.27 Mamantopoulos et al.,58 in two different animal facilities, dismissed a role of NLRP6 in maintaining homeostasis in the gut microbiota at steady state. However, it has been proposed that NLRP6 does contribute to microbiota balance, but this contribution is dependent on microbiota community structure (and especially in the presence of pathobionts such as Helicobacter spp.). Both of these studies point to familial transmission of microbiota rather than NLRP6 impairment for the observed changes in previous studies.59, 60 How the cells activate NLRP6 and whether NLRP6 forms an active inflammasome are still not fully answered questions. No direct evidence shows NLRP6 forming an inflammasome but Nlrp6 −/− studies mimic the results of Caspase‐1 −/− and Asc −/−, suggesting that this might be the case.61, 62 Recently, taurine, an amino acid present in bile acid and shown to regulate gut microbiota, has been proposed to be the activating signal for NLRP6 as it induces IL‐18 secretion in wild‐type mice but not in Nlrp6 −/− mice.62, 63 Although this finding needs to be further tested and investigated, it would be highly interesting as the gut microbiota in return has also been shown to regulate bile acid metabolism and especially taurine.64 Further research is needed to elucidate the molecular events leading to NLRP6 activation.
A highly prevalent condition of the gut is inflammatory bowel disease (IBD, including Crohn's disease and ulcerative colitis). IBD is a chronic inflammation of the digestive system associated with the alterations of the gut microbiota.65 The most common mouse model of IBD is to induce colitis using dextran sodium sulphate (DSS), which damages the epithelium, increasing gut permeability.66 Inflammasome responses in experimental models of colitis are well characterized, but mainly in monocytes and macrophages.67 In such models, NLRP3 inhibition ameliorates DSS‐induced colitis.68, 69, 70 However, these studies fail to show a specific role for epithelial cells, despite these being the main cell type affected by IBD. In fact, recent studies suggest that NLRP3 is not expressed by epithelial cells in the colon of mice, placing tissue‐resident macrophages as the principal contributors to this NLRP3‐dependent response.71 These studies suggest that although NLRP3 is a main actor in IBD, the epithelial cell layer's contribution to IBD might be NLRP3 independent. Surprisingly, epithelial cell‐specific depletion of capase‐1 induces decreased IL‐18 levels and decreased pathology in a DSS‐induced colitis model, pointing to the involvement of another inflammasome‐forming receptor.72 This is consistent with the finding that during colitis, IL‐18 produced by epithelial cells exacerbates pathology. Interestingly, epithelial IL‐18 signalling during colitis targets epithelial cells themselves as Il‐18R −/− in haematopoietic cells fails to rescue mice from colitis, whereas specific Il‐18R −/− in epithelial cells protects mice from DSS‐induced colitis.73 Recent studies also point to a role of AIM2 in DSS‐induced colitis. AIM2 knockout mice are more severely affected by colitis and present a higher bacterial burden; however, how AIM2 is activated or how DNA is delivered in the epithelial cells during colitis is unknown. In fact, authors suggest that the role of AIM2 in colitis is to control the anti‐microbial peptide production (such as α‐ and β‐defensins) in IEC, and to regulate tissue repair, activating an IL‐18‐dependent pathway.74, 75
All these reports together show an increasingly recognized role of inflammasomes in innate immunity driven by gut epithelial cells and places the inflammasome response as a coordinated process with several outcomes. However, a more profound study using IEC or myeloid‐cell‐specific knockout mice would be useful to better understand the role of epithelial cells in barrier defence.
Lung epithelium
Due to the extended area of contact between internal and external stimuli, airway epithelium is a primary target for pathogens and damaging agents. Lung epithelial cells have been shown to activate the inflammasome and release IL‐1β and IL‐18 upon a diverse range of stimuli such as influenza virus, ozone, particulate matter, or asbestos.76, 77, 78, 79 However, despite the importance of the release of IL‐1β and IL‐18 in this organ, the events leading to their release from epithelial cells are still not well understood and are the subject of controversy, probably partly due to different cell types used in different studies. For instance, the involvement of caspase‐1 is not clear because some studies show that epithelial cell lines (Beas2Bs, 16HBE, Calu‐3) and also primary cells (human bronchial epithelial cells) express little to no caspase‐1 in an infection model, whereas others show a role for caspase‐1 in epithelial cells (for instance in Beas2Bs, mouse tracheobronchial epithelial cells and A549).80, 81, 82, 83 It has been suggested that as some of these cells do not express the inflammasome component, caspase‐11 is responsible for the inflammasome response to infections, probably by direct biding of LPS.84 These differential results highlight the importance of analysing the expression of inflammasome components in the lung epithelial cell lines before conducting inflammasome experiments and the need to work in mouse and human primary cells.
As in the gut, the lungs constitute an area that is heavily colonized by commensal bacteria. However, in contrast to the gut, the lung epithelium is also heavily exposed to agents causing sterile inflammation such as particulate matter present in tobacco smoke or urban pollution.
Despite the fact that airway macrophages respond strongly to particulate matter due to their increased ability to phagocytose, epithelial cells also respond to air pollutants.85 Human bronchial epithelial cells (Beas2B) have been shown to up‐regulate, process and release IL‐1β in response to crystal silica exposure, which is found in the air and taken up by epithelial cells.83 Interestingly, in the same study, they also detect release of High mobility group box 1 protein (HMGB1) from epithelial cells after silica exposure. HMGB1 is a potent danger signal also released by A549 cells under infection.82 These observations are relevant because the crosstalk among epithelial cells and between epithelial cells and immune cells is important for tissue repair. Other urban particulate matter (particulate matter < 10 µm) can also activate the inflammasome in an NLRP3‐dependent manner.79 This could be particularly important in urban or industrial areas. This particulate matter uptake by epithelial cells and inflammasome activation could worsen already existing conditions or cause new ones. In fact, it has been suggested that following periods of poor air quality (characterized by higher levels of particulate matter), inflammasome activity could be contributing to the exaggerated response to viruses such as influenza and allergy.21, 86
Another major agent for lung dysfunction, cigarette smoke, increases the release of IL‐1β in human lung epithelial cells, and this increase is even higher in patients with chronic obstructive pulmonary disease (COPD).87 Indeed, IL‐1β has been shown to be a marker of COPD severity as individuals with COPD present higher levels of IL‐1β in serum.88 In contrast, another study shows that cigarette smoke reduces the inflammatory response in mice treated with another NLRP3‐activating crystal: asbestos.89 Although contradictory, the reduction of the inflammatory response could be explained by the promotion of the proteasomal degradation of NLRP3 mediated by its ubiquitination.90
In addition to sterile agents, the lung is also exposed to pathogens. A condition where the lung epithelium integrity is compromised is during aspergillosis. Being inhaled every day, the spores of Aspergillus fumigatus invade the lung of not only immunocompromised patients but also patients with COPD.91 Inflammasomes have long been involved with the innate immune response to A. fumigatus, and recent studies show that inflammasome activation occurs early during infection and is key for macrophage anti‐fungal activity.92, 93 Interestingly, β‐glucan, a component of the A. fumigatus cell wall, upon dectin‐1‐binding activates NLRP3 inflammasome in bronchoepithelial cells.94 In fact, Syk tyrosine kinase, a kinase activated through dectin‐1, has been shown to be involved in inflammasome activation by A. fumigatus in monocytes, reinforcing the role of β‐glucan in inflammasome activation in lung epithelial cells.95 Both studies point at reactive oxygen species, a major mediator of inflammasome activation, as a key factor during aspergillosis.
The complexity of the lung together with the distinct environments and stimuli that lung epithelial cells face makes the study of the epithelial inflammasome in this organ quite challenging. However, these studies show the inflammasome in the lung as a clear therapeutic target to ameliorate a broad range of pathologies with an inflammatory component.
Oral epithelium
An entry gate for nutrients, the oral mucosa is a heavily exposed environment where particulate matter, and bacterial and fungal agents constantly challenge the homeostasis of the epithelium.
Gingivitis is inflammation of the gums, where the gum pulls away from the bone, creating cavities that are colonized by pathogens, which if left untreated can lead to periodontitis. One of the main pathogens colonizing these cavities and causing the disease is Porphyromonas gingivalis. Periodontitis has been linked to increased expression of inflammasome‐sensing proteins NLRP3 and AIM2, mainly in the epithelial layer.22, 96 Moreover, a polymorphism in other NLR family member, NLRC5, has been linked to increased susceptibility to periodontitis,97 although how it contributes to this pathology has not been investigated. NLRC5 has a caspase recruitment domain but it has not been shown to form inflammasomes on its own. In fact, NLRC5 can associate with NLRP3 in response to different PAMPs and DAMPs to cooperatively activate the inflammasome.98 This would suggest that the polymorphisms of NLRC5 induced to increased susceptibility to periodontitis are the consequence of an aberrant assistance to NLRP3 to form inflammasomes. Other roles for NLRC5 have been described including negative regulation of nuclear factor‐κB,99 interferon signalling99, 100 and regulation of major histocompatibility complex class I genes,101 all of which could also explain its contribution to periodontitis.
As expected, infection of gingival epithelial cells with P. gingivalis induced an increase in Il‐1β gene expression. However, IL‐1β was left unprocessed and accumulated intracellularly. Interleukin‐1β was only processed and released after the activation of the ATP‐gated receptor P2X7, which led to NLRP3 inflammasome formation. The P2X7R is also involved in NLRP3 inflammasome formation and release of mature IL‐1β from salivary epithelial cells, as the release of this pro‐inflammatory cytokine is dampened in P2X7R knockout mice.102, 103 Why IL‐1β remains uncleaved and is intracellular during P. gingivalis infection remains an interesting question. We could speculate that the immune machinery of epithelial cells might reduce the activation of caspase‐1 during chronic infection. This mechanism could potentially dampen immune reactions to commensal bacteria and prevent pyroptosis to maintain the integrity of the epithelial layer. In fact, in macrophages, it has been shown that IL‐1β can be released via exosomes in a mechanism dependent on NLRP3 and ASC but independent of caspase‐1.104, 105 Potentially, IL‐1β produced by epithelial cells could be stored and released on exosomes to have two potential effects. (i) Be processed by other cells (potentially immune cells) to avoid activating caspase‐1 and compromising the epithelial layer in epithelial cells. (ii) Have an effect in areas distant to the site of infection. Although epithelial cells have been shown to release cytokines trapped in exosomes, it is unknown whether IL‐1β could follow the same pathway.106
Interestingly, another NLR family member, NLRX1, has recently been shown to potentiate the inflammasome response to ATP in gingival epithelial cells through the generation of mitochondrial reactive oxygen species. However, how NLRX1 (not predicted to form inflammasomes) potentiates the response to ATP is not well understood and might assist NLRP3 in its function in a similar manner to NLRC5.107
Although inflammasome activity is required for optimal control and clearance of pathogens, over‐activation could lead to unwanted outcomes. Excessive levels of IL‐1β and of pyroptosis have been linked to bone loss during periodontitis and to malignancy during oral carcinogenesis.108, 109 Epithelial cells can avoid this and reduce the extent of inflammasome activity during acute infection through the stress‐induced enzyme haem oxygenase‐1. This enzyme has anti‐oxidant and anti‐inflammatory effects, and can dampen LPS signalling and inflammasome activity in oral epithelial cells, although the exact mechanism by which this occurs is not clear.110
Skin epithelial cells
The skin is the main physical barrier that protects our body from the external environment, therefore it is active in the immune control of pathogen invasion. Immune homeostasis in the skin is essential to maintain skin integrity and its role as a barrier.
Although the role of the inflammasome in skin is not well characterized, mutations in different genes coding for NLR‐forming inflammasomes have been associated with skin conditions. Even before the discovery of the inflammasome, mutations in the NLRP3 gene were found to cause a spectrum of diseases known as cryopyrin‐associated periodic syndromes (or CAPS), which are associated with skin inflammation and rashes.111 Rashes typically disappear, or are controlled, after anti‐IL‐1β therapy, showing a role for IL‐1β signalling in skin rashes and in CAPS in general.112
Recently the first genetic evidence connecting inflammasome signalling to non‐fever skin diseases has been found. Gain of function mutations in the NLRP1 sensor protein cause two overlapping skin disorders: multiple self‐healing palmoplantar carcinoma and familial keratosis lichenoides chronica. These mutations lead to spontaneous inflammasome activation and paracrine IL‐1 signalling in keratinocytes from these patients.24 Polymorphisms of NLRP1 have also been associated with the autoimmune disease vitiligo, a condition characterized by the development of white patches in the skin due to lack of melanin.113 This is believed to occur through higher IL‐1β production and NLRP1 activation, although the specific mechanism by which the NLRP1 inflammasome shows greater functional activity is not known.114
Inflammasome has also been involved in another chronic inflammatory syndrome affecting the skin. Deregulated levels of IL‐1β and AIM2 inflammasome activity by keratinocytes have been linked to psoriatic disease. Abnormally high levels of IL‐1β could potentiate a T helper type 17 response with up‐regulation of IL‐17 and excessive infiltration of neutrophils, characteristics of psoriatic diseases.115 It is not surprising that AIM2, a receptor involved in the recognition of cytosolic DNA, is involved in psoriatic diseases as cytosolic DNA has been shown to be responsible for inflammasome activation in keratinocytes during psoriasis.116
Interestingly, a recent study points to a role in AIM2 in immune memory of epithelial cells.30 Using epithelial cell stem cells they determined that these cells possess immune memory, enabling a rapid second response after a previous exposure to inflammation. This striking capacity of skin epithelial stem cells is dependent on AIM2 and downstream of IL‐1β as Il‐1β −/− and Il‐1r1 −/− but not Il‐18 −/− failed to induce immune memory. Given the above‐mentioned role of AIM2 in recurrent skin diseases such as psoriasis, we could speculate that both mechanisms might be linked, suggesting major implications for immune memory of epithelial cells in chronic inflammatory diseases.
Finally, the role of IL‐18 in skin has been widely studied. In fact, IL‐18 has been linked with psoriatic disease, where increased caspase‐1 activity has been reported.117 Variations in the IL‐18 gene have also been associated with increased risk of atopic eczema, a chronic inflammatory condition of the skin.118
Final remarks
In this review we have presented the involvement of inflammasome activation in the role of epithelial cells as mediators of the immune response. We have reviewed how epithelial cells are able to release IL‐1β and IL‐18. However, despite the relatively low concentration of these cytokines released by epithelial cells compared with macrophages, it is important to mention that in healthy conditions epithelial cells are relatively more abundant in barriers than immune cells. Therefore, the epithelial compartment could contribute greatly to the inflammatory response to pathogens, at least in early infection when not many immune cells have been recruited.
We have highlighted the diversity of responses and of sensor proteins involved in the inflammasome response. The differential expression of sensor molecules and inflammasome components and mediators in the different types of epithelial cells will shape the immune response according to the needs and threats of each specific site. Similarly, the types of pathogens and threats that each barrier will face vary from one epithelium to another, raising the question of whether there is a general mechanism leading to inflammasome activation in epithelial cells or the response is specific to each epithelium and threat. The increasing body of publications in the field points in the direction of several types of inflammasomes involved in different epithelia, making the response tissue‐dependent. We conclude that, in order to better understand this question, the field would benefit from a broad study comparing the involvement of different inflammasomes in the different epithelia. Even more, it is not always clear and differs from one cell line model to another what inflammasome components are present in each epithelium. In our review we have highlighted several examples of differential expression of inflammasome components or pro‐inflammatory cytokines in different cell types or tissues. We could hypothesize that within one organ, different types of epithelial cells will express different inflammasome components, including cytosolic sensors and effector caspases. That would ensure a diverse, adapted and targeted response and prevent a much feared loss of epithelial integrity by massive activation of inflammasomes. It is also important to highlight that most of the available data on the role of inflammasomes in epithelial cell immunity has been obtained using either human cell lines, or primary epithelial cells from mouse. Key differences between human and murine inflammasomes have already been described (for instance NLRC4 is not activated by the same components of bacterial flagellum in mouse and humans and DNA inflammasome activation in human monocytes depends on NLRP3 instead of AIM2119, 120). Therefore, more studies are needed in human lung epithelial cells to confirm the findings mentioned in this review.
Gasdermin‐D, as mentioned before, is the executor of pyroptosis. However, its role and function in non‐haematopoietic cells is still not well characterized. We could speculate that after inflammasome activation in epithelial cells, the triggering of pyroptosis could have two possible outcomes, both of them highlighting the need for a tight control of pyroptosis in epithelial cells. Pyroptosis during infection could be detrimental (and would be somehow silenced or dampened in epithelial cells) by causing membrane integrity loss. This result is in line with the previously mentioned report showing accumulation of pro‐IL‐1β during P. gingivalis infection of the oral mucosa. Nevertheless, pyroptosis could also be beneficial during epithelium colonization, as could a mechanism of containing the infection and avoiding further invasion of internal layers of the epithelium. To reinforce this idea, during Salmonella infection caspase‐1 and gasdermin‐D cleavage are required for IEC pyroptosis but not for IEC expulsion.41 In fact, in that study, the authors demonstrate that during the death and expulsion of IEC, the epithelial integrity is maintained through the rearrangement of neighbouring cells. This result also links with the discovery that small amounts of caspases‐1 are required for gasdermin‐D cleavage (meaning that even a weak activation of inflammasome produces cell death) while detecting active IL‐1β requires higher levels of processed caspases‐1.121 In such a scenario, weak invasion of the epithelial layer would lead to epithelial cell death and expulsion but still no immune response as the threshold to release IL‐1β is much higher than for cell death. This mechanism, potentially occurring with low infection rates where few cells are being infected, could be important to maintain the equilibrium with normal microbiota.
The recent discovery of inflammasome inhibitors such as MCC950 or boron‐based compounds is opening the path to the future treatment of inflammasome‐related pathologies with specific drugs.122, 123 However, while the field is moving towards better drugs, there is also an increasing body of evidence that inflammasome activity is key in the immune response of many cell types. The potential specific targeting of either immune cells or epithelial cells in different inflammasome‐related pathologies would be beneficial. Such a specificity would allow the targeting of the over‐activation of inflammasome in cells where its activity is detrimental while allowing the normal inflammasome function in other cells.
In conclusion, the role of the inflammasomes in epithelium has been extensively reported. We hypothesize that their contribution to the triggering of inflammation, in opposition to the common belief, is far from being negligible. This combination of findings provides some support for the conceptual premise that targeting inflammasome activity in epithelial cells might be essential for further treatment of a wide range of inflammatory disorders. There is then, abundant room for further progress in elucidating the mechanism of inflammasome activation in epithelial cells and investigating whether it is similar to that in macrophages or not.
Disclosures
The authors have declared that no conflict of interest exists.
Acknowledgements
This work was supported by a Sir Henry Dale Fellowship Jointly Funded by the Wellcome Trust and the Royal Society (Grant number 104192/Z/14/Z) to GL‐C and by the Manchester Collaborative Centre for Inflammation Research, A Joint Initiative of the University of Manchester, AstraZeneca and GlaxoSmithKline (PP‐R).
References
- 1. The lungs at the frontlines of immunity. Nat Immunol 2015; 16:17. [DOI] [PubMed] [Google Scholar]
- 2. Tate MD, Pickett DL, van Rooijen N, Brooks AG, Reading PC. Critical role of airway macrophages in modulating disease severity during influenza virus infection of mice. J Virol 2010; 84:7569–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ritter M, Mennerich D, Weith A, Seither P. Characterization of Toll‐like receptors in primary lung epithelial cells: strong impact of the TLR3 ligand poly(I:C) on the regulation of Toll‐like receptors, adaptor proteins and inflammatory response. J Inflamm (Lond) 2005; 2:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lund JM, Alexopoulou L, Sato A, Karow M, Adams NC, Gale NW et al Recognition of single‐stranded RNA viruses by Toll‐like receptor 7. Proc Natl Acad Sci USA 2004; 101:5598–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hornung V, Ablasser A, Charrel‐Dennis M, Bauernfeind F, Horvath G, Caffrey DR et al AIM2 recognizes cytosolic dsDNA and forms a caspase‐1‐activating inflammasome with ASC. Nature 2009; 458:514–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Moldoveanu B, Otmishi P, Jani P, Walker J, Sarmiento X, Guardiola J et al Inflammatory mechanisms in the lung. J Inflamm Res 2009; 2:1–11. [PMC free article] [PubMed] [Google Scholar]
- 7. Chen C‐J, Kono H, Golenbock D, Reed G, Akira S, Rock KL. Identification of a key pathway required for the sterile inflammatory response triggered by dying cells. Nat Med 2007; 13:851–6. [DOI] [PubMed] [Google Scholar]
- 8. He W, Wan H, Hu L, Chen P, Wang X, Huang Z et al Gasdermin D is an executor of pyroptosis and required for interleukin‐1β secretion. Cell Res 2015; 25:1285–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Van Opdenbosch N, Gurung P, Vande Walle L, Fossoul A, Kanneganti T‐D, Lamkanfi M. Activation of the NLRP1b inflammasome independently of ASC‐mediated caspase‐1 autoproteolysis and speck formation. Nat Commun 2014; 5:3209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Case CL, Shin S, Roy CR. Asc and Ipaf Inflammasomes direct distinct pathways for caspase‐1 activation in response to Legionella pneumophila . Infect Immun 2009; 77:1981–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Case CL, Roy CR. Asc modulates the function of NLRC4 in response to infection of macrophages by Legionella pneumophila . mBio 2011; 2:e00117‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Juliana C, Fernandes‐Alnemri T, Kang S, Farias A, Qin F, Alnemri ES. Non‐transcriptional priming and deubiquitination regulate NLRP3 inflammasome activation. J Biol Chem 2012; 287:36617–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kayagaki N, Warming S, Lamkanfi M, Vande Walle L, Louie S, Dong J et al Non‐canonical inflammasome activation targets caspase‐11. Nature 2011; 479:117–21. [DOI] [PubMed] [Google Scholar]
- 14. Chung H, Vilaysane A, Lau A, Stahl M, Morampudi V, Bondzi‐Simpson A et al NLRP3 regulates a non‐canonical platform for caspase‐8 activation during epithelial cell apoptosis. Cell Death Differ 2016; 23:1331–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Shi J, Zhao Y, Wang Y, Gao W, Ding J, Li P et al Inflammatory caspases are innate immune receptors for intracellular LPS. Nature 2014; 514:187–92. [DOI] [PubMed] [Google Scholar]
- 16. Rühl S, Broz P. Caspase‐11 activates a canonical NLRP3 inflammasome by promoting K+ efflux. Eur J Immunol 2015; 45:2927–36. [DOI] [PubMed] [Google Scholar]
- 17. Sutton C, Brereton C, Keogh B, Mills KHG, Lavelle EC. A crucial role for interleukin (IL)‐1 in the induction of IL‐17‐producing T cells that mediate autoimmune encephalomyelitis. J Exp Med 2006; 203:1685–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lee J‐K, Kim S‐H, Lewis EC, Azam T, Reznikov LL, Dinarello CA. Differences in signaling pathways by IL‐1β and IL‐18. Proc Natl Acad Sci USA 2004; 101:8815–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Harrison OJ, Srinivasan N, Pott J, Schiering C, Krausgruber T, Ilott NE et al Epithelial‐derived IL‐18 regulates Th17 cell differentiation and Foxp3+ Treg cell function in the intestine. Mucosal Immunol 2015; 8:1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Eckmann L, Kagnoff MF, Fierer J. Epithelial cells secrete the chemokine interleukin‐8 in response to bacterial entry. Infect Immun 1993; 61:4569–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hastie AT, Everts KB, Cho SK, Zangrilli J, Shaver JR, Pollice MB et al IL‐1β release from cultured bronchial epithelial cells and bronchoalveolar lavage cells from allergic and normal humans following segmental challenge with ragweed. Cytokine 1996; 8:730–8. [DOI] [PubMed] [Google Scholar]
- 22. Kummer JA, Broekhuizen R, Everett H, Agostini L, Kuijk L, Martinon F et al Inflammasome components NALP 1 and 3 show distinct but separate expression profiles in human tissues suggesting a site‐specific role in the inflammatory response. J Histochem Cytochem 2007; 55:443–52. [DOI] [PubMed] [Google Scholar]
- 23. Xue F, Shu R, Xie Y. The expression of NLRP3, NLRP1 and AIM2 in the gingival tissue of periodontitis patients: RT‐PCR study and immunohistochemistry. Arch Oral Biol 2015; 60:948–58. [DOI] [PubMed] [Google Scholar]
- 24. Zhong FL, Mamaï O, Sborgi L, Boussofara L, Hopkins R, Robinson K et al Germline NLRP1 mutations cause skin inflammatory and cancer susceptibility syndromes via inflammasome activation. Cell 2016; 167:187–202.e17. [DOI] [PubMed] [Google Scholar]
- 25. Quan J‐H, Huang R, Wang Z, Huang S, Choi I‐W, Zhou Y et al P2X7 receptor mediates NLRP3‐dependent IL‐1β secretion and parasite proliferation in Toxoplasma gondii‐infected human small intestinal epithelial cells. Parasit Vectors 2018; 11:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hasegawa T, Nakashima M, Suzuki Y. Nuclear DNA damage‐triggered NLRP3 inflammasome activation promotes UVB‐induced inflammatory responses in human keratinocytes. Biochem Biophys Res Commun 2016; 477:329–35. [DOI] [PubMed] [Google Scholar]
- 27. Wlodarska M, Thaiss CA, Nowarski R, Henao‐Mejia J, Zhang J‐P, Brown EM et al NLRP6 inflammasome orchestrates the colonic host–microbial interface by regulating goblet cell mucus secretion. Cell 2014; 156:1045–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zhu S, Ding S, Wang P, Wei Z, Pan W, Palm NW et al Nlrp9b inflammasome restricts rotavirus infection in intestinal epithelial cells. Nature 2017; 546:667–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Nordlander S, Pott J, Maloy KJ. NLRC4 expression in intestinal epithelial cells mediates protection against an enteric pathogen. Mucosal Immunol 2014; 7:775–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Naik S, Larsen SB, Gomez NC, Alaverdyan K, Sendoel A, Yuan S et al Inflammatory memory sensitizes skin epithelial stem cells to tissue damage. Nature 2017; 550:475–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Abdelaziz DHA, Gavrilin MA, Akhter A, Caution K, Kotrange S, Khweek AA et al Asc‐dependent and independent mechanisms contribute to restriction of Legionella pneumophila infection in murine macrophages. Front Microbiol 2011; 2:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Gringhuis SI, Kaptein TM, Wevers BA, Theelen B, van der Vlist M, Boekhout T et al Dectin‐1 is an extracellular pathogen sensor for the induction and processing of IL‐1β via a noncanonical caspase‐8 inflammasome. Nat Immunol 2012; 13:246–54. [DOI] [PubMed] [Google Scholar]
- 33. Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H et al Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 2015; 526:660–5. [DOI] [PubMed] [Google Scholar]
- 34. Kayagaki N, Stowe IB, Lee BL, O'Rourke K, Anderson K, Warming S et al Caspase‐11 cleaves gasdermin D for non‐canonical inflammasome signalling. Nature 2015; 526:666–71. [DOI] [PubMed] [Google Scholar]
- 35. Liu X, Zhang Z, Ruan J, Pan Y, Magupalli VG, Wu H et al Inflammasome‐activated gasdermin D causes pyroptosis by forming membrane pores. Nature 2016; 535:153–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Aglietti RA, Estevez A, Gupta A, Ramirez MG, Liu PS, Kayagaki N et al GsdmD p30 elicited by caspase‐11 during pyroptosis forms pores in membranes. Proc Natl Acad Sci USA 2016; 113:7858–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ding J, Wang K, Liu W, She Y, Sun Q, Shi J et al Pore‐forming activity and structural autoinhibition of the gasdermin family. Nature 2016; 535:111–6. [DOI] [PubMed] [Google Scholar]
- 38. Sborgi L, Rühl S, Mulvihill E, Pipercevic J, Heilig R, Stahlberg H et al GSDMD membrane pore formation constitutes the mechanism of pyroptotic cell death. EMBO J 2016; 35:1766–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Evavold CL, Ruan J, Tan Y, Xia S, Wu H, Kagan JC. The pore‐forming protein gasdermin D regulates interleukin‐1 secretion from living macrophages. Immunity 2017; 48:35–44.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Tamura M, Tanaka S, Fujii T, Aoki A, Komiyama H, Ezawa K et al Members of a novel gene family, Gsdm, are expressed exclusively in the epithelium of the skin and gastrointestinal tract in a highly tissue‐specific manner. Genomics 2007; 89:618–29. [DOI] [PubMed] [Google Scholar]
- 41. Rauch I, Deets KA, Ji DX, von Moltke J, Tenthorey JL, Lee AY et al NAIP‐NLRC4 inflammasomes coordinate intestinal epithelial cell expulsion with eicosanoid and IL‐18 release via activation of caspase‐1 and ‐8. Immunity 2017; 46:649–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Karmakar M, Katsnelson M, Malak HA, Greene NG, Howell SJ, Hise AG et al Neutrophil IL‐1β processing induced by pneumolysin is mediated by the NLRP3/ASC inflammasome and caspase‐1 activation and is dependent on K+ efflux. J Immunol 2015; 194:1763–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Martín‐Sánchez F, Martínez‐García JJ, Muñoz‐García M, Martínez‐Villanueva M, Noguera‐Velasco JA, Andreu D et al Lytic cell death induced by melittin bypasses pyroptosis but induces NLRP3 inflammasome activation and IL‐1β release. Cell Death Dis 2017; 8:e2984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Estes JD, Harris LD, Klatt NR, Tabb B, Pittaluga S, Paiardini M et al Damaged intestinal epithelial integrity linked to microbial translocation in pathogenic simian immunodeficiency virus infections. PLoS Pathog 2010; 6:e1001052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kolinska J, Lisa V, Clark JA, Kozakova H, Zakostelecka M, Khailova L et al Constitutive expression of IL‐18 and IL‐18R in differentiated IEC‐6 cells: effect of TNF‐α and IFN‐γ treatment. J Interferon Cytokine Res 2008; 28:287–96. [DOI] [PubMed] [Google Scholar]
- 46. Rouabhia M, Ross G, Pagé N, Chakir J. Interleukin18 and γ interferon production by oral epithelial cells in response to exposure to Candida albicans or lipopolysaccharide stimulation. Infect Immun 2003; 70:7073–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Gewirtz AT, Navas TA, Lyons S, Godowski PJ, Madara JL. Cutting edge: bacterial flagellin activates basolaterally expressed TLR5 to induce epithelial proinflammatory gene expression. J Immunol 2001; 167:1882–5. [DOI] [PubMed] [Google Scholar]
- 48. Radema SA, van Deventer SJ, Cerami A. Interleukin 1β is expressed predominantly by enterocytes in experimental colitis. Gastroenterology 1991; 100(5 Pt 1):1180–6. [PubMed] [Google Scholar]
- 49. Takeuchi M, Nishizaki Y, Sano O, Ohta T, Ikeda M, Kurimoto M. Immunohistochemical and immuno‐electron‐microscopic detection of interferon‐γ‐inducing factor (“interleukin‐18”) in mouse intestinal epithelial cells. Cell Tissue Res 1997; 289:499–503. [DOI] [PubMed] [Google Scholar]
- 50. Liu Z, Zaki MH, Vogel P, Gurung P, Finlay BB, Deng W et al Role of inflammasomes in host defense against Citrobacter rodentium infection. J Biol Chem 2012; 287:16955–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Song‐Zhao GX, Srinivasan N, Pott J, Baban D, Frankel G, Maloy KJ. Nlrp3 activation in the intestinal epithelium protects against a mucosal pathogen. Mucosal Immunol 2014; 7:763–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Becker HM, Apladas A, Scharl M, Fried M, Rogler G. Probiotic Escherichia coli Nissle 1917 and commensal E. coli K12 differentially affect the inflammasome in intestinal epithelial cells. Digestion 2014; 89:110–8. [DOI] [PubMed] [Google Scholar]
- 53. Hu B, Elinav E, Huber S, Booth CJ, Strowig T, Jin C et al Inflammation‐induced tumorigenesis in the colon is regulated by caspase‐1 and NLRC4. Proc Natl Acad Sci USA 2010; 107:21635–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Allen IC, TeKippe EM, Woodford R‐MT, Uronis JM, Holl EK, Rogers AB et al The NLRP3 inflammasome functions as a negative regulator of tumorigenesis during colitis‐associated cancer. J Exp Med 2010; 207:1045–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Müller AA, Dolowschiak T, Sellin ME, Felmy B, Verbree C, Gadient S et al An NK cell perforin response elicited via IL‐18 controls mucosal inflammation kinetics during Salmonella gut infection. PLoS Pathog 2016; 12:e1005723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Knodler LA, Crowley SM, Sham HP, Yang H, Wrande M, Ma C et al Noncanonical inflammasome activation of caspase‐4/caspase‐11 mediates epithelial defenses against enteric bacterial pathogens. Cell Host Microbe 2014; 16:249–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Sellin ME, Müller AA, Felmy B, Dolowschiak T, Diard M, Tardivel A et al Epithelium‐intrinsic NAIP/NLRC4 inflammasome drives infected enterocyte expulsion to restrict Salmonella replication in the intestinal mucosa. Cell Host Microbe 2014; 16:237–48. [DOI] [PubMed] [Google Scholar]
- 58. Mamantopoulos M, Ronchi F, Van Hauwermeiren F, Vieira‐Silva S, Yilmaz B, Martens L et al Nlrp6‐ and ASC‐dependent inflammasomes do not shape the commensal gut microbiota composition. Immunity 2017; 47:339–48.e4. [DOI] [PubMed] [Google Scholar]
- 59. Lemire P, Robertson SJ, Maughan H, Tattoli I, Streutker CJ, Platnich JM et al The NLR protein NLRP6 does not impact gut microbiota composition. Cell Rep 2017; 21:3653–61. [DOI] [PubMed] [Google Scholar]
- 60. Gálvez EJC, Iljazovic A, Gronow A, Flavell R, Strowig T. Shaping of intestinal microbiota in Nlrp6‐ and Rag2‐deficient mice depends on community structure. Cell Rep 2017; 21:3914–26. [DOI] [PubMed] [Google Scholar]
- 61. Elinav E, Strowig T, Kau AL, Henao‐Mejia J, Thaiss CA, Booth CJ et al NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell 2011; 145:745–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Levy M, Thaiss CA, Zeevi D, Dohnalová L, Zilberman‐Schapira G, Mahdi JA et al Microbiota‐modulated metabolites shape the intestinal microenvironment by regulating NLRP6 inflammasome signaling. Cell 2015; 163:1428–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Yu H, Guo Z, Shen S, Shan W. Effects of taurine on gut microbiota and metabolism in mice. Amino Acids 2016; 48:1601–17. [DOI] [PubMed] [Google Scholar]
- 64. Sayin SI, Wahlström A, Felin J, Jäntti S, Marschall H‐U, Bamberg K et al Gut microbiota regulates bile acid metabolism by reducing the levels of tauro‐β‐muricholic acid, a naturally occurring FXR antagonist. Cell Metab 2013; 17:225–35. [DOI] [PubMed] [Google Scholar]
- 65. De Mattos BRR, Garcia MPG, Nogueira JB, Paiatto LN, Albuquerque CG, Souza CL et al Inflammatory bowel disease: an overview of immune mechanisms and biological treatments. Mediators Inflamm 2015; 2015:493012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Laroui H, Ingersoll SA, Liu HC, Baker MT, Ayyadurai S, Charania MA et al Dextran sodium sulfate (DSS) induces colitis in mice by forming nano‐lipocomplexes with medium‐chain‐length fatty acids in the colon. PLoS One 2012; 7:e32084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Zaki MH, Boyd KL, Vogel P, Kastan MB, Lamkanfi M, Kanneganti T‐D. The NLRP3 inflammasome protects against loss of epithelial integrity and mortality during experimental colitis. Immunity 2010; 32:379–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Guo W, Hu S, Elgehama A, Shao F, Ren R, Liu W et al Fumigaclavine C ameliorates dextran sulfate sodium‐induced murine experimental colitis via NLRP3 inflammasome inhibition. J Pharmacol Sci 2015; 129:101–6. [DOI] [PubMed] [Google Scholar]
- 69. Zhou W, Liu X, Zhang X, Tang J, Li Z, Wang Q et al Oroxylin A inhibits colitis by inactivating NLRP3 inflammasome. Oncotarget 2017; 8:58903–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Perera AP, Fernando R, Shinde T, Gundamaraju R, Southam B, Sohal SS et al MCC950, a specific small molecule inhibitor of NLRP3 inflammasome attenuates colonic inflammation in spontaneous colitis mice. Sci Rep 2018; 8:8618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Yao X, Zhang C, Xing Y, Xue G, Zhang Q, Pan F et al Remodelling of the gut microbiota by hyperactive NLRP3 induces regulatory T cells to maintain homeostasis. Nat Commun 2017; 8:1896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Błażejewski AJ, Thiemann S, Schenk A, Pils MC, Gálvez EJC, Roy U et al Microbiota normalization reveals that canonical caspase‐1 activation exacerbates chemically induced intestinal inflammation. Cell Rep 2017; 19:2319–30. [DOI] [PubMed] [Google Scholar]
- 73. Nowarski R, Jackson R, Gagliani N, de Zoete MR, Palm NW, Bailis W et al Epithelial IL‐18 equilibrium controls barrier function in colitis. Cell 2015; 163:1444–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Hu S, Peng L, Kwak Y‐T, Tekippe EM, Pasare C, Malter JS et al The DNA sensor AIM2 maintains intestinal homeostasis via regulation of epithelial antimicrobial host defense. Cell Rep 2015; 13:1922–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Ratsimandresy RA, Indramohan M, Dorfleutner A, Stehlik C. The AIM2 inflammasome is a central regulator of intestinal homeostasis through the IL‐18/IL‐22/STAT3 pathway. Cell Mol Immunol 2017; 14:127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Michaudel C, Couturier‐Maillard A, Chenuet P, Maillet I, Mura C, Couillin I et al Inflammasome, IL‐1 and inflammation in ozone‐induced lung injury. Am J Clin Exp Immunol 2016; 5:33–40. [PMC free article] [PubMed] [Google Scholar]
- 77. Hillegass JM, Miller J, Macpherson M, Westbom C, Sayan M, Thompson J et al Asbestos and erionite prime and activate the NLRP3 inflammasome that stimulates autocrine cytokine release in human mesothelial cells. Part Fibre Toxicol 2013; 10:39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Allen IC, Scull MA, Moore CB, Holl EK, McElvania‐TeKippe E, Taxman DJ et al The NLRP3 inflammasome mediates in vivo innate immunity to influenza A virus through recognition of viral RNA. Immunity 2009; 30:556–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Hirota JA, Hirota SA, Warner SM, Stefanowicz D, Shaheen F, Beck PL et al The airway epithelium nucleotide‐binding domain and leucine‐rich repeat protein 3 inflammasome is activated by urban particulate matter. J Allergy Clin Immunol 2012; 129:1116–25.e6. [DOI] [PubMed] [Google Scholar]
- 80. Gillette DD, Shah PA, Cremer T, Gavrilin MA, Besecker BY, Sarkar A et al Analysis of human bronchial epithelial cell proinflammatory response to Burkholderia cenocepacia infection: inability to secrete IL‐1β . J Biol Chem 2013; 288:3691–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Bauer RN, Brighton LE, Mueller L, Xiang Z, Rager JE, Fry RC et al Influenza enhances caspase‐1 in bronchial epithelial cells from asthmatics and is associated with pathogenesis. J Allergy Clin Immunol 2012; 130:958–67.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Furugen M, Higa F, Hibiya K, Teruya H, Akamine M, Haranaga S et al Legionella pneumophila infection induces programmed cell death, caspase activation, and release of high‐mobility group box 1 protein in A549 alveolar epithelial cells: inhibition by methyl prednisolone. Respir Res 2008; 9:39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Peeters PM, Perkins TN, Wouters EFM, Mossman BT, Reynaert NL. Silica induces NLRP3 inflammasome activation in human lung epithelial cells. Part Fibre Toxicol 2013; 10:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Wang J, Sahoo M, Lantier L, Warawa J, Cordero H, Deobald K et al Caspase‐11‐dependent pyroptosis of lung epithelial cells protects from melioidosis while caspase‐1 mediates macrophage pyroptosis and production of IL‐18. PLoS Pathog 2018; 14:e1007105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Alexis NE, Lay JC, Zeman KL, Geiser M, Kapp N, Bennett WD. In vivo particle uptake by airway macrophages in healthy volunteers. Am J Respir Cell Mol Biol 2006; 34:305–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Hirota JA, Marchant DJ, Singhera GK, Moheimani F, Dorscheid DR, Carlsten C et al Urban particulate matter increases human airway epithelial cell IL‐1β secretion following scratch wounding and H1N1 influenza A exposure in vitro . Exp Lung Res 2015; 41:353–62. [DOI] [PubMed] [Google Scholar]
- 87. Rusznak C, Mills PR, Devalia JL, Sapsford RJ, Davies RJ, Lozewicz S. Effect of cigarette smoke on the permeability and IL‐1β and sICAM‐1 release from cultured human bronchial epithelial cells of never‐smokers, smokers, and patients with chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol 2000; 23:530–6. [DOI] [PubMed] [Google Scholar]
- 88. Zou Y, Chen X, Liu J, Zhou DB, Kuang X, Xiao J et al Serum IL‐1β and IL‐17 levels in patients with COPD: associations with clinical parameters. Int J Chron Obstruct Pulmon Dis 2017; 12:1247–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Morris GF, Danchuk S, Wang Y, Xu B, Rando RJ, Brody AR et al Cigarette smoke represses the innate immune response to asbestos. Physiol Rep 2015; 3:e12652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Han S, Jerome JA, Gregory AD, Mallampalli RK. Cigarette smoke destabilizes NLRP3 protein by promoting its ubiquitination. Respir Res 2017; 18:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Kousha M, Tadi R, Soubani AO. Pulmonary aspergillosis: a clinical review. Eur Respir Rev 2011; 20:156–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Karki R, Man SM, Malireddi RKS, Gurung P, Vogel P, Lamkanfi M et al Concerted activation of the AIM2 and NLRP3 inflammasomes orchestrates host protection against Aspergillus infection. Cell Host Microbe 2015; 17:357–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Caffrey AK, Lehmann MM, Zickovich JM, Espinosa V, Shepardson KM, Watschke CP et al IL‐1α signaling is critical for leukocyte recruitment after pulmonary Aspergillus fumigatus challenge. PLoS Pathog 2015; 11:e1004625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Huang Y, Hua M, Cui X. Fungal β‐Glucan activates the NLRP3 inflammasome in human bronchial epithelial cells through ROS production. Inflammation 2017; 41:164–73. [DOI] [PubMed] [Google Scholar]
- 95. Saïd‐Sadier N, Padilla E, Langsley G, Ojcius DM. Aspergillus fumigatus stimulates the NLRP3 inflammasome through a pathway requiring ROS production and the Syk tyrosine kinase. PLoS One 2010; 5:e10008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Bostanci N, Emingil G, Saygan B, Turkoglu O, Atilla G, Curtis MA et al Expression and regulation of the NALP3 inflammasome complex in periodontal diseases. Clin Exp Immunol 2009; 157:415–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Zupin L, Navarra CO, Robino A, Bevilacqua L, Di Lenarda R, Gasparini P et al NLRC5 polymorphism is associated with susceptibility to chronic periodontitis. Immunobiology 2017; 222:704–8. [DOI] [PubMed] [Google Scholar]
- 98. Davis BK, Roberts RA, Huang MT, Willingham SB, Conti BJ, Brickey WJ et al Cutting edge: NLRC5‐dependent activation of the inflammasome. J Immunol 2011; 186:1333–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Cui J, Zhu L, Xia X, Wang HY, Legras X, Hong J et al NLRC5 negatively regulates the NF‐κB and type I interferon signaling pathways. Cell 2010; 141:483–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Kuenzel S, Till A, Winkler M, Häsler R, Lipinski S, Jung S et al The nucleotide‐binding oligomerization domain‐like receptor NLRC5 is involved in IFN‐dependent antiviral immune responses. J Immunol 2010; 184:1990–2000. [DOI] [PubMed] [Google Scholar]
- 101. Meissner TB, Li A, Biswas A, Lee K‐H, Liu Y‐J, Bayir E et al NLR family member NLRC5 is a transcriptional regulator of MHC class I genes. Proc Natl Acad Sci USA 2010; 107:13794–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Yilmaz O, Sater AA, Yao L, Koutouzis T, Pettengill M, Ojcius DM. ATP‐dependent activation of an inflammasome in primary gingival epithelial cells infected by Porphyromonas gingivalis . Cell Microbiol 2010; 12:188–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Khalafalla MG, Woods LT, Camden JM, Khan AA, Limesand KH, Petris MJ et al P2X7 receptor antagonism prevents IL‐1β release from salivary epithelial cells and reduces inflammation in a mouse model of autoimmune exocrinopathy. J Biol Chem 2017; 292:16626–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Qu Y, Franchi L, Nunez G, Dubyak GR. Nonclassical IL‐1β secretion stimulated by P2X7 receptors is dependent on inflammasome activation and correlated with exosome release in murine macrophages. J Immunol 2007; 179:1913–25. [DOI] [PubMed] [Google Scholar]
- 105. Qu Y, Ramachandra L, Mohr S, Franchi L, Harding CV, Nunez G et al P2X7 receptor‐stimulated secretion of MHC class II‐containing exosomes requires the ASC/NLRP3 inflammasome but is independent of caspase‐1. J Immunol 2009; 182:5052–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Borges FT, Melo SA, Özdemir BC, Kato N, Revuelta I, Miller CA et al TGF‐β1‐containing exosomes from injured epithelial cells activate fibroblasts to initiate tissue regenerative responses and fibrosis. J Am Soc Nephrol 2013; 24:385–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Hung S‐C, Huang P‐R, Almeida‐da‐Silva CLC, Atanasova KR, Yilmaz O, Ojcius DM. NLRX1 modulates differentially NLRP3 inflammasome activation and NF‐κB signaling during Fusobacterium nucleatum infection. Microbes Infect 2017; doi: 10.1016/j.micinf.2017.09.014. [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Cheng R, Feng Y, Zhang R, Liu W, Lei L, Hu T. The extent of pyroptosis varies in different stages of apical periodontitis. Biochim Biophys Acta 2018; 1864:226–37. [DOI] [PubMed] [Google Scholar]
- 109. Lee C‐H, Chang JS‐M, Syu S‐H, Wong T‐S, Chan JY‐W, Tang Y‐C et al IL‐1β promotes malignant transformation and tumor aggressiveness in oral cancer. J Cell Physiol 2015; 230:875–84. [DOI] [PubMed] [Google Scholar]
- 110. Li H, Zhou X, Zhang J. Induction of heme oxygenase‐1 attenuates lipopolysaccharide‐induced inflammasome activation in human gingival epithelial cells. Int J Mol Med 2014; 34:1039–44. [DOI] [PubMed] [Google Scholar]
- 111. Hoffman HM, Mueller JL, Broide DH, Wanderer AA, Kolodner RD. Mutation of a new gene encoding a putative pyrin‐like protein causes familial cold autoinflammatory syndrome and Muckle–Wells syndrome. Nat Genet 2001; 29:301–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Ramos E, Aróstegui JI, Campuzano S, Rius J, Bousoño C, Yagüe J. Positive clinical and biochemical responses to anakinra in a 3‐yr‐old patient with cryopyrin‐associated periodic syndrome (CAPS). Rheumatology (Oxford) 2005; 44:1072–3. [DOI] [PubMed] [Google Scholar]
- 113. Jin Y, Mailloux CM, Gowan K, Riccardi SL, LaBerge G, Bennett DC et al NALP1 in vitiligo‐associated multiple autoimmune disease. N Engl J Med 2007; 356:1216–25. [DOI] [PubMed] [Google Scholar]
- 114. Levandowski CB, Mailloux CM, Ferrara TM, Gowan K, Ben S, Jin Y et al NLRP1 haplotypes associated with vitiligo and autoimmunity increase interleukin‐1β processing via the NLRP1 inflammasome. Proc Natl Acad Sci USA 2013; 110:2952–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Kim HJ, Kim SH, Park J, Lee M, Kim DS, Lee M‐G. Up‐regulation of receptor antagonist interleukin‐1 family members in psoriasis and their regulation by pro‐inflammatory cytokines. J Dermatol Sci 2016; 82:204–6. [DOI] [PubMed] [Google Scholar]
- 116. Dombrowski Y, Peric M, Koglin S, Kammerbauer C, Göß C, Anz D et al Cytosolic DNA triggers inflammasome activation in keratinocytes in psoriatic lesions. Sci Transl Med 2011; 3:82ra38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Johansen C, Moeller K, Kragballe K, Iversen L. The activity of caspase‐1 is increased in lesional psoriatic epidermis. J Invest Dermatol 2007; 127:2857–64. [DOI] [PubMed] [Google Scholar]
- 118. Novak N, Kruse S, Potreck J, Maintz L, Jenneck C, Weidinger S et al Single nucleotide polymorphisms of the IL18 gene are associated with atopic eczema. J Allergy Clin Immunol 2005; 115:828–33. [DOI] [PubMed] [Google Scholar]
- 119. Yang J, Zhao Y, Shi J, Shao F. Human NAIP and mouse NAIP1 recognize bacterial type III secretion needle protein for inflammasome activation. Proc Natl Acad Sci USA 2013; 110:14408–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Gaidt MM, Ebert TS, Chauhan D, Ramshorn K, Pinci F, Zuber S et al The DNA inflammasome in human myeloid cells is initiated by a STING‐cell death program upstream of NLRP3. Cell 2017; 171:1110–24.e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Dick MS, Sborgi L, Rühl S, Hiller S, Broz P. ASC filament formation serves as a signal amplification mechanism for inflammasomes. Nat Commun 2016; 7:11929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Coll RC, Robertson AAB, Chae JJ, Higgins SC, Muñoz‐Planillo R, Inserra MC et al A small‐molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat Med 2015; 21:248–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Baldwin AG, Rivers‐Auty J, Daniels MJD, White CS, Schwalbe CH, Schilling T et al Boron‐based inhibitors of the NLRP3 inflammasome. Cell Chem Biol 2017; 24:1321–35.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]