

Abstract
Lactate dehydrogenase A (LDHA) has appropriately received attention as a therapeutic target for the treatment of a broad spectrum of tumor types, yet little is known regarding intrinsic resistance to LDHA inhibitors. Pathria et al (2018) now establish that ATF4‐dependent control of enzymes that direct amino acid metabolism confers resistance to LDHA inhibitors in melanoma and identify chokepoints that can be exploited to overcome metabolic compensation, setting the stage for trials with such combinations.
Subject Categories: Cancer, Metabolism
Cancer cells exhibit the “Warburg Effect”, a metabolic state characterized by marked increases in glucose consumption and amino acid transport, and by high rates of glycolytic flux despite sufficient oxygen, which together provide metabolic intermediates needed to sustain the anabolic, rapidly dividing cancer cell. This results in the abundant production of lactate via lactate dehydrogenase A (LDHA), which directs the last step of glycolysis, by converting pyruvate to lactate and oxidizing NADH to NAD+. LDHA expression is normally restricted to skeletal muscle and is highly expressed in many cancers, thus making it an attractive therapeutic target (Fantin et al, 2006; Ward & Thompson, 2012). However, LDHA inhibitors, or drugs that disrupt other glycolytic enzymes or glucose or lactate transporters, trigger compensatory shifts in metabolism toward glutaminolysis and increased oxidative phosphorylation (OXPHOS), or to the induction of transporters that compromise the efficacy of such inhibitors (Doherty et al, 2014; Boudreau et al, 2016; Martinez‐Outschoorn et al, 2017). However, how these compensatory metabolic shifts are controlled is largely unknown and targeting these bypass mechanisms is important if such agents are going to show clinical benefit. Here, the rigorous studies of Pathria et al (2018) and the signaling savvy of the Ronai laboratory resolve this mystery, that is at least for compensatory responses manifest ex vivo in melanoma cells following treatment with LDHA inhibitors.
Using mostly BRAF mutant melanoma cell lines, the authors used silencing and LDHA‐specific inhibitors to show that LDHA activity is dispensable for proliferation and anchorage‐independent growth, yet is necessary under hypoxia (which enforces a glycolytic phenotype). As seen for other agents that disrupt glycolysis (Doherty et al, 2014), blocking LDHA disables flux through the distal half of glycolysis, here likely due to reductions in NAD+ levels produced by LDHA that are necessary for GAPDH activity. This leads to a domino effect, where increases in proximal glycolytic substrates lead to reductions in hexokinase activity and glucose uptake, and in a shift to OXPHOS. Using tracing studies, the authors show that OXPHOS induced upon LDHA inhibition is supported by elevated uptake and utilization of glutamine (Gln), and by, interestingly, increased expression of SLC1A5, a Gln transporter (Fig 1). In turn, elevated intracellular levels of Gln augment exchange and import of essential amino acids via SLC7A5 and these induce the nutrient sensing kinase mTOR to support cell growth. Accordingly, targeting SLC1A5 disrupts Gln utilization and dual inhibition of LDHA and SLC1A5 provokes apoptosis, underscoring the key roles of the Gln shift in compensatory responses.
Figure 1. Compensatory ATF4‐dependent amino acid metabolism confers resistance to LDHA inhibition.
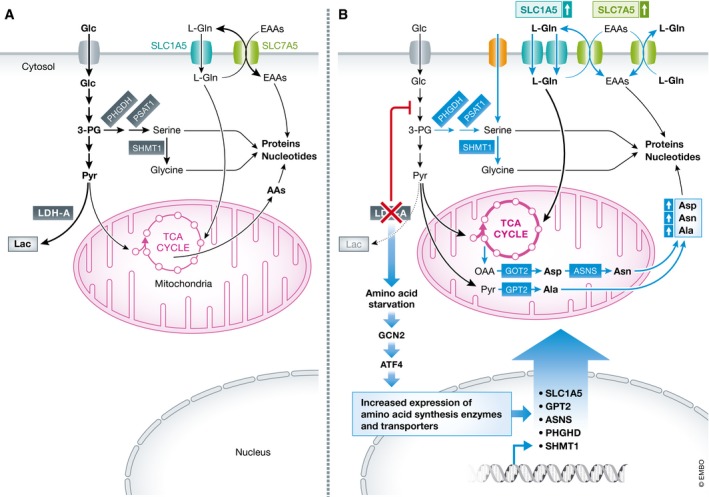
(A) Melanoma cells primarily rely on glycolysis to sustain the anabolic pathways crucial for proliferation. (B) LDHA inhibition leads to increased utilization and uptake (via SLC1A5) of Gln that supports OXPHOS, and to reductions in Ser and Asp that induce the metabolic stress response directed by the serine/threonine kinase GCN2 and the transcription factor ATF4. In turn, ATF4 induces its target genes that include metabolic enzymes and transporters that direct biosynthesis and transport (green arrows) of key non‐essential and essential amino acids.
As LDHA inhibition induces SLC1A5 mRNA levels, the authors deduced that a transcription factor directs the compensatory response and expectations were this was the metabolic regulator MYC, as SLC1A5 is a direct MYC transcription target (Wise et al, 2008). While the authors showed MYC contributes to basal levels of SLC1A5 expression in melanoma cells, they clearly demonstrated that SLC1A5 induction following LDHA inhibition was MYC independent. Here, Ronai and colleagues deduced that LDHA inhibition would trigger deficits in amino acids and activate the GCN2‐ATF4 stress circuit that induces genes involved in amino acid transport (Zhang et al, 2002; Kilberg et al, 2009). Indeed, the authors showed GCN2‐dependent upregulation of ATF4 expression precedes and was necessary for the induction of SLC1A5 following LDHA inhibition, and that dual inhibition of LDHA and ATF4, or of LDHA and GCN2, abolished transport of essential amino acids and blocked melanoma cell growth.
Interestingly, Pathria et al (2018) established that a second layer of compensation also involves ATF4 control of amino acid metabolism. Specifically, LDHA inhibition was shown to lead to marked reductions in intracellular serine (Ser) and aspartate (Asp), and that Ser reductions were due to depletion of the glycolytic substrate 3‐phosphoglycerate, which is diverted to de novo serine biosynthesis in cancer cells (Ye et al, 2012). Pathria et al (2018) reasoned that bypass here was also orchestrated by ATF4 and showed that ATF4 induces genes involved in transport and synthesis of serine, and those that catalyze production of Asp, asparagine, and alanine (GPT2, ASNS, SHMT1, and PSAT1, Fig 1). ATF4 control is in this case independent of Gln inputs, as the expression of these enzymes is not affected by knockdown of SLC1A5, and in accord with key roles of these components of the compensatory response, simultaneous targeting of LDHA and ASNS, or of LDHA and PHGDH, led to a superior anti‐melanoma response. Intriguingly, reductions in intracellular Ser and Asp following LDHA inhibition precede ATF4 induction, and supplementing media with excess Ser and Asp blocks activation of the GCN2‐ATF4 circuit following LDHA inhibition. Thus, LDHA activity sustains amino acid homeostasis and dampens the GCN2‐ATF4 circuit.
To test the potential clinical relevance of their findings, Pathria et al (2018) chose to assess the effects of inhibiting mTOR with rapamycin in conjunction with inducible silencing of LDHA. While efficient LDHA silencing had little effect on tumor growth, dual LDHA/mTOR inhibition did lead to reductions in tumor volume due to increases in apoptosis that could reflect reduced adaptability to hypoxia. Further, as expected, analysis of the LDHA‐silenced tumors demonstrated elevated levels of ATF4, SLC1A5, and mTOR activity, indicating this circuit is operational in vivo, and this is also suggested by the authors analyses of TCGA melanoma datasets, which established an inverse correlation between the expression of LDHA and mTOR.
Finally, and importantly, Pathria et al present studies suggesting that ATF4 rewires amino acid metabolism in melanoma patients treated with MAPK inhibitors. Specifically, treatment of BRAF mutant melanomas with these agents ex vivo impairs glycolysis, lactate production, and LDHA expression and leads to increases in Gln uptake. Further, RNAseq analysis of melanoma specimens from patients treated with BRAF inhibitors revealed upregulation of ATF4, GPT2, ASNS, and PSAT1, indicating that targeting ATF4 activation or Ser or Asp biosynthesis may augment the therapeutic benefit of BRAF or MEK inhibitors.
The surging efforts of academia and the pharmaceutical industry toward developing drugs that disable tumor metabolism require a detailed understanding of the potential hurdles that are inherent to such strategies. In this regard, the studies of Pathria et al provide a preclinical roadmap for how one should interrogate the effects of such drugs to identify metabolic chokepoints that can be co‐exploited to reap therapeutic benefit. Having said this, there are several technical hurdles that must be met. First, such studies should carefully consider the type of media used to test the efficacy of compounds targeting metabolic pathways, particularly since those used in the field fail to recapitulate the nutritional state of normal human serum (Cantor et al, 2017), and efforts should be made to move such studies into 3‐D platforms, preferably organoids. Second, measuring global dynamic changes in metabolism, in real time and in vivo, is currently not feasible. Third, the compensatory metabolic pathways identified ex vivo with any model may be a far cry from those operational in the patient, and they fail to consider dynamic changes manifest in the tumor microenvironment, where for example changes in levels of one or more amino acids could profoundly affect the efficacy of therapeutics designed to target a metabolic enzyme. Finally, they fail to address effects of such agents on nutrient competition with other cells in the tumor milieu, and on tumor vascularization and immune surveillance, or for that matter the effects of the metabolic state of the patient. Nonetheless, the efforts of Pathria et al are excellent first steps into understanding how to actualize such metabolic targeting agents.
Acknowledgements
J.L.C. is supported by the Cortner‐Couch Endowed Chair for Cancer Research of the University of South Florida and by NCI Center Core Support Grant P30‐CA076292. M.R.F. is supported by NRSA Award F32 CA203217.
The EMBO Journal (2018) 37: e100600
See also: https://doi.org/10.15252/embj.201899735 (October 2018)
References
- Boudreau A, Purkey HE, Hitz A, Robarge K, Peterson D, Labadie S, Kwong M, Hong R, Gao M, Del Nagro C, Pusapati R, Ma S, Salphati L, Pang J, Zhou A, Lai T, Li Y, Chen Z, Wei B, Yen I et al (2016) Metabolic plasticity underpins innate and acquired resistance to LDHA inhibition. Nat Chem Biol 12: 779–786 [DOI] [PubMed] [Google Scholar]
- Cantor JR, Abu‐Remaileh M, Kanarek N, Freinkman E, Gao X, Louissaint A Jr, Lewis CA, Sabatini DM (2017) Physiologic medium rewires cellular metabolism and reveals uric acid as an endogenous inhibitor of UMP synthase. Cell 169: 258–272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doherty JR, Yang C, Scott KE, Cameron MD, Fallahi M, Li W, Hall MA, Amelio AL, Mishra JK, Li F, Tortosa M, Genau HM, Rounbehler R, Lu Y, Dang CV, Kumar GK, Butler AA, Bannister TD, Hooper AT, Unsal‐Kacmaz K et al (2014) Blocking lactate export by inhibiting the Myc target MCT1 disables glycolysis and glutathione synthesis. Cancer Res 74: 908–920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fantin VR, St‐Pierre J, Leder P (2006) Attenuation of LDH‐A expression uncovers a link between glycolysis, mitochondrial physiology, and tumor maintenance. Cancer Cell 9: 425–434 [DOI] [PubMed] [Google Scholar]
- Kilberg MS, Shan J, Su N (2009) ATF4‐dependent transcription mediates signaling of amino acid limitation. Trends Endocrinol Metab 20: 436–443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez‐Outschoorn UE, Peiris‐Pages M, Pestell RG, Sotgia F, Lisanti MP (2017) Cancer metabolism: a therapeutic perspective. Nat Rev Clin Oncol 14: 11–31 [DOI] [PubMed] [Google Scholar]
- Pathria G, Scott DA, Feng Y, Lee JS, Fujita Y, Zhang G, Sahu AD, Ruppin E, Herlyn M, Osterman AL, Ronai ZA (2018) Targeting the Warburg Effect via LDHA inhibition engages ATF4 signaling for cancer cell survival. EMBO J 37: e99735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward PS, Thompson CB (2012) Metabolic reprogramming: a cancer hallmark even Warburg did not anticipate. Cancer Cell 21: 297–308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wise DR, DeBerardinis RJ, Mancuso A, Sayed N, Zhang XY, Pfeiffer HK, Nissim I, Daikhin E, Yudkoff M, McMahon SB, Thompson CB (2008) Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc Natl Acad Sci USA 105: 18782–18787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye J, Mancuso A, Tong X, Ward PS, Fan J, Rabinowitz JD, Thompson CB (2012) Pyruvate kinase M2 promotes de novo serine synthesis to sustain mTORC1 activity and cell proliferation. Proc Natl Acad Sci USA 109: 6904–6909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P, McGrath BC, Reinert J, Olsen DS, Lei L, Gill S, Wek SA, Vattem KM, Wek RC, Kimball SR, Jefferson LS, Cavener DR (2002) The GCN2 eIF2alpha kinase is required for adaptation to amino acid deprivation in mice. Mol Cell Biol 22: 6681–6688 [DOI] [PMC free article] [PubMed] [Google Scholar]