

Summary
Microglia and non‐professional immune cells (endothelial cells, neurons) participate in the recognition and removal of pathogens and tissue debris in the injured central nervous system through major pro‐inflammatory processes. However, the mechanisms involved in regulating these responses remain ill‐characterized. We herein show that CD93, also known as complement C1qRp/AA4 stem cell marker, has an important role in the regulation of inflammatory processes. The role of CD93 was evaluated in two models of neuroinflammation. We used the MOG‐experimental autoimmune encephalomyelitis (EAE) model and the antibody‐dependent EAE (ADEAE), which were induced in wild‐type and CD93 knockout mice. We found that CD93 was highly expressed by neurons, endothelial cells and microglia (ramified >> amoeboid). Astrocytes and oligodendrocytes did not to express CD93. We further observed that CD93‐deficient (CD93−/−) mice presented a more robust brain and spinal cord inflammation in EAE and ADEAE. Encephalitis in CD93−/− was characterized by increased numbers of infiltrating M1 macrophages (CD11c+ CD206−) and amoeboid microglia exhibiting a more activated phenotype (Tomato Lectinhigh Cox2high). Damage to and leakage through the blood–brain barrier was increased in CD93−/− animals and was associated with a more robust neuronal injury when compared with wild‐type EAE mice. We propose that CD93 is an important neuro‐immune regulator to control central nervous system inflammation.
Keywords: CD93, complement, innate immunity, multiple sclerosis, neuro‐immune regulator, neuroinflammation, neurons
Introduction
The perpetuation of an uncontrolled innate immune response in the central nervous system (CNS) is an inherent risk because the subsequent local inflammation may cause collateral tissue damage with necrosis, possibly leading to autoimmunity.1 Hence, non‐resolving inflammation is a major driver of chronic diseases including Alzheimer's disease as well as multiple sclerosis, as exemplified in several experimental autoimmune encephalomyelitis (EAE) mouse models.2, 3
Innate immune complement proteins and other soluble pattern recognition receptors have the ability to recognize equally pathogen‐associated molecular patterns and host‐derived damage‐associated molecular patterns (DAMPs or alarmins).4, 5 It is critical that this initial recognition event signals appropriate effector‐function responses with the phagocytosis of the debris but in a non‐phlogistic manner.6
Several years ago, in an effort to identify markers of differentiation events in the murine haematopoietic cell lineages, a monoclonal antibody, clone AA4, was raised against a cell surface antigen.7 The target amino acid sequence deduced from the cDNA predicted a type 1 glycosylated transmembrane protein with distinctive structural motifs including a C‐type lectin‐like domain (CTLD) and a series of five epidermal growth factor‐like domains.8 The domain composition and organization of the mouse homologue AA4 are identical to those of complement C1qRp (now renamed CD93 for both molecules) and bear a striking resemblance to those of thrombomodulin (CD141)9 and endosialin (CD248, also known as tumour endothelial marker number one, TEM1).10 All three proteins are now grouped in a family known as the group XIV CTLD family.11 More recently, the CTLD group XIV member A (also known as Clec14A) was identified based upon nucleotide sequences showing high homology to CD93, CD248 and CD141.12
Of critical note, the N‐terminal CTLD part of CD141 has potent anti‐inflammatory activity. This domain was shown to orchestrate the suppression of endothelial and neutrophil cell activation through mitogen‐activated protein kinase and nuclear factor‐κB pathways.13 It has also been demonstrated that CD141 can sequester and limit the pro‐inflammatory alarmin HMGB1 (high‐mobility group‐B1 DNA‐binding protein).14
It has now been proposed that CD93 has functions beyond those of a myeloid and stem cell marker.15 Its expression on myeloid cells (including microglia) is consistent with a role in the phagocytic response,16 but, in view of its expression in non‐immune cells (endothelial cells, platelets and neurons) and its domain composition, it has been proposed that CD93 may be a critical regulator of inflammation and tissue remodelling.17
The protective anti‐inflammatory role of CD93 has been evidenced previously in two different models of chronic inflammation, thioglycollate‐induced peritonitis and CNS stroke, but whether CD93 could also control adaptive immunity in the CNS remains to be characterized.18, 19
We set out to specifically address the role of CD93 in autoimmune inflammatory responses in the CNS using the EAE and antibody‐dependent (AD) EAE models in wild‐type (WT) and CD93 knockout (KO) animals. Our results demonstrate for the first time that CD93 has a broad role in the control of brain autoimmunity and inflammation leading otherwise to neuronal injuries. We believe that CD93 behaves as a key neuroimmune regulatory protein in a manner reminiscent of CD200, fractalkine, intercellular adhesion molecule‐5 and complement factor‐H.20, 21, 22, 23
Materials and methods
Animals
CD93−/− (backcrossed onto C57BL/6) mice were as described elsewhere.16 Age‐, strain‐ and sex‐matched mice were used as controls in each experiment. Animals were maintained in specific pathogen‐free conditions. All animal procedures were in accordance with institutional and UK Home Office guidelines. Ten‐ to twelve‐week‐old mice were used in all experiments, with 8–11 mice in each experimental condition.
Induction of CNS inflammation (EAE and ADEAE)
For EAE, on day 0, mice were immunized subcutaneously at a single site close to the base of the tail with 200 μl of an emulsion consisting of a 1 : 1 mixture of complete Freund's adjuvant (CFA) and phosphate‐buffered saline (PBS) containing 2·5 mg/ml Mycobacterium tuberculosis H37 Ra and 0·5 mg/ml recombinant mouse MOG, consisting of amino acids 1–117 of the extracellular immunoglobulin domain (rMOG; kindly provided by Dr Hugh Reid, Monash University, Australia). CFA and M. tuberculosis H37 Ra (MtbH37) were from Difco (Epsom, UK). Mice also received 200 ng pertussis toxin (Sigma, Poole, UK) intraperitoneally in PBS on days 0 and 2. On day 11, ADEAE animals additionally received an intraperitoneal injection of Z12 complement activating anti‐MOG monoclonal antibody (0·5 mg in PBS). Mice were weighed daily and monitored for signs of clinical disease, which was scored as follows: 0, no disease; 1, tail atony; 2, hind‐limb weakness; 3, hind‐limb paralysis; 4, moribund; 5, dead. Animals that attained a clinical score of 3 or 4 at monitoring were killed immediately to conform to Home Office guidelines.
Tissue processing and analysis
Animals were killed while under terminal anaesthesia and the brains and spinal cords were removed. The brain was cut sagitally to divide the two hemispheres and the spinal cord was quartered approximating to the cervical, thoracic, lumbar and sacral regions. One half of the brain and the cervical and lumbar spinal cord sections were immediately frozen in isopentane on dry ice. The remaining CNS tissue was post‐fixed in 4% paraformaldehyde in PBS for paraffin wax embedding and microtome sectioning.
Analysis and quantification of inflammation and demyelination were performed by an independent observer as described previously.24 Paraffin sections (6 µm) were cut and stained with haematoxylin & eosin for analysis of inflammation. Inflammatory cells in subpial and perivascular areas were separately scored on arbitrary scales. Neuronal injury was further evaluated by counting the number of immune cells attracted by and in close contact with dying (necrotic) pyramidal neurons in the anterior horn of the spinal cord. Demyelination was assessed for each experimental group in luxol fast blue/cresyl violet‐stained sections; the area (in mm2) of myelin loss and total white matter area were measured using a graticule and the percentage demyelination was reported on a four‐point scale from zero through to 4 (0, no demyelination; 2, perivascular demyelination; 4, extensive perivascular and subpial demyelination with formation of confluent plaques).
Immunohistochemical staining and analyses of tissue sections for neuroinflammation
All primary antibodies used were rat anti‐mouse antigens from Serotec (Oxford UK) unless otherwise stated. The rat monoclonal antibodies used were against CD3 (MCA500GA, T cells), CD4 (MCA1767GA, T cells), CD8α (MCA609GA, T cells), CD11b (BD Pharmingen, Franklin Lakes, NJ, USA; MAC 1 Integrin α M chain, macrophages/microglia), CD18 (MCA1032G, macrophages/microglia), CD19 (MCA1439GA, B lymphocytes), CD45 (BD Pharmingen; Ly5, leucocyte common antigen), CD68 (MCA1957), F4/80 (MCA497) both for activated macrophages/amoeboid microglia, and anti CD34 (MCA1825GA, endothelial cells, microglia progenitors). The 5D3 rat anti‐mouse macrophage mannose receptor CD206 was used to stain for CNS M2 macrophages/microglia (kind gift of Dr Luisa Martinez‐Pomares, Oxford, UK). CD11c/CR4+ macrophages were identified using the hamster antibody (BD 550283). In mouse, M1 parenchymal macrophages were identified as F4/80+, CD18+, CD11c+, CD206−, in contrast to M2 polarized macrophages, which were F4/80+, CD18+, CD11c−, CD206+.25
To screen for BBB damage, we stained for parenchymal deposits of fibrinogen using the goat polyclonal anti‐fibrinogen (AB6666; Abcam, Cambridge, UK). To further ascertain the phenotypic activation (or polarization) of macrophage/microglia we stained tissue sections for nitric oxide synthase‐I (rabbit antibody AB1632; Chemicon, Millipore, Fischer Scientific, Loughbo rough, UK), cyclooxygenase‐2 (Cox2, Rabbit AB5118; Chemicon). Pericytes were identified as NG2+ perivascular cells (Rabbit 5320; Chemicon).
Biotinylated tomato lectin (TL) (Lycopersicon esculentum; Vector Labs, Burlingame, CA) was used to label macrophages/microglia of different functional activation stages and morphologies. TL has high affinity for poly‐N acetyl lactosamine residues highly expressed by reactive macrophages/microglia.26, 27 Other antibodies used to label sections included rabbit anti‐mouse CD93 against full‐length fusion protein.28 Immunocytochemical staining was performed for C3 (rabbit anti‐human C3c, strongly cross‐reactive with mouse C3, A0062; Dako, Glostrup, Denmark) and rabbit anti‐human C1q, cross‐reacts with mouse C1q (Behring, Haywards Heath, UK), for analysis of complement deposition. Neurons were labelled using NeuN (Chemicon International, MAB377, Millipore, Fischer Scientific, Loughbo rough, UK). Single‐strand DNA monoclonal antibody (Chemicon International; MAB3299) was used to detect late apoptosis and staining of apoptotic blebs.29 Due to interference by monoclonal Z12 antibody, used to induce ADEAE pathology, the ssDNA monoclonal antibody was biotin‐labelled (NHS‐biotin, B2643; Sigma UK) to quantify apoptosis in ADEAE sections.
For all immunofluorescence, fresh frozen sections were left to air dry for 1 hr before fixation in acetone chilled to −20° for 5 min. Sections were then allowed to dry before marking with a pap pen and washing in PBS and blocking in PBS/bovine serum albumin for 30 min. Sections were then incubated with the relevant primary antibody for 2 hr at room temperature using the manufacturer's recommended dilutions, before further repeated washes and blocking, and then incubating in secondary antibody, which was either conjugated to fluorescein isothiocyanate or tetramethylrhodamine, at 1 : 200 dilution. Sections were simultaneously incubated with 4′,6‐diamidino‐2‐phenylindole at 1 : 1000 to stain nuclei. Following an overnight incubation, sections were washed and mounted with Vectorshield (H‐1000; Vector Labs). In the case of TL, which was biotinylated, following the washes and blocking step after the primary antibody incubation, sections were incubated with Streptavidin Alexa 647 (1 : 1000).
The numbers of apoptotic cells were counted in ADEAE sections following incubation with the biotinylated ssDNA antibody. Sections were washed, then incubated with the Vector ABC elite peroxidase kit (PK‐6100). The signal was developed using AEC as the chromogen (SK‐4200) according to the manufacturer's instructions. Sections were then washed in running water, and lightly counterstained with haematoxylin, before rinsing in running tap water and coverslipping with Faramount aqueous mounting medium (Dako Cytomation, S3025).
The degree of staining in immunocytochemistry was quantified using the openlab 3.5 image analysis system (Improvision, Coventry, UK). In order to quantify CD68‐positive cells and C3/C1q deposition, density slices were taken from the same regions of white and grey matter in all sections, and the degree of staining was measured, with reference to fixed background cut‐off values. Multiple fields in three sections were analysed for each animal and a mean value of relative area stained, corrected for secondary control labelling, was generated. To count the number of apoptotic cells, the entire section was density sliced and positively labelled cells were counted.
Statistical analysis
All comparisons of non‐parametric data (clinical score and demyelination) were performed using Mann–Whitney non‐parametric U‐tests; the exact two‐tailed P‐value corrected for ties is quoted throughout. Student's t‐test was applied to parametric data (inflammation) and Fisher's exact test was used for comparison of disease incidence. Two‐tailed P‐values are quoted throughout.
Results
CD93 is expressed constitutively on microglia, endothelial cells and neurons in mouse brain
In the CD93‐sufficient naive mouse, vascular endothelial cells stained for CD34 were strongly positive for CD93 throughout the brain. Ramified CD11b‐positive microglia also expressed high levels of CD93 (Fig. 1a). Expression of CD93 was also observed on neurons double stained for NeuN in the hippocampus. CD93 was present at the neuronal cell membrane but also in cytoplasmic vesicles, (Fig. 1a(vi), white arrow). In contrast, endothelial CD93 staining was solely at the cell membrane (Fig. 1a(vi), arrowhead). CD93‐deficient mice showed no specific staining with our in‐house affinity‐purified anti‐CD93 antibody (Fig. 1c(iii)).
Figure 1.
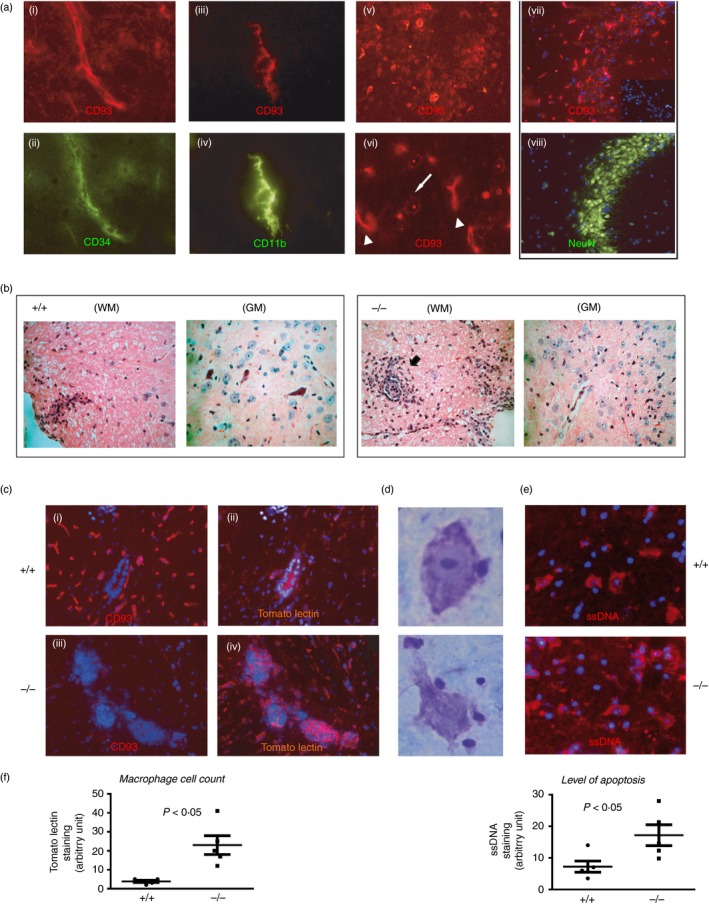
Lack of CD93 confers increased bystander neuronal injury and inflammation in the spinal cord of experimental autoimmune encephalomyelitis (EAE) mice. (a) In a naive C57BL/6 mouse brain, CD93 was expressed on endothelial cells (CD34+), ramified microglia (CD11b+) and neurons (NeuN+ cells). CD93 was also localized in cytoplasmic vesicles in neurons (white arrow in (vi)) compared with a more robust membrane‐restricted CD93 staining in endothelium (arrowhead in (vi)). Representative data obtained from tissue sections from the brains of two animals. (b) Histological haematoxylin analysis of semi‐serial paraffin sections from the spinal cord of representative CD93+/+ and CD93−/− mice after EAE. Cellular infiltrates were prominent in white matter (WM) lesions of CD93+/+ mice whereas these infiltrates where more numerous and localized at the border between the WM and the grey matter (GM) in CD93−/− mice. (c) Immunostaining of EAE (days 15–18) spinal cords. Tomato lectin (TL) identified the infiltrating and reactive macrophages/microglia in CD93+/+ (b) in response to EAE and these cells were more numerous and aggregated at the border between the WM/GM in CD93−/− spinal cords following EAE (d). (d, e) Necrotic‐like Nissl+ neurons (surrounded by many lymphocyte‐like cells) were more numerous in CD93−/− sections and may correspond to cells in late apoptosis as detected by the anti‐ssDNA antibody (e). (f). Histograms: image and statistical analyses were performed on five sections of three different mice. TL stain was used as an indication of macrophage infiltration and ssDNA staining as a marker of apoptosis. Panels (ii–vi), representative data obtained from tissue sections from three different brains from either wild‐type (WT) or knockout (KO) animals of comparable clinical scores.
More abundant immune infiltrating cells and more robust apoptosis in CD93−/− compared to WT
Experimental autoimmune encephalomyelitis was induced by the injection of recombinant MOG + CFA; EAE animals were killed on days 15–17 unless previously euthanized because of the severity of disease. Spinal cord was taken from all mice and sectioned for staining. The clinical score and weight loss were evaluated during the course of the disease and only the latter was more significantly impacted in CD93−/− mice when compared with WT animals (see Supplementary material, Figure S1a,b). The degree of inflammatory infiltrate was semi‐quantitatively assessed in haematoxylin & eosin‐stained cervical cord sections by observers blinded to the sample identity and was judged as significantly more pronounced in KO animal white matter (WM) when compared with WT animals (Fig. 1b; P = 0·021). LFV/CV staining of the spinal cord sections revealed that the level of demyelination was more pronounced in the KO animals but did not reach statistical significance (P = 0·058; see Supplementary material, Figure S1c). Perivascular cuffing of immune cells at the border between the WM and the grey matter (GM) of the spinal cord was more prominent in CD93−/− mice when compared with WT (Fig. 1b, WM picture). Compared with WT, perivascular cuffing in CD93−/− mice contained more reactive macrophages (TL+ cells, P = 0·032, Fig. 1c) which were also stained for CD68 (not shown).
Experimental autoimmune encephalomyelitis has long been known to be accompanied by neuronal injury possibly caused by cytotoxic cytokines and reactive macrophages and T cells.30 In the GM photomicrographs (haematoxylin & eosin stain, Fig. 1b) of CD93−/− mice, we observed more dying neurons with pyknotic nuclei when compared with viable neurons with pale nuclei in WT mice. We found that these injured neurons (Nissl+) were surrounded by many lymphocyte‐like cells (Fig. 1d). Numbers of late apoptotic neuron‐like cells (stained for ssDNA) were increased in CD93−/− GM (P = 0·047; Fig. 1). These data reinforced the notion that CD93 is involved in the control of tissue inflammation in response to brain autoimmunity.
CD93 controls BBB integrity and the polarization of infiltrating macrophages during CNS inflammation
The elevated recruitment of immune cells into the brain parenchyma may be facilitated by a more severely disrupted BBB. Indeed, we found that fibrinogen transudation into the brain parenchyma was more prominent in the WM of CD93−/− compared with WT mice (Fig. 2a,b). Fibrinogen deposits were also more abundant in the GM of CD93−/− animals (Fig 2a and data analysis Fig 2c).
Figure 2.
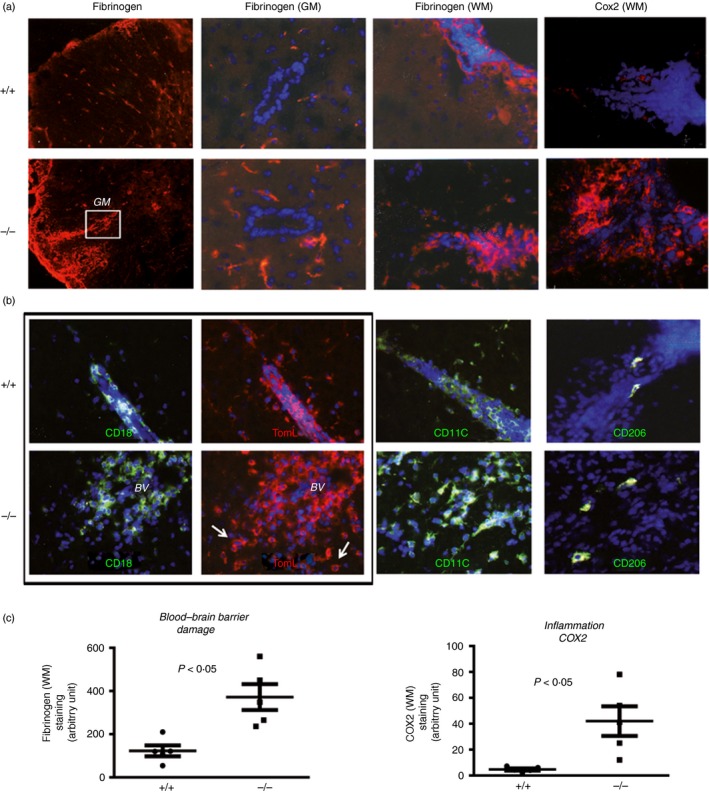
Absence of CD93 exacerbates blood–brain barrier (BBB) damage and accumulation of polarized M1 macrophages (Cox2+, TL+, CD11c+ and CD206−) in experimental autoimmune encephalomyelitis (EAE). (a) Fibrinogen staining in the parenchyma (white or grey matters, WM/GM) was used as an indicator of BBB disruption and allowing transudation of other neurotoxic serum‐derived molecules (thrombin, complement). Robust Cox2 staining in perivascular inflammatory cells was found in CD93−/− EAE mice compared with discrete staining in wild‐type (WT) mice. (b) Double (CD18/TL) and single (CD11c or CD206) immunostaining was used to identify cytotoxic/pro‐inflammatory M1 versus regulatory/anti‐inflammatory M2 macrophage/microglia in EAE WT or CD93 mice. (c) Quantification of staining and image analysis were performed on five sections from three mice. Representative data obtained from tissue sections from three different brains from either WT or knockout (KO) animals with comparable clinical scores.
We could not detect inducible nitric oxide synthase expression during the early time‐points of our EAE model but found that Cox2, in contrast, was expressed by infiltrating myeloid cells (Fig. 2a). More importantly, Cox2+ infiltrating immune cells were significantly more numerous in CD93−/− EAE when compared with WT EAE mice (Fig. 2a, and data analysis in Fig. 2c).
We further analysed the phenotype of infiltrating immune cells as described previously25 and found that M1‐like macrophages (double‐stained CD18+, TLhigh perivascular cells, Fig. 2b) were more abundant in CD93−/− mice, when compared with EAE‐induced WT animals. CD11c+ M1‐like macrophages were also more abundant in CD93−/− when compared with WT (Fig. 2b). CD206+ M2 cells were detected only sparsely in EAE and equally when we compared the distribution in WT and KO animals.
We also addressed the role of CD93 in ADEAE mice. We did not find any significant statistical differences between the WT and KO animals when we compared the clinical scores and weight loss (data not shown). However, in CD93−/− sections, we found increased infiltration of TL+ immune cells associated with marked deposition of C1q at the sites of deposits of the Z12 anti‐MOG antibody (Fig. 3). C1q and Z12 stainings in CD93−/− mice were more prominent around blood vessels, a possible consequence of a damaged BBB. These vascular areas contained more infiltrating cells that were strongly positive for TL (Fig. 3b) in CD93−/− ADEAE mice when compared with WT ADEAE animals. Single‐stranded DNA antibody staining of sections was used to measure late apoptosis and DNA‐enriched vesicles released by necrotic cells. The number of late apoptotic cells imaged in the spinal cord sections of ADEAE CD93−/− mice was significantly higher compared with ADEAE CD93‐sufficient mice (17·37 ± 1·77 versus 11·64 ± 0·53, P = 0·02, Table 1). In contrast, the levels of CD68 staining in WM and GM of both types of mice were not significantly different. The ADEAE model is a more acute model because of the robust complement activation triggered by exogenous Z12 anti‐MOG antibody binding to myelin sheaths. Staining with the C3 antibody in ADEAE demonstrated increased complement activation in WM of CD93‐deficient when compared with sufficient mice (Table 1, P = 0·029).
Figure 3.
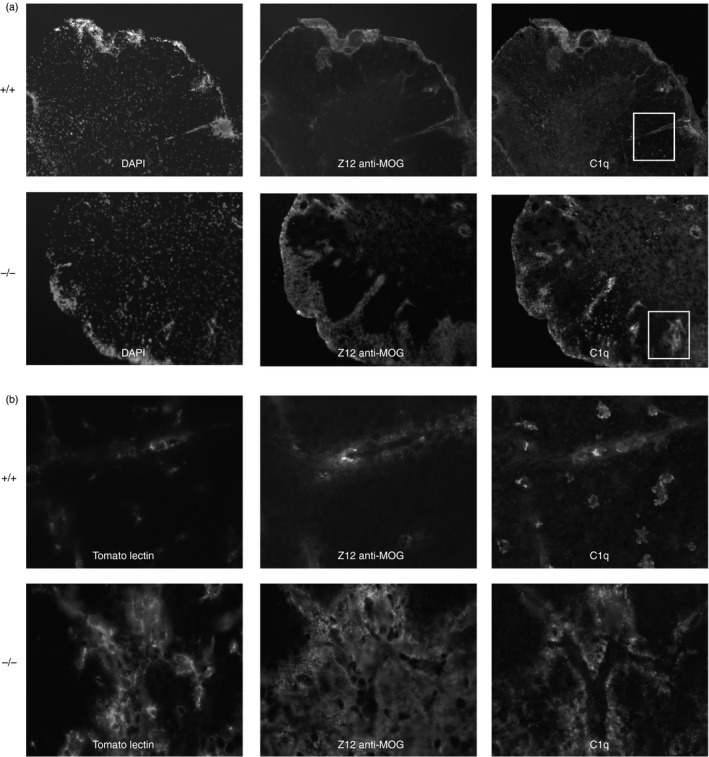
Increased complement activation and macrophage activation in the spinal cords of CD93‐deficient mice treated for antibody‐dependent autoimmune encephalomyelitis (ADEAE). (a) The ADEAE is more severe and acute than EAE and through the capacity of the Z12 monoclonal antibody drives a robust activation of the complement system. We stained for C3 (see Table 1) and for C1q and found a more robust complement activation/opsonization in CD93−/− mice when compared with wild‐type (WT). Triple immunostaining for Z12, tomato lectin (TL) and C1q. Magnification at 200 × in top six panels, and × 600 for bottom six panels depicting the inset areas (b). Panels (a, b) representative data obtained from tissue sections from three different brains from either WT or knockout (KO) animals of comparable clinical scores.
Table 1.
Immunocytochemical parameters in antibody‐dependent autoimmune encephalomyelitis (ADEAE) in CD93−/− mice
| Apoptosis | C3 staining | CD68 staining | |||
|---|---|---|---|---|---|
| White matter | Grey matter | White matter | Grey matter | ||
| ADEAE CD93−/− | 17·36 ± 1·77* | 38·64 ± 2·94** | 25·42 ± 3·06 | 27·37 ± 1·9 | 20·34 ± 1·4 |
| ADEAE CD93+/+ | 11·64 ± 0·53 | 30·9 ± 1·52 | 24·12 ± 1·38 | 27·23 ± 1·2 | 19·83 ± 0·83 |
All values are mean ± SD. Statistical analysis revealed significant differences between CD93‐deficient and CD93‐sufficient groups for the following listed parameters.
*P = 0·02, ADEAE CD93−/− versus CD93+/+.
**P = 0·029, ADEAE CD93−/− white matter versus CD93+/+ white matter.
Discussion
The essence of the innate immune system in the CNS is not only to act as a defence mechanism against pathogens but also to mediate normal physiological processes. Innate immunity can contribute to tissue remodelling after damage and during development, transport of blood lipids and scavenging of apoptotic/necrotic cells and toxic entities such as amyloid fibrils.31, 32 There is a growing list of DAMPs/alarmins derived from the host such as the HMGB1, hyaluronan, heat‐shock proteins and β−A4 peptide, many of which will signal through the same receptors as those used for the recognition of pathogens.33 In contrast to the great wealth of information about the receptors (e.g. Toll‐like receptors 3, 4, 7, 9, retinoic acid inducible gene / melanoma differentiation‐associated protein 5, nucleotide oligomerization domain, multiple lectins) involved in the activation of innate immunity against pathogen‐associated molecular patterns and DAMPs, little is known about the regulatory and protective innate immune mechanisms involved in the resolution of inflammation.33, 34
C1qRp/CD93 was first characterized by Nepomuceno et al. in 1997 and Petrenko et al. in 1999 focusing on the receptor involved in myeloid phagocytosis and on the identification of a haematopoietic stem cell marker, respectively.8, 35 Our new data further indicate that CD93 had a broad and ubiquitous expression in the brain and may be involved in the control of innate and adaptive immunity in the CNS.
First, CD93 was found on endothelial cells. Endothelial cells (as confirmed by CD34 and FVIII stainings) but not perivascular pericytes (CD248+ NG2+) were shown to express high levels of CD93. CD93−/− mice failed to maintain the integrity of the BBB, leading to the accumulation of the neurotoxic fibrinogen in the spinal cord parenchyma. These data are in agreement with the observation that the lack of vascular integrity in a model of peritoneal inflammation was more prominent in CD93−/− versus WT mice.19 The BBB was more impaired at the level of both WM and GM of the spinal cord, and this may have contributed to the massive influx of leucocytes as observed in CD93−/− tissues.
Second, the notable expression of CD93 on neurons confirmed the previous observations that a subset of pyramidal neurons in human brain tissue sections expressed CD93.36 Strong expression of mouse CD93 was particularly prominent in hippocampus regions and this was confirmed by immunocytochemical and in situ hybridization stainings (data not shown). We have noted that some of the CD93+ hippocampal cells were also ki67+ and doublecortin+ and may correspond to neuronal stem cells. However, neuronal stem cells in one of the other most prominent stem cell niches in adult brain (i.e. the subventricular zone) were not stained for CD93 (data not shown). CD93 staining was localized at the cell membrane but was also located in vesicles, which may represent an intracellular pool to be mobilized in inflammatory settings.
Third, we found that microglia (ramified) and infiltrating monocytes/macrophages expressed CD93. More importantly, we also found that the absence of CD93 (KO mice data) led to a polarization of myeloid cells towards an M1‐like phenotype (CD18+, CD11c+, CD206−, TLhigh) associated with high pro‐inflammatory (Cox2high) properties. Interestingly, differential expression of mouse CD93 on monocyte subsets has been described by Zhu et al.37 The authors reported a unique population of CD11b+, Ly‐6Chigh, F4/80+, arginase 1+ and CD93high cells, which were highly immunosuppressive for activated CD4+ and CD8+ mouse T cells.37 These Ly‐6Chigh CD93 high monocytes have been shown to induce T‐cell apoptosis and to protect against EAE. It could be hypothesized that CD93 contribute to the immunosuppressive functions of this unique subset of monocytes in the brain and that its deficiency may facilitate T‐cell aggression in EAE.
In humans, others have found that high expression of CD93 on monocytes clustered within M2 myeloid cell subsets (CD1a+, CD1b+, DNAM‐1/CD226+) that also express higher levels of CCL22, CCL18, and lectin DCIR (Clec4e) when compared with M1 pro‐inflammatory monocytes.38
Taking into consideration these published phenotyping data and our new data, we can hypothesize that the loss of CD93 (as in KO animals) may fail to control monocyte activation and may grant a full activation spectrum towards a more aggressive innate and adaptive immune cell reaction involved in neurocytopathic activities.
Another indication of a more robust CNS inflammation in our CD93−/− EAE mouse model was revealed by the level of complement activation (C3 and C1q deposition) in the brain parenchyma. There was a statistically significant increase in C3 (and C1q) deposition in the WM of CD93‐deficient mice with EAE and ADEAE when compared with WT mice. As C3 and C1q are mainly present in blood, elevated levels in the WM of CD93‐deficient mice might also suggest a leakier BBB. We also found elevated levels of apoptotic cells in CD93‐deficient compared with WT EAE and ADEAE mice. Although this may reflect a ‘failure of clearance’ of apoptotic cells due to the absence of CD93 as described previously,16 it may also indicate elevated levels of apoptosis exerted against brain cells by recruited innate and adaptive immune cells.
Overall, we have demonstrated that CD93 controls the severity of inflammation, apoptosis and bystander neuronal injury in EAE/ADEAE and, hence, the resolution of the autoimmune process. Experiments are now warranted to ascertain the pathways that may be mobilized specifically through CD93‐dependent protective functions and the relative importance of the different cellular contributions.
Disclosures
The authors declare that they have no financial and commercial conflicts of interest.
Supporting information
Figure S1. (a) Weight loss of mice over the time course of MOG‐EAE (P = 0·085). (b) Clinical score of the animals (P = 0·24). (c) Luxol fast blue/cresyl violet stain to assess the level of demyelination in the spinal cord.
Acknowledgements
Fundings were secured from grants to PG from the MRC‐UK (G120‐188). The authors thank Dr Hugh Reid, Monash University, Australia who kindly provided rMOG.
References
- 1. Nathan C, Ding A. Nonresolving inflammation. Cell 2010; 140:871–82. [DOI] [PubMed] [Google Scholar]
- 2. Ransohoff RM, Brown MA. Innate immunity in the central nervous system. J Clin Invest. 2012; 122:1164–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Stephenson J, Nutma E, van der Valk P, Amor S. Inflammation in CNS neurodegenerative diseases. Immunology 2018; 154:204–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Akira S, Takeda K. Toll‐like receptor signalling. Nat Rev Immunol 2004; 4:499–511. [DOI] [PubMed] [Google Scholar]
- 5. Gasque P. Complement: a unique innate immune sensor for danger signals. Mol Immunol 2004; 41:1089–98. [DOI] [PubMed] [Google Scholar]
- 6. Savill J, Fadok V. Corpse clearance defines the meaning of cell death. Nature 2000; 407:784–8. [DOI] [PubMed] [Google Scholar]
- 7. McKearn JP, Baum C, Davie JM. Cell surface antigens expressed by subsets of pre‐B cells and B cells. J Immunol 1984; 132:332–9. [PubMed] [Google Scholar]
- 8. Petrenko O, Beavis A, Klaine M, Kittappa R, Godin I, Lemischka IR. The molecular characterization of the fetal stem cell marker AA4. Immunity 1999; 10:691–700. [DOI] [PubMed] [Google Scholar]
- 9. Van de Wouwer M, Conway EM. Novel functions of thrombomodulin in inflammation. Crit Care Med 2004; 32:S254–61. [DOI] [PubMed] [Google Scholar]
- 10. Rettig WJ, Garin‐Chesa P, Healey JH, Su SL, Jaffe EA, Old LJ. Identification of endosialin, a cell surface glycoprotein of vascular endothelial cells in human cancer. Proc Natl Acad Sci USA 1992; 89:10832–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Drickamer K. C‐type lectin‐like domains. Curr Opin Struct Biol 1999; 9:585–90. [DOI] [PubMed] [Google Scholar]
- 12. Rho S‐S, Choi H‐J, Min J‐K, Lee H‐W, Park H, Park H et al Clec14a is specifically expressed in endothelial cells and mediates cell to cell adhesion. Biochem Biophys Res Commun 2011; 404:103–8. [DOI] [PubMed] [Google Scholar]
- 13. Conway EM, Van de Wouwer M, Pollefeyt S, Jurk K, Van Aken H, De Vriese A et al The lectin‐like domain of thrombomodulin confers protection from neutrophil‐mediated tissue damage by suppressing adhesion molecule expression via nuclear factor κB and mitogen‐activated protein kinase pathways. J Exp Med 2002; 196:565–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Abeyama K, Stern DM, Ito Y, Kawahara K, Yoshimoto Y, Tanaka M et al The N‐terminal domain of thrombomodulin sequesters high‐mobility group‐B1 protein, a novel antiinflammatory mechanism. J Clin Invest 2005; 115:1267–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Greenlee‐Wacker MC, Galvan MD, Bohlson SS. CD93: recent advances and implications in disease. Curr Drug Targets 2012; 13:411–20. [DOI] [PubMed] [Google Scholar]
- 16. Norsworthy PJ, Fossati‐Jimack L, Cortes‐Hernandez J, Taylor PR, Bygrave AE, Thompson RD et al Murine CD93 (C1qRp) contributes to the removal of apoptotic cells in vivo but is not required for C1q‐mediated enhancement of phagocytosis. J Immunol 2004; 172:3406–14. [DOI] [PubMed] [Google Scholar]
- 17. McGreal E, Gasque P. Structure–function studies of the receptors for complement C1q. Biochem Soc Trans 2002; 30:1010–4. [DOI] [PubMed] [Google Scholar]
- 18. Harhausen D, Prinz V, Ziegler G, Gertz K, Endres M, Lehrach H et al CD93/AA4.1: a novel regulator of inflammation in murine focal cerebral ischemia. J Immunol 2010; 184:6407–17. [DOI] [PubMed] [Google Scholar]
- 19. Greenlee‐Wacker MC, Briseño C, Galvan M, Moriel G, Velázquez P, Bohlson SS. Membrane‐associated CD93 regulates leukocyte migration and C1q‐hemolytic activity during murine peritonitis. J Immunol 2011; 187:3353–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hoek RM, Ruuls SR, Murphy CA, Wright GJ, Goddard R, Zurawski SM et al Down‐regulation of the macrophage lineage through interaction with OX2 (CD200). Science 2000; 290:1768–71. [DOI] [PubMed] [Google Scholar]
- 21. Cardona AE, Pioro EP, Sasse ME, Kostenko V, Cardona SM, Dijkstra IM et al Control of microglial neurotoxicity by the fractalkine receptor. Nat Neurosci 2006; 9:917–24. [DOI] [PubMed] [Google Scholar]
- 22. Tian L, Lappalainen J, Autero M, Hanninen S, Rauvala H, Gahmberg CG. Shedded neuronal ICAM‐5 suppresses T‐cell activation. Blood 2008; 111:3615–25. [DOI] [PubMed] [Google Scholar]
- 23. Griffiths MR, Neal JW, Fontaine M, Das T, Gasque P. Complement factor H, a marker of self protects against experimental autoimmune encephalomyelitis. J Immunol 2009; 182:4368–77. [DOI] [PubMed] [Google Scholar]
- 24. Mead RJ, Singhrao SK, Neal JW, Lassmann H, Morgan BP. The membrane attack complex of complement causes severe demyelination associated with acute axonal injury. J Immunol 2002; 168:458–65. [DOI] [PubMed] [Google Scholar]
- 25. Lumeng CN, Bodzin JL, Saltiel AR. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J Clin Invest 2007; 117:175–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Acarin L, Vela JM, Gonzalez B, Castellano B. Demonstration of poly‐N‐acetyl lactosamine residues in ameboid and ramified microglial cells in rat brain by tomato lectin binding. J Histochem Cytochem 1994; 42:1033–41. [DOI] [PubMed] [Google Scholar]
- 27. Peng ZC, Kristensson K, Bentivoglio M. Distribution and temporal regulation of the immune response in the rat brain to intracerebroventricular injection of interferon‐γ . Exp Neurol 1998; 154:403–17. [DOI] [PubMed] [Google Scholar]
- 28. Dean YD, McGreal EP, Gasque P. Endothelial cells, megakaryoblasts, platelets and alveolar epithelial cells express abundant levels of the mouse AA4 antigen, a C‐type lectin‐like receptor involved in homing activities and innate immune host defense. Eur J Immunol 2001; 31:1370–81. [DOI] [PubMed] [Google Scholar]
- 29. Elward K, Griffiths M, Mizuno M, Harris CL, Neal JW, Morgan BP et al CD46 plays a key role in tailoring innate immune recognition of apoptotic and necrotic cells. J Biol Chem 2005; 280:36342–54. [DOI] [PubMed] [Google Scholar]
- 30. Bjartmar C, Trapp BD. Axonal and neuronal degeneration in multiple sclerosis: mechanisms and functional consequences. Curr Opin Neurol 2001; 14:271–8. [DOI] [PubMed] [Google Scholar]
- 31. Hauwel M, Furon E, Canova C, Griffiths M, Neal J, Gasque P. Innate (inherent) control of brain infection, brain inflammation and brain repair: the role of microglia, astrocytes, “protective” glial stem cells and stromal ependymal cells. Brain Res Brain Res Rev 2005; 48:220–33. [DOI] [PubMed] [Google Scholar]
- 32. Rivest S. Regulation of innate immune responses in the brain. Nat Rev Immunol 2009; 9:429–39. [DOI] [PubMed] [Google Scholar]
- 33. Hoarau J‐J, Krejbich‐Trotot P, Jaffar‐Bandjee M‐C, Das T, Thon‐Hon G‐V, Kumar S et al Activation and control of CNS innate immune responses in health and diseases: a balancing act finely tuned by neuroimmune regulators (NIReg). CNS Neurol Disord Drug Targets 2011; 10:25–43. [DOI] [PubMed] [Google Scholar]
- 34. Popovich PG, Longbrake EE. Can the immune system be harnessed to repair the CNS? Nat Rev Neurosci 2008; 9:481–93. [DOI] [PubMed] [Google Scholar]
- 35. Nepomuceno RR, Henschen‐Edman AH, Burgess WH, Tenner AJ. cDNA cloning and primary structure analysis of C1qR(P), the human C1q/MBL/SPA receptor that mediates enhanced phagocytosis in vitro . Immunity 1997; 6:119–29. [DOI] [PubMed] [Google Scholar]
- 36. Fonseca MI, Carpenter PM, Park M, Palmarini G, Nelson EL, Tenner AJ. C1qR(P), a myeloid cell receptor in blood, is predominantly expressed on endothelial cells in human tissue. J Leukoc Biol 2001; 70:793–800. [PubMed] [Google Scholar]
- 37. Zhu B, Bando Y, Xiao S, Yang K, Anderson AC, Kuchroo VK et al CD11b+Ly‐6Chi suppressive monocytes in experimental autoimmune encephalomyelitis. J Immunol 2007; 179:5228–37. [DOI] [PubMed] [Google Scholar]
- 38. Beyer M, Mallmann MR, Xue J, Staratschek‐Jox A, Vorholt D, Krebs W et al High‐resolution transcriptome of human macrophages. PLoS ONE 2012; 7:e45466. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. (a) Weight loss of mice over the time course of MOG‐EAE (P = 0·085). (b) Clinical score of the animals (P = 0·24). (c) Luxol fast blue/cresyl violet stain to assess the level of demyelination in the spinal cord.