

Abstract
Uncontrolled cell division is a hallmark of cancer. Deregulation of Wnt components has been linked to aberrant cell division by multiple mechanisms, including Wnt‐mediated stabilisation of proteins signalling, which was notably observed in mitosis. Analysis of Wnt components revealed an unexpected role of B‐cell CLL/lymphoma 9 (BCL9) in maintaining mitotic Wnt signalling to promote precise cell division and growth of cancer cell. Mitotic interactome analysis revealed a mechanistic role of BCL9 in inhibiting clathrin‐mediated degradation of LRP6 signalosome components by interacting with clathrin and the components in Wnt destruction complex; this function was further controlled by CDK1‐driven phosphorylation of BCL9 N‐terminal, especially T172. Interestingly, T172 phosphorylation was correlated with cancer patient prognosis and enriched in tumours. Thus, our results revealed a novel role of BCL9 in controlling mitotic Wnt signalling to promote cell division and growth.
Keywords: BCL9, BCL9 phosphorylation, CDK1, cell division, clathrin, mitosis, spindle, Wnt signalling
Subject Categories: Cancer, Cell Cycle, Signal Transduction
Introduction
One of the notable properties of cancer is uncontrolled and/or improper chromosome segregation during mitosis (Jallepalli & Lengauer, 2001; Funk et al, 2016), which is primarily due to the deregulation of cell division regulators and/or mitotic signalling pathways (Johnson & Dent, 2013; Dominguez‐Brauer et al, 2015). As the nuclear envelope breaks down at the beginning of mitosis, some mitotic‐associated complexes are reassembled by the mixture of nuclear and cytoplasmic factors, which probably directly regulate mitotic events to promote normal cell division and sustain proliferation. Elucidation of the cancer‐associated mitotic signalling network and corresponding regulators would provide valuable therapeutic targets for cancer diagnosis or drug discovery. Canonical Wnt signalling plays a crucial and complex role in cancers (Polakis, 2012; Zhan et al, 2017). Wnt signalling is activated in a spatiotemporal manner in current models (Clevers & Nusse, 2012; Nusse & Clevers, 2017). Wnt ligand binding to two co‐receptors, Frizzled (Fzd) and LRP5 or LRP6, promotes the phosphorylation of the cytoplasmic tail of LRP6 by casein kinase 1 (CK1) and later GSK3, which are recruited together with axis inhibition protein (Axin) from a cytosolic “destruction” complex (consisting of APC, Axin, CK1 and GSK3, normally to degrade β‐catenin). This further triggers polymerisation of dishevelled (DVL) and LRP6 on the plasma membrane and subsequent endocytosis of the Wnt receptor complex to form the LRP6 signalosome, which is crucial for Wnt signalling activation (Bilic et al, 2007; Gammons et al, 2016). In contrast, Dickkopf 1 (DKK1), which inhibits Wnt‐dependent stabilisation of β‐catenin, induces the internalisation of LRP6 via clathrin‐mediated endocytosis (CME) to promote LRP6 receptor complex degradation. These changes result in failure to induce signalosome formation and β‐catenin stabilisation, leading to Wnt signalling inhibition (Semenov et al, 2008; Yamamoto et al, 2008).
Wnt signalling peaks in the G2/M phase due to priming of low‐density lipoprotein receptor‐related 6 (LRP6) phosphorylation by cyclin Y and CDK14 (Davidson et al, 2009; Davidson & Niehrs, 2010) indicating a potential mitotic role of Wnt signalling. In cancer cells, mitotic Wnt signalling has been shown to be involved in the stabilisation of proteins (STOP; Acebron et al, 2014), including well‐known oncogenic drivers such as c‐Myc and β‐catenin. Basal mitotic Wnt signalling has been further implicated in ensuring spindle assembly and precise chromosome segregation to promote embryogenesis and cancer cell proliferation (Huang et al, 2015; Stolz et al, 2015). Moreover, Wnt signalling components have been detected in mitotic spindle, centrosome, kinetochore and microtubule ends to modulate the spindle orientation (Schlesinger et al, 1999; Kikuchi et al, 2010; Segalen et al, 2010; Morin & Bellaiche, 2011), chromosome alignment and precise cell division (Kaplan et al, 2001; Hadjihannas et al, 2006; Kikuchi et al, 2010; Ong et al, 2010; Niehrs & Acebron, 2012; Poulton et al, 2013; Stolz et al, 2015), eventually promoting efficient cancer cell growth. However, the intrinsic mechanism and potential regulators underlying mitotic Wnt signalling are still poorly understood.
BCL9/legless (LGS, a Drosophila homologue of human BCL9) has been identified as an essential component in canonical Wnt signalling at the level of nuclear β‐catenin to promote Wnt target transcription (Kramps et al, 2002; Sampietro et al, 2006). BCL9 is highly amplified and expressed in many types of cancers described in The Cancer Genome Atlas, TCGA (TCGA cbioportal, http://www.cbioportal.org/). BCL9 has been implicated in several cancers primarily via its HD2 domain‐mediated transcription (de la Roche et al, 2012; Takada et al, 2012). Beyond this function, the mechanical role of BCL9 in human cancers is poorly understood. Here, BCL9 was shown to have an unexpected role in sustaining mitotic Wnt signalling to stabilise proteins by modulating the interaction with LRP6/Axin1 and the clathrin complex, which is further modulated by CDK1‐driven N‐terminal phosphorylation on BCL9. Thus, BCL9 would further ensure the high efficiency of cancer cell division and promote tumorigenesis.
Results
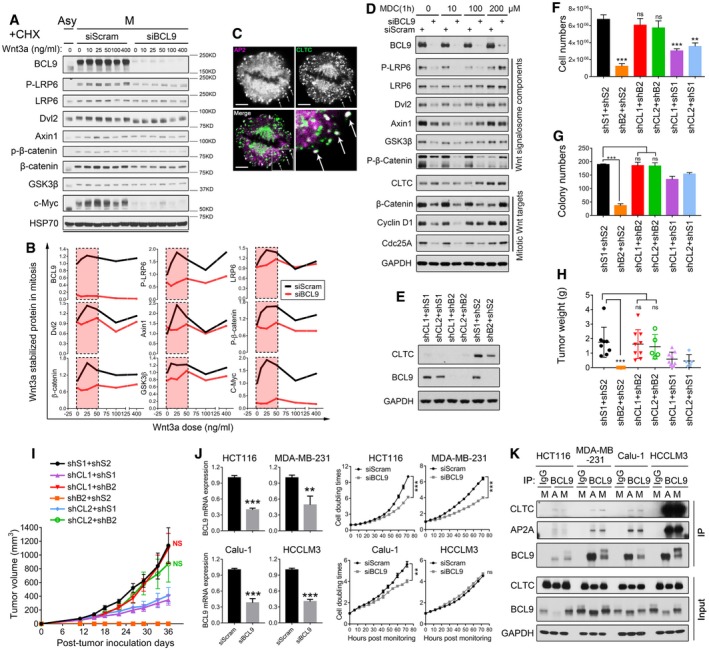
Identification of BCL9 in mitotic Wnt signalling to promote tumorigenesis
Deregulation of mitosis and Wnt signalling are both frequently observed in human cancer (Polakis, 2012; Dominguez‐Brauer et al, 2015; Funk et al, 2016; Zhan et al, 2017). Recently, a new role of Wnt signalling in mitosis has been reported and plays a crucial role in cancer cells (Acebron et al, 2014; Zhan et al, 2017); however, its regulatory mechanism and potential regulators are largely unknown. To identify mitotic Wnt signalling regulators, we employed a mini‐screen involving siRNAs targeting 47 reported Wnt signalling regulators expressed in HeLa cells (MacDonald et al, 2009; Davidson, 2010; Clevers, 2012; Fig EV1A and B, and [Link], [Link]), a cell line confirmed with active mitotic Wnt signalling (Acebron et al, 2014). To monitor the signalling activity, we determined the expression of well‐established mitotic Wnt signalling targets, such as c‐Myc and β‐catenin (Acebron et al, 2014), using immunoblotting (Figs 1A and EV1C), and further validated our findings by enzyme‐linked immunosorbent assays (ELISAs; Fig 1B) in both mitotic and asynchronised cells. As expected, we found common regulators for mitotic Wnt signalling and canonical Wnt signalling, such as Axin1, APC and DKK1, indicating the similarity between these two signalling pathways. Unexpectedly, we revealed that BCL9, a downstream nuclear regulator of β‐catenin‐mediated transcription, strongly regulated c‐Myc and β‐catenin proteins, especially in mitotic cells (Figs 1A and B, and EV1C). To exclude the off‐target effect of BCL9 Smartpool siRNAs, we employed another independent siRNA pool (siGenome pool) for subsequent assays (Fig 1C and D, and Table EV1). Moreover, to exclude the nocodazole effect on mitotic Wnt target stability, we assessed natural mitotic cells generated by the shake‐off method (Fig EV1D) and incubated them with cycloheximide (CHX) to inhibit protein translation. BCL9 depletion significantly inhibited mitotic Wnt signalling target proteins stability, notably in mitotic cells (Fig 1C and D), which was further confirmed in stable cells using previously reported BCL9 shRNA sequences (Fig EV1E; Mani et al, 2009). To explore whether BCL9 regulates mitotic Wnt signalling by inhibiting GSK3, we monitored GSK3 activity via a GFP‐based biosensor, which responds well to Wnt3a in HeLa cells (Taelman et al, 2010; Acebron et al, 2014). Live‐cell imaging analysis showed that BCL9 knockdown significantly impaired the Wnt3a‐stabilised GSK3 biosensor, especially during G2M phase (Fig 1E and Movie EV1), indicating that BCL9 promotes mitotic Wnt signalling by inhibiting GSK3. To investigate whether BCL9 regulates mitotic Wnt target transcription, we analysed the transcriptome by RNA sequencing in the cells treated with the BCL9 siRNA pools described above and/or Wnt3a treatment for 4 h, a validated time point with peak expression of Axin2, a transcriptional target of Wnt signalling (Fig EV1F). We confirmed that this mitotic Wnt target gene expression was not markedly inhibited by BCL9 knockdown by transcriptome and real‐time PCR assays (Table EV3 and Fig EV1G). Moreover, a fifteen Wnt signalling transcriptional targeting gene signature controlled by Axin1, APC and Wnt3a was identified (Fig 1F and Table EV3). Unexpectedly, BCL9 regulated none of these genes (Fig 1F and Table EV3), indicating that BCL9 has a potential role in Wnt signalling beyond transcription in HeLa cells. As BCL9 stabilises the β‐catenin and c‐Myc proteins, we employed a gene set enrichment analysis (GSEA) based on the transcriptome of cells with BCL9 knockdown to further evaluate the β‐catenin and c‐Myc activities (Figs 1G and EV1H). The results further confirmed that BCL9 knockdown significantly inhibited β‐catenin and c‐Myc activities.
Figure EV1. BCL9 regulates mitotic Wnt signalling to stabilise proteins and promote tumorigenesis (related to Fig 1).
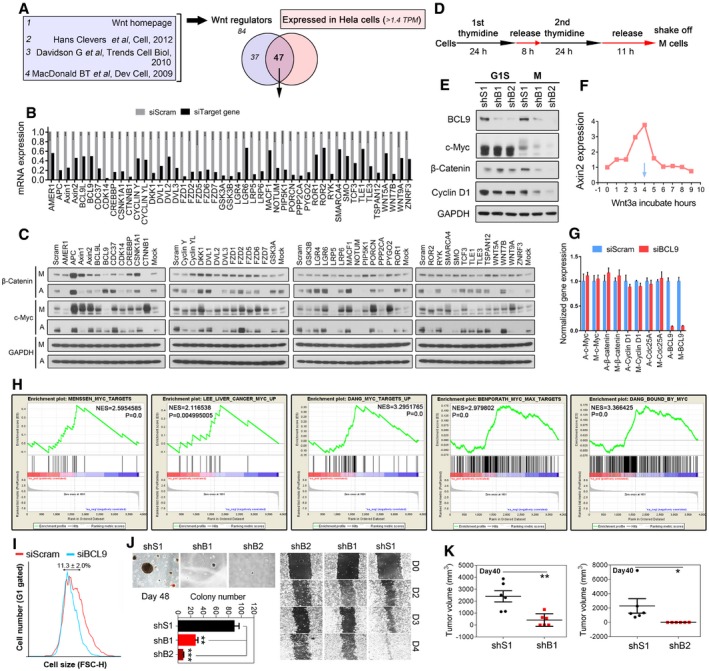
- The selection of 47 Wnt signalling regulators expressed in HeLa cells for siRNA study.
- Validation of siRNA knockdown efficiency by real‐time PCR.
- Immunoblotting analysis of Wnt‐stabilised protein targets β‐catenin and c‐Myc in mitosis (M) and asychronised (A) HeLa cells treated with siRNA.
- The synchronisation protocol to collect nature mitotic cells.
- Immunoblotting analysis of Wnt‐stabilised proteins expression in BCL9 stable knockdown mitotic or G1S HeLa cells.
- Time‐dependent induction of the expression of the Wnt transcriptional target Axin2 in HeLa cells by Wnt3a protein (100 ng/ml).
- Gene expression by real‐time PCR of four mitotic Wnt targets in non‐sychronised or mitotic cells. A: asynchronised cell; M: mitotic cells.
- Gene enrichment analysis of c‐Myc activity in different dataset by using BCL9 regulated gene.
- Knockdown of BCL9 decreases G1 cell size. Forward scatter (FSC‐H) profile of G1‐gated HeLa cells treated with BCK9 or scramble siRNA 72 h post‐transfection.
- Oncogenic soft agar colony formation and wound healing analysis in BCL9 stable knockdown cells.
- Effect of BCL9 knockdown on xenograft weight in tumorigenesis assay on day 40 post‐tumour inoculation. Significance was measured by two‐tailed unpaired t‐test, *P < 0.05, **P < 0.01. All data are means ± SD, n = 6 for shS1 vs. shB1, n = 5 for shS1 vs. shB2.
Source data are available online for this figure.
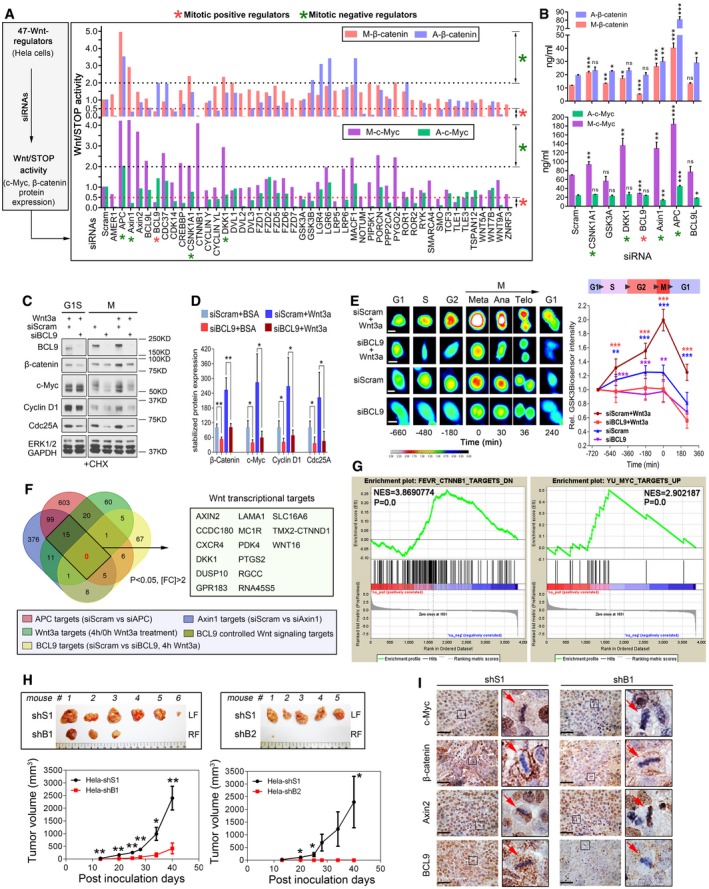
Figure 1. Identification of BCL9 as a novel mitotic Wnt signalling regulator to stabilise proteins and promote oncogenesis.
-
ANormalised Wnt/STOP signalling activity by quantification of the protein expression of two Wnt/STOP target proteins, β‐catenin and c‐Myc, in mitotic (M) and asynchronised cells (A) after silencing 47 Wnt signalling regulators.
-
BELISA validation of selected Wnt/STOP signalling regulators (n = 3).
-
C, D(C) Immunoblotting and (D) quantification of the stability of four STOP target proteins in both G1S and natural mitotic cells treated with BCL9 siGenome pools and CHX (n = 3).
-
ERepresentative pictures (left panel) and the normalised biosensor intensity based on live‐cell imaging of the cells with a GSK3‐GFP biosensor following different treatments before and after metaphase (n = 10). Scale bars represent 10 μm.
-
FVenn diagram analysis of the number of Axin1, APC, and Wnt3a signalling and/or BCL9‐regulated transcriptional targets.
-
GMyc and β‐catenin target gene enrichment analysis based on the transcriptome targets regulated by BCL9.
-
HThe representative xenograft tumours (top panel) and tumour growth analysis (bottom panel) of scramble and BCL9 stable knockdown cells. LF: left flank of mouse, RF: right flank. n = 6 for shS1 vs. shB1, n = 5 for shS1 vs. shB2.
-
IImmunohistochemical staining of the xenograft; the red arrows indicate the representative mitotic cells. Scale bars represent 200 μm.
To address BCL9's roles in HeLa cells, we investigated the effect of BCL9 on cell size which could be regulated by mitotic Wnt signalling (Acebron et al, 2014), and we confirmed that BCL9 knockdown decreased the cell size in G1 phase HeLa cells (Fig EV1I). Moreover, we constructed stable BCL9 knockdown cells and found that loss of BCL9 substantially impaired soft agar colony formation, wound healing migration (Fig EV1J) and tumorigenesis of the cells (Figs 1H and EV1K). Protein expression of c‐Myc, β‐catenin or Axin2 was further examined in xenograft by immunohistochemistry (IHC) staining, which indicated that BCL9 knockdown inhibited c‐Myc and β‐catenin expression in mitotic cells and in interphase cells to a lesser extent but not Axin2 expression (Fig 1I). These results indicated that BCL9 regulates mitotic Wnt signalling beyond the activation of Wnt signalling transcription to promote cancer cells growth and tumorigenesis.
BCL9 regulates the mitotic spindle and cell division partially by modulating mitotic Wnt signalling
Wnt signalling components have been implicated in mitosis via regulation of the mitotic spindle orientation, chromosome alignment and/or precise cell division as shown previously (Hadjihannas et al, 2006; Kaplan et al, 2001; Kikuchi et al, 2010; Ong et al, 2010; Niehrs, 2012; Poulton et al, 2013; Stolz et al, 2015). To examine BCL9's roles in mitosis, we first synchronised cells (Fig EV2A) and analysed BCL9 or cyclin B1 protein (Fig 2A) and RNA (Fig EV2B) level from G1 to M phase. Notably, a significant band shift in addition to the BCL9 major band was detected specifically in mitosis and was verified by phosphorylated histone H3 (p‐H3S10) and flow cytometric assays (Fig 2A). These assays showed that there were mitotic modifications on BCL9, which were most likely phosphorylation. After incubation of mitotic lysate with lambda protein phosphatase (λ‐PPase), the shifted band disappeared (Fig EV2C), indicating mitotic phosphorylation on BCL9. To identify the potential kinase(s) involved, we first investigated CDK1, a master kinase in mitosis. A CDK1‐specific inhibitor (RO3306) completely abolished the phosphorylation of both endogenous and exogenous BCL9 (Fig EV2D), indicating that CDK1 potentially regulates mitotic BCL9 phosphorylation. To confirm that CDK1 phosphorylating BCL9 is a direct effect of CDK1, we perform in vitro kinase assay by incubating CDK1/cyclin B1 and BCL9 proteins with or without ATP (Fig EV2E). We found that indeed CDK1 could directly induce BCL9 band shift (Fig EV2E), indicating CDK1 phosphorylating BCL9 protein directly.
Figure EV2. BCL9 is phosphorylated and localises on mitotic spindle (related to Fig 2).
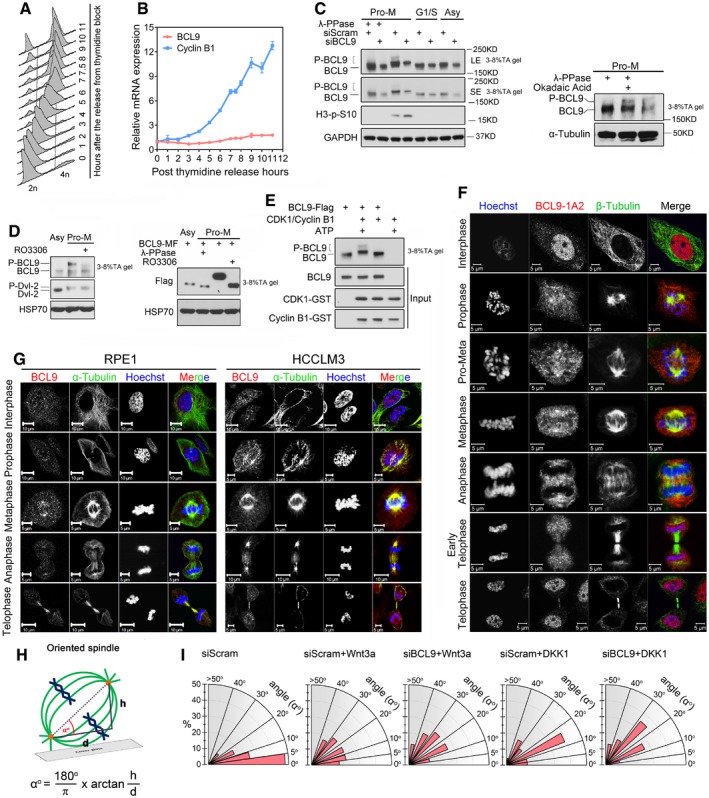
- Flow cytometry cell cycle analysis of thymidine synchronised HeLa cells.
- Cyclin B1 and BCL9 gene expression in synchronised cells.
- Immunoblotting analysis of lambda phosphatase effect on BCL9 phosphorylation in pro‐metaphase (Pro‐M), G1S and asynchronised HeLa cells (left panel), or the effect of phosphatase inhibitor okadaic acid, alone or combined with lambda phosphatase, on BCL9 phosphorylation in Pro‐M HeLa cells (right panel). 3–8% TA gel refers to NuPAGE 3–8% Tris–acetate protein gels.
- Immunoblotting analysis of CDK1‐specific inhibitor RO3306 inhibiting both endogenous (left panel) and exogenous (right panel) BCL9 phosphorylation. 3–8% TA gel refers to NuPAGE 3–8% Tris‐acetate protein gels.
- CDK1/cyclin B1 kinase assay to detect the direct phosphorylation band shift of BCL9 protein purified by wheat germ translation system. 3–8% TA gel refers to NuPAGE 3–8% Tris–acetate protein gels.
- Immunofluorescence staining of BCL9 subcellular localisation during cell cycle by a mouse monoclonal antibody in HeLa cells. Scale bars represent 5 μm.
- Immunofluorescence staining of BCL9 subcellular localisation during cell cycle in RPE1 and HCCLM3 cells. Scale bars represent 5 or 10 μm as indicated in each figure.
- A diagram of instruction for how to calculate the spindle angle.
- Radial histograms showing the distribution of spindle angles in cells treated with Wnt3a or DKK1 purified proteins (n = 23).
Source data are available online for this figure.
Figure 2. The novel role of BCL9 in maintaining mitotic Wnt signalling to regulate proper cell division.
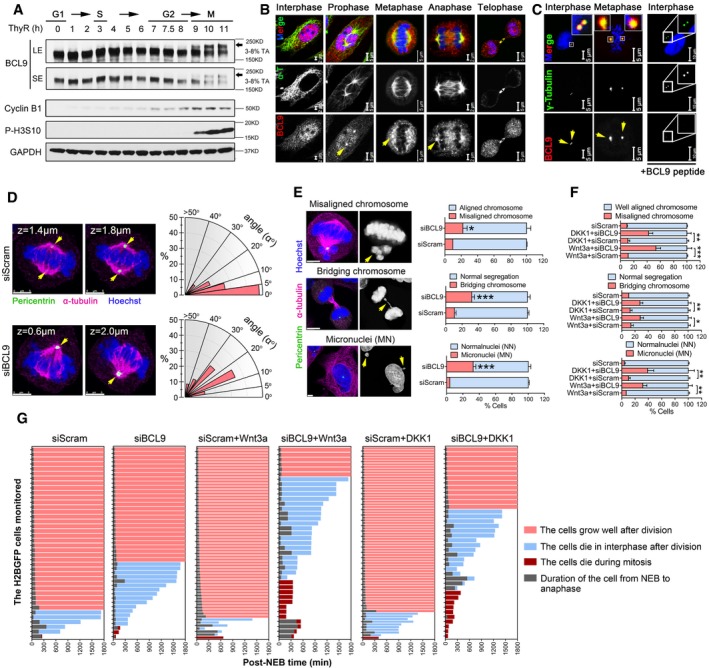
- Immunoblotting analysis of BCL9 and cell cycle protein expression in double thymidine synchronised cells; the black arrow indicates the delayed band of mitotic BCL9.
- Immunofluorescence confocal analysis of BCL9 localisation on the spindle by antibody staining. Yellow arrows indicate the spindle pole. Scale bars represent: 2 μm for interphase and anaphase; 5 μm for prophase, metaphase and telophase.
- Immunofluorescence confocal analysis of BCL9 localisation on the centrosome. Yellow arrows indicate the centrosome. Scale bars represent: 5 μm for control treatment, 10 μm for BCL9 peptide treatment.
- Confocal microscope 3D analysis and radial histograms of the spindle angle in the cells with siRNA treatment; yellow arrows indicate the centrosome. Scale bars represent 5 μm.
- Confocal (left panel) and live‐cell imaging (right panel) analysis of chromosome segregation in H2BGFP‐cells following siRNA treatment; yellow arrows indicate the typical abnormal chromosomes by BCL9 knockdown. Twenty‐three H2BGFP‐mitotic cells were monitored for each time experiment; data are shown from three independent experiments. Scale bars represent 5 μm.
- Quantification of aberrant chromosome segregation rate by live‐cell imaging of H2BGFP cells upon Wnt3a or DKK1 treatment. Twenty‐three H2BGFP‐mitotic cells were monitored for each time experiment; data are shown from three independent experiments.
- Quantification of the duration of NEB to ANA (NEB: nuclear envelope breakdown, ANA: anaphase), NEB to cell death in interphase after cell division (blue colour bar) or in mitosis (dark red bar) or growth well in 30 h (pink bar) by live‐cell imaging analysis in monitored H2BGFP cells with different treatments.
As Wnt components often localise to the mitotic spindle and/or centrosome (Niehrs, 2012), BCL9 subcellular localisation was evaluated by two commercial antibodies in HeLa cells (Figs 2B and EV2F). BCL9 was detected on the spindle and spindle pole during mitosis (Figs 2B and EV2F), and this localisation was further confirmed in two additional human cell lines (Fig EV2G). BCL9 was also detected on the centrosome by methanol fixation (Fig 2C). These data indicated that BCL9 might have a function in regulating mitotic spindle. To explore this role, we knocked down BCL9 and investigated the spindle orientation (SO; Figs 2D and EV2H), a notable Wnt‐associated spindle function (Niehrs, 2012; Habib et al, 2013). By calculating two spindle pole distances in the x‐axis, y‐axis and z‐axis, we could determine the angle between, which reflected the SO (Fig EV2H). These data indicated that BCL9 knockdown induced aberrant SO by increasing the spindle angle compared to scramble knockdown (Fig 2D), suggesting that BCL9 regulates SO. To determine the effect on cell division, we used H2B‐GFP cells or immunofluorescence (IF) staining for live‐cell imaging or confocal analysis. BCL9 knockdown significantly induced aberrant cell division with misaligned chromosomes (MCs), bridging chromosomes (BCs) and subsequent micronuclei (MN) formation in interphase cells (Fig 2E and Movie EV2).
To examine the interplay of Wnt signalling and BCL9 with SO and cell division, we pre‐incubated BCL9‐ or scramble‐knockdown cells with Wnt3a or DKK1 to activate or inhibit endogenous Wnt signalling, and the spindle angle and cell division were further monitored and analysed. Our data indicated that both Wnt3a and DKK1 treatment could increase the spindle angle (Fig EV2I), indicating that aberrant activation or inhibition of mitotic Wnt signalling would cause abnormal SO. Silencing BCL9 in cells with Wnt3a or DKK1 resulted in even larger spindle angles (Fig EV2I) and led to a rigorous increase of the rate of aberrant chromosome segregation (Fig 2F). However, Wnt3a or DKK1 treatment in scramble‐knockdown cells did not significantly increase the aberrant cell division rate compared to no treatment scramble cells (Fig 2F). These data indicated that there should be an intrinsic mechanism for balancing excess or inhibited mitotic Wnt signalling to ensure proper cell division, and this requires BCL9.
To further investigate the impact of aberrant cell division on cell death or growth after BCL9 knockdown, we monitored and analysed the cell status 30 h post‐nuclear envelope breakdown (NEB). BCL9 knockdown led to cell death primarily after cell division (Fig 2G and Movie EV3). Wnt3a or DKK1 treatment appeared to have no significant effect on cell death, cell growth and the duration of cell division from NEB to anaphase (Fig 2G and Movie EV3), while BCL9 knockdown together with Wnt3a or DKK1 treatment induced substantial increases in both interphase and mitotic cell death and aberrant duration of cell division from NEB to anaphase (Fig 2G and Movie EV3). These data are consistent with the SO phenotype and cell division analysis described above, indicating that BCL9 promoted cell proliferation partially by balancing mitotic Wnt signalling to modulate the mitotic spindle and cell division precisely.
Elucidation of the BCL9 interactome in mitosis
To elucidate the mechanism of BCL9 in mitosis, we analysed the mitotic BCL9 interactome by the stable isotope labelling with amino acids in cell culture (SILAC) proteomic approach (Ong et al, 2002). Using SILAC analysis (Fig EV3A), we identified 19 of the top BCL9 interactors (normalised H/L ratio > 2) in mitotic cells (Fig 3A and Table EV4). To predict the functions or pathways of the BCL9 interactome, we performed DAVID functional annotation analysis (Fig 3B and Table EV4) and ingenuity pathway analysis (IPA; Fig EV3B) based on the potential interactors (normalised H/L ratio > 1.5). Interestingly, three major functions or pathways enriched were associated with Wnt signalling and the destruction complex, mitotic spindle and CME (Fig 3B and Table EV4). Notably, the interactors included many core components of the Wnt destruction complex, including APC, Axin1, AMER1, CK1 and β‐catenin; spindle regulators such as HMMR, FAM83D (also named CHICA) and CKAP5 (also named ch‐Tog); and endocytosis components such as clathrin heavy chain (CLTC), α‐adaptin (AP2A) and intersectin 1 (ITSN1; Table EV4).
Figure EV3. Identification of mitotic interactome of BCL9 (related to Fig 3).
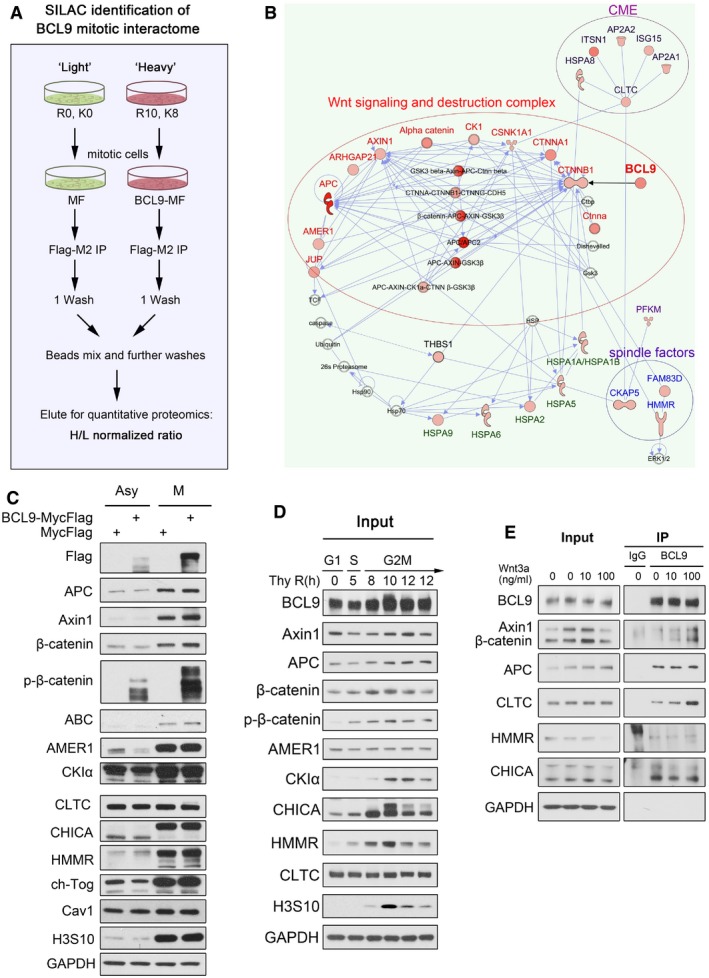
- A schematic figure of SILAC approach to identify BCL9 interactome in HeLa cells.
- Ingenuity pathway analysis of potential mitotic BCL9 interactome in HeLa cells. Thirty‐five potential interactors (normalised H/L > 1.5) were input for direct or indirect interaction analysis.
- The input for immunoprecipitation analysis in Fig 3C.
- The input for immunoprecipitation analysis in Fig 3D.
- The immunoprecipitation analysis of the effect of Wnt3a on endogenous mitotic BCL9 interactors for 1.5 h.
Source data are available online for this figure.
Figure 3. Dissecting the mitotic interactome of BCL9.
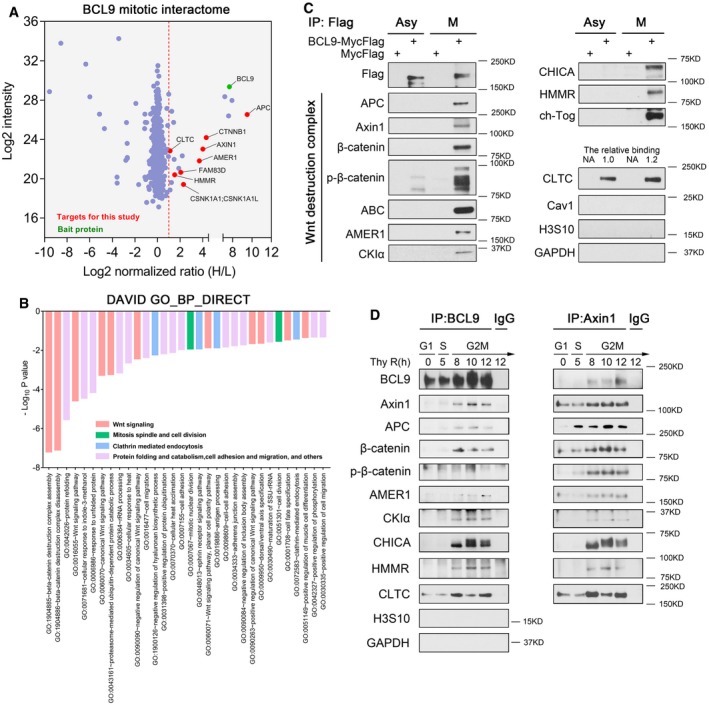
- Volcano plot of the BCL9 mitotic interactome by the SILAC proteomics approach. Red dashed line indicates a strict cut off by a normalised SILAC H/L ratio > 2.
- DAVID functional annotation analysis of enriched biological process (BP) by the potential mitotic BCL9 interactome (normalised SILAC H/L ratio > 1.5).
- An independent immunoprecipitation assay for validation of potential BCL9 interactors in both asynchronised and mitotic cells. The relative binding of CLTC with BCL9 is quantified and labelled on top of the CLTC blot.
- Immunoprecipitation assay of the endogenous BCL9 and Axin1 interaction in synchronised HeLa cells, from G1S to G2M. P‐H3S10, a mitotic marker indicating mitosis.
We further validated most of the interactors by independent immunoprecipitation (IP) assays with lysates from both asynchronised and mitotic cells (Figs 3C and EV3C). The majority of the interactors show mitotic specificity, with the exception of clathrin (Figs 3C and EV3C). Moreover, to evaluate the dynamic interaction of endogenous BCL9 and the destruction complex during the cell cycle, we further immunoprecipitated and validated the interaction in synchronised cells, from G1 to M phase, using a BCL9 antibody or an Axin1 antibody (Figs 3D and EV3D). We confirmed that BCL9 interacted with these interactors primarily in G2M phase, again except for CLTC (Figs 3D and EV3D), and BCL9 was confirmed to be involved in the mitotic destruction complex (Figs 3D and EV3D). To investigate whether these interactions were regulated by Wnt signalling, we treated synchronised mitotic cells with different dose of Wnt3a for 1.5 h and found that Wnt3a could promote the BCL9 interaction with destruction complex and CLTC but not with spindle regulators such as HMMR and CHICA (Fig EV3E).
BCL9 regulates the mitotic destruction complex and competes with clathrin for the LRP6/Axin1 complex
To explore the potential roles of BCL9 in the mitotic destruction complex, we performed endogenous IP assays with Axin1, APC or IgG antibodies using lysates of synchronised scramble‐ or BCL9‐knockdown cells from G1S to G2M (Fig EV4A). The relative immunoprecipitated proteins were detected and quantified by normalisation to the corresponding input, target protein and bait protein immunoprecipitated at the starting time point of release (Fig EV4B). The quantification indicated that BCL9 depletion reinforced the sequestration of β‐catenin in the mitotic Axin1 and APC destruction complex (Fig EV4B), indicating a shielded role of BCL9 on β‐catenin in mitosis. Interestingly, BCL9 depletion substantially promoted CLTC binding to the destruction complex (Fig EV4B), demonstrating that BCL9 blocked clathrin and destruction complex binding. To further verify this, we performed Duolink in situ interaction assays to quantify relative protein–protein interactions (Fig 4A–C) and confirmed that BCL9 depletion significantly enhanced the Axin1/β‐catenin and Axin1/CLTC interactions (Fig 4A and B), similar to the phenotype observed by IP analysis. Meanwhile, the BCL9 and CLTC interaction Duolink signal could be detected both on the spindle and out of it, and this signal was significantly impaired in BCL9‐depleted cells (Fig 4C). To exclude the off‐target effect of BCL9 siRNA knockdown on the interactions, we optimised the transfection condition and re‐expressed BCL9‐Flag in BCL9‐knockdown cells in mitosis (Fig EV4C). It was demonstrated that BCL9 knockdown could still promote Axin1 interaction with β‐catenin or CLTC which was attenuated obviously after re‐expression of BCL9‐Flag in the cells (Fig EV4C). This confirmed that BCL9 had direct effect on regulating the interactions of destruction complex with β‐catenin or CLTC in mitosis. BCL9 and CLTC interaction was further confirmed by a BCL9‐null HAP1 cell line (Fig EV4D).
Figure EV4. BCL9 regulates mitotic destruction complex and competes with clathrin for LRP6/Axin1 complex binding (related to Fig 4).
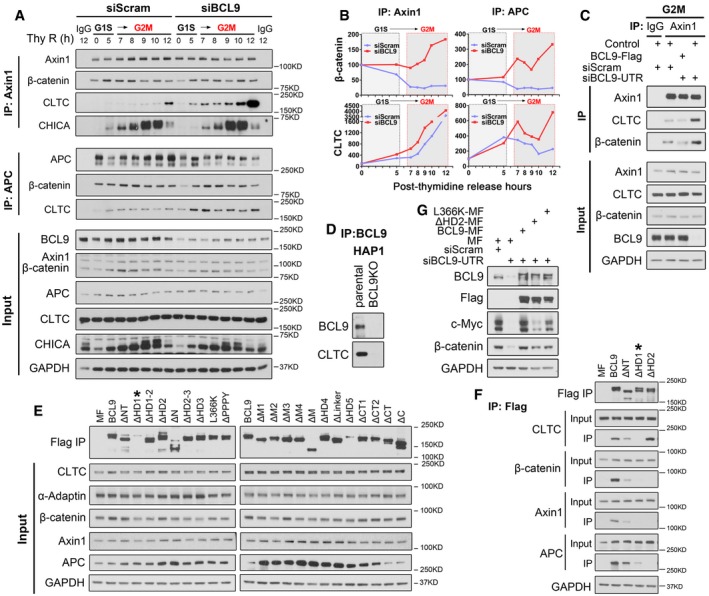
- Immunoprecipitation analysis of endogenous Axin1 or APC interaction with CLTC or β‐catenin during different stages of cell cycle by thymidine synchronisation. Thy R (h) indicates hours post‐double thymidine release.
- The relative binding was determined for (A).
- A rescue experiment by investigating the effect of re‐expression of BCL9‐Flag in BCL9 knockdown cells on the Axin1 interaction with CLTC or β‐catenin.
- The immunoprecipitation analysis of BCL9 and CLTC in HAP1 parental and BCL9 KO cells.
- The input for immunoprecipitation analysis in Fig 4E.
- An additional data for the validation of ΔHD1 which is important for interaction with destruction complex, but is not well confirmed in (E).
- Immunoblotting analysis of the effect of BCL9 and two truncates re‐expression on mitotic Wnt signalling activity by detecting mitotic Wnt target proteins.
Source data are available online for this figure.
Figure 4. BCL9 regulates the mitotic destruction complex and competes with clathrin for Axin1 complex binding.
-
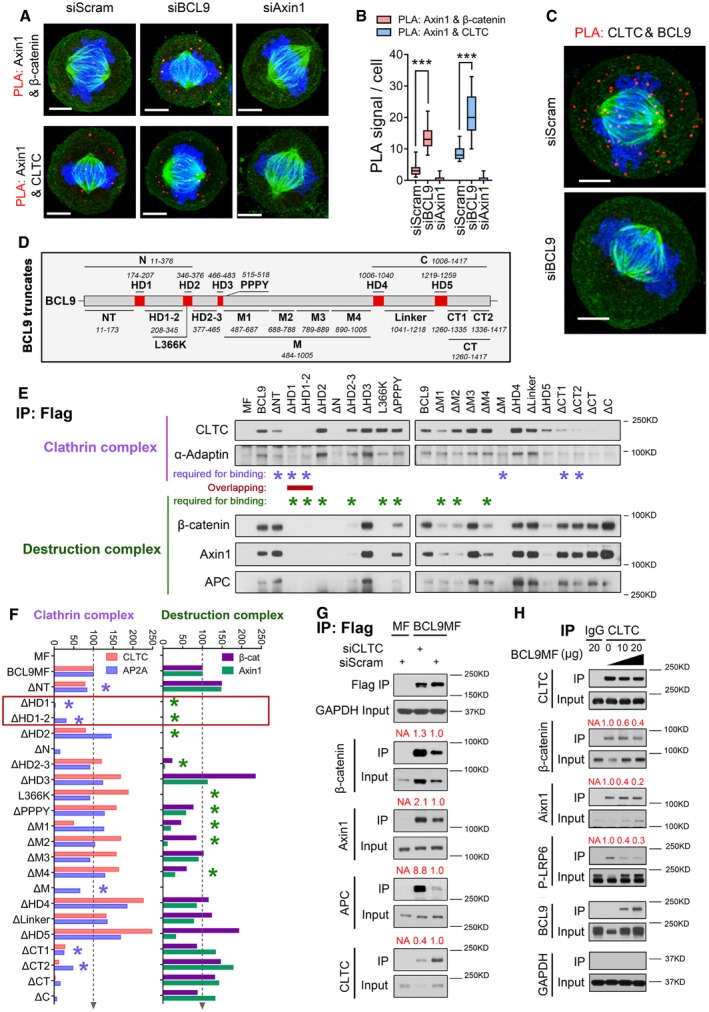
A, BDuolink analysis (A) and its quantification (B) of the interaction by assessing the PLA complex in mitotic cells after BCL9 or Axin1 knockdown (n = 20). Scale bars represent 5 μm. Significance was measured by two‐tailed unpaired t‐test, ***P < 0.001, the horizontal lines in boxplot indicate the medium reading of the data, box ranges from Min to Max, all data are the mean ± SD.
-
CDuolink analysis of the interaction of BCL9 and CLTC in mitotic cells. Scale bars represent 5 μm.
-
DSchematic diagram of 20 truncated BCL9 forms designed for subsequent immunoprecipitation analysis.
-
EImmunoprecipitation analysis of the interaction of truncated BCL9 forms in mitosis; the purple stars indicate the domain required for clathrin complex binding, and the green stars indicate the domains for β‐catenin complex binding. The domain required for both clathrin and β‐catenin complexes binding is indicated by a red line.
-
FQuantification of the relative binding of the truncated forms with the clathrin complex or β‐catenin complex; the red frame indicates the common domains of BCL9 for binding of both complexes.
-
GImmunoprecipitation analysis of the BCL9‐MF interaction in CLTC‐knockdown mitotic cells.
-
HImmunoprecipitation analysis of the endogenous clathrin interaction with LRP6 signalosome components in BCL9 MF‐overexpressing mitotic cells.
BCL9 is a large scaffold protein with many uncharacterised domains. To precisely analyse the interacting protein domains of BCL9, we cloned 20 truncated deletion forms of BCL9, including ΔNT, ΔHD1, ΔHD1‐2, ΔHD2, ΔHD2‐3, ΔN, ΔHD3, ΔPPPY, ΔM1, ΔM2, ΔM3, ΔM4, ΔM, ΔHD4, ΔLinker, ΔHD5, ΔCT1, ΔCT2, ΔCT, ΔC and L366K (Fig 4D and Table EV2), a mutant reported to be deficient for β‐catenin binding (de la Roche et al, 2008). We further examined the domains for clathrin complex interaction or β‐catenin complex interaction in mitosis (Figs 4E and F, and EV4E and F). The interaction of BCL9 and the clathrin complex primarily required N‐terminal domains (NT, HD1 and HD1‐2) and C‐terminal domains (CT1, CT2) of BCL9 (Figs 4E and F, and EV4E and F). The mitotic interaction of BCL9 and the β‐catenin complex required other N‐terminal domains in addition to HD2, including HD1, HD1‐2, HD2‐3 and the M domains to a lesser extent (Figs 4E and F, and EV4E and F).
As an overlapping region including the HD1 and HD1‐2 domains was required for the binding of both the clathrin and β‐catenin complexes with BCL9 (Figs 4E and F, and EV4E and F), we hypothesised that a competitive type of interaction might exist among clathrin, the β‐catenin complex and BCL9. To further assess this hypothesis, we depleted CLTC by siRNA, which resulted in substantial enhancement of BCL9 binding to the β‐catenin destruction complex (Fig 4G). These results showed that clathrin competes with BCL9 for binding to the destruction complex. To confirm this observation, we performed an IP assay with a clathrin antibody using a mitotic lysate with elevated exogenous BCL9 expression (Fig 4H). Interestingly, elevated BCL9 expression notably increased endogenous protein expression of β‐catenin, Axin1 and APC as well as Ser1490‐phosphorylated‐LRP6 (p‐LRP6; Fig 4H), a core component of the active LRP6 signalosome (Bilic et al, 2007). Our results indicated a potential role of BCL9 in stabilising the mitotic LRP6/Axin1 complex, also referred to as the LRP6 signalosome (Bilic et al, 2007). By quantifying immunoprecipitated proteins, we found that BCL9 expression substantially inhibited clathrin binding to components of the LRP6 signalosome (Fig 4H), further confirming that BCL9 competes with clathrin for the LRP6/Axin1 complex. To investigate whether the reported HD2 domain of BCL9 for β‐catenin binding is required for mitotic Wnt signalling activity, we monitored β‐catenin and c‐Myc protein expression (Fig EV4G). BCL9 depletion could attenuate these two mitotic Wnt target expression which could completely reverse by re‐expression of BCL9 but only partially reversed by re‐expression of ΔHD2 or L366K truncate (Fig EV4G). This indicates that the interaction of BCL9‐HD2 domain with β‐catenin is required for BCL9 activity in promoting mitotic Wnt signalling; nevertheless, at present we could not rule out the involvement of other BCL9 domains.
BCL9 stabilises LRP6 signalosome components by inhibiting clathrin
To verify the effect of BCL9 on LRP6 signalosome stabilisation, we stimulated scramble‐ or BCL9‐knockdown synchronised mitotic cells with Wnt3a protein (dose ranging from 0 to 400 ng/ml) and treated the cells with CHX to inhibit protein translation. The stability of signalosome components was evaluated (Fig EV5A) and further normalised for quantification (Fig EV5B). Our data indicated that Wnt3a with a dose ranging from 0 to 50 ng/ml could stabilise the signalosome components as well as c‐Myc and β‐catenin in scramble‐knockdown mitotic cells at 1 h post‐treatment (Fig EV5A and B). However, BCL9 depletion significantly attenuated Wnt3a‐induced stabilisation of these components (Fig EV5A and B), indicating that BCL9 is essential for stabilisation of the LRP6 signalosome complex to further activate mitotic Wnt signalling. As clathrin competed with BCL9 for signalosome components (Fig 4G and H), we hypothesised that clathrin might play an opposite role in mitotic LRP6 signalosome stabilisation. Thus, we silenced BCL9, CLTC or both of them in synchronised mitotic cells and monitored signalosome component stability by CHX (Fig 5A and B). As expected, BCL9 depletion decreased the stability of LRP6 signalosome components, while CLTC depletion in BCL9 knockdown mitotic cells could reverse the BCL9 knockdown effect on signalosome component stabilisation (Fig 5A and B), indicating that clathrin is involved in Wnt/BCL9 signalling‐mediated protein stabilisation in mitotic cells.
Figure EV5. BCL9 stabilises LRP6 signalosome components and STOP targets via inhibiting clathrin (related to Fig 5).
-
AImmunoblotting analysis of Wnt3a‐stabilised protein stability in natural mitotic cells with CHX.
-
BQuantification analysis of Wnt3a‐stabilised protein stability in natural mitotic cells with CHX; the effective doses of Wnt3a to stabilise proteins in 1 h are labelled by a pink frame with black dashed lines.
-
CImmunofluorescence analysis of clathrin and adaptor protein AP2 in mitotic cells, the arrow indicates the clathrin pits. Scale bars represent 5 μm.
-
DImmunoblotting analysis of BCL9 knockdown effect on signalosome components and STOP targets by immunoblotting in nature mitotic cells treated with DMSO or MDC, an inhibitor for clathrin‐mediated endocytosis, for 1 h.
-
E–H(E) Immunoblotting analysis, (F) normalised cell growth assay, (G) soft agar colony formation assay, (H) the mice tumours weight of stable knockdown cells on day 36 post‐inoculation of BCL9 and CLTC expression in stable knockdown cells.
-
IXenograft tumour growth is monitored for stable knockdown cells by calculating the tumour volume (n = 8 for shS1 + shS2 or shB2 + shS2, n = 9 for shCL1 + shS1 or shCL1 + shB2, n = 5 for shCL1 + shS1 o shCL1 + shB2). Data are the mean ± SEM.
-
JReal‐time PCR and cell growth analysis of BCL9 knockdown by siRNA in four cancer cell lines. Data are the mean ± SD.
-
KImmunoprecipitation analysis of BCL9 interaction with clathrin complex in non‐synchronised or mitotic cell lysate of four cancer cell lines.
Figure 5. BCL9 stabilises LRP6 signalosome components to maintain oncogenicity of the cancer cells by inhibiting clathrin.
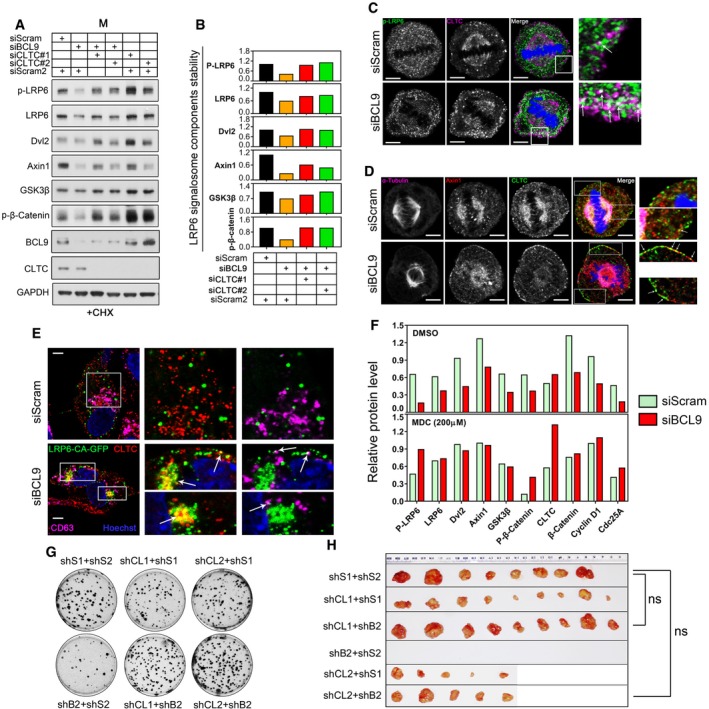
- Immunoblotting analysis of signalosome components by immunoblotting in BCL9, CLTC or in both BCL9 and CLTC‐knockdown mitotic cells with CHX.
- Quantification of the stability of several LRP6 signalosome components by immunoblotting of mitotic cells with CHX.
- Immunofluorescence confocal analysis of Ser1490‐phosphorylated LRP6 and CLTC co‐localisation in mitotic HeLa cells; white arrows indicate the co‐localisation. Scale bars represent 5 μm.
- Immunofluorescence analysis of α‐tubulin, Axin1 and CLTC triple‐staining in mitotic cells. The white arrows indicate the co‐localisation. Scale bars represent 5 μm.
- Immunofluorescence analysis of co‐localisation of LRP6CAGFP and CLTC or CD63 in interphase cells. The white arrows indicate the co‐localisation. Scale bars represent 5 μm.
- Quantification of signalosome components and mitotic Wnt target expression level by immunoblotting in natural mitotic cells treated for 1 h with DMSO or MDC, an inhibitor of clathrin‐mediated‐endocytosis.
- A representative image of soft agar oncogenic colony formation assays for stable knockdown cells.
- A representative image of the mouse xenograft tumours for stable knockdown cells at the end point of the experiment.
When Wnt signalling was inhibited, clathrin could mediate the degradation of Wnt receptor and LRP6 signalosome by a physiological process called CME (Semenov et al, 2008; Yamamoto et al, 2008). As in the natural synchronised mitotic HeLa cells, CME is still active and functional (Boucrot & Kirchhausen, 2007; Tacheva‐Grigorova et al, 2013), we thus hypothesised that BCL9 depletion might promote CME of the LRP6 signalosome for degradation. To confirm this, we first examined clathrin‐coated pits (CCP) by double staining CLTC and α‐adaptin (AP2), which is the core adaptor protein for functional CCP formation and was also detected in our SILAC interactome list as a potential interactor (Fig 4E and Table EV4). The staining indicated that clathrin and AP2 co‐localised close to the membrane and formed CCP in mitotic cells (Fig EV5C).
To verify whether BCL9 is involved in signalosome components sorted to CCP, we performed a confocal microscopy analysis and confirmed that BCL9 depletion caused p‐LRP6 or Axin1 detachment from the spindle to the clathrin complex close to the plasma membrane of mitotic cells (Fig 5C and D), indicating that LRP6 signalosome endocytosis by clathrin might be inhibited by BCL9. Moreover, BCL9 depletion also caused the LRP6CA‐GFP protein, a constitutively active LRP6 co‐receptor, to form signalosome and undergo sequestration to the clathrin complex or even lysosome (indicated by CD63 staining) in the cells after cytokinesis (Fig 5E). To confirm that CME regulates signalosome component destabilisation, we incubated the synchronised mitotic cells depleted with BCL9 or scramble with the CME inhibitor monodansylcadaverine (MDC) for 1 h, and we found that 200 μM MDC completely reversed the effect of BCL9 depletion on destabilised Wnt signalosome components and mitotic Wnt targets (Figs 5F and EV5D). These findings showed that BCL9 inhibits CME of the LRP6 signalosome to potentiate mitotic Wnt signalling.
To examine whether clathrin modulates BCL9 function, we performed cell growth, soft agar colony formation and tumorigenic assays by scramble‐, BCL9‐, CLTC‐knockdown or BCL9/CLTC double‐knockdown stable cells (Fig EV5E). BCL9 knockdown significantly abolished cell growth (Fig EV5F), colony formation (Figs 5G and EV5G) and tumorigenic (Figs 5H, and EV5H and I) properties of HeLa cells as mentioned above. However, BCL9/CLTC double knockdown completely reversed the BCL9 knockdown effect on cell growth, colony formation and mouse tumorigenic assays (Figs 5G and H, and EV5F–I) and maintained the oncogenic potential of HeLa cells, indicating that clathrin inhibited BCL9 oncogenic function.
To investigate whether clathrin binds to and inhibits BCL9 function in a common or a cell‐type‐dependent manner, we further employed four more cancer cell lines: a colon cancer cell line—HCT116, a breast cancer cell line—MB‐MDA‐231, a lung cancer cell line—Calu1 and a liver cancer cell line—HCCLM3. We found that knockdown of BCL9 in HCT116, MDA‐MB‐231 and Calu1 cells inhibited cell growth (Fig EV5J); however, knockdown of BCL9 in HCCLM3 cells showed no significant inhibition on cell growth (Fig EV5J). By further investigating the interaction of BCL9 and clathrin in both asychronised and mitotic cells, we found that the binding of clathrin to BCL9 was much stronger in HCCLM3 cells than in the other three cell lines (Fig EV5K). These data confirm that clathrin inhibits BCL9‐mediated cancer cell growth via modulating the interaction with BCL9, which is a common regulatory mechanism in cancer cancers.
Our data thus confirmed that BCL9 stabilises LRP6 signalosome components by inhibiting clathrin, which is required to maintain mitotic Wnt signalling and for BCL9 oncogenic activation.
CDK1‐driven phosphorylation of BCL9 regulates clathrin and β‐catenin complex interaction
To address why BCL9 acts in mitosis, we further examined mitotic‐associated phosphorylation of BCL9, which was mentioned above (Figs 2A and EV2C–E). To identify mitotic BCL9 phosphorylation sites, we immunoprecipitated endogenous and overexpressed BCL9 by an antibody or Flag beads. Mass spectrum (MS) analysis revealed that there were at least two common phosphorylation sites at the N‐terminal in both endogenous and exogenous mitotic BCL9, although there are many randomly phosphorylated sites in N‐terminal. By further comparison with previous reported mitotic proteome phosphorylation in HeLa cells (Dephoure et al, 2008), only one phosphorylation candidate site, threonine (Thr) 172, was identified (Fig 6A). Alignment analysis of the sequence around T172 identified a CDK1 consensus sequence (S/T*‐P‐x‐K/R; Holt et al, 2009; Fig 6B). T172 was adjacent to HD1 (amino acid 174‐207) which has been demonstrated crucial for both mitotic clathrin complex and β‐catenin complex binding (Fig 4D). These findings indicated that CDK1 probably phosphorylates BCL9 to regulate its interaction. To confirm this, we generated two T172 phosphorylation polyclonal antibodies (Fig EV6A), and one of them was used for further analysis (Fig EV6B–E). We confirmed that this antibody could recognise the phosphorylation of BCL9 at T172 (p‐T172‐BCL9). Then, we in vitro translated and purified BCL9‐Flag and its phosphorylation mutant T172A‐Flag and incubated them with active CDK1/cyclin B1 proteins and/or ATP for kinase assays (Fig 6C), which confirmed that CDK1/cyclin B1 phosphorylates BCL9 at T172.
Figure 6. CDK1‐driven T172 phosphorylation stabilises BCL9 and regulates its interaction with clathrin.
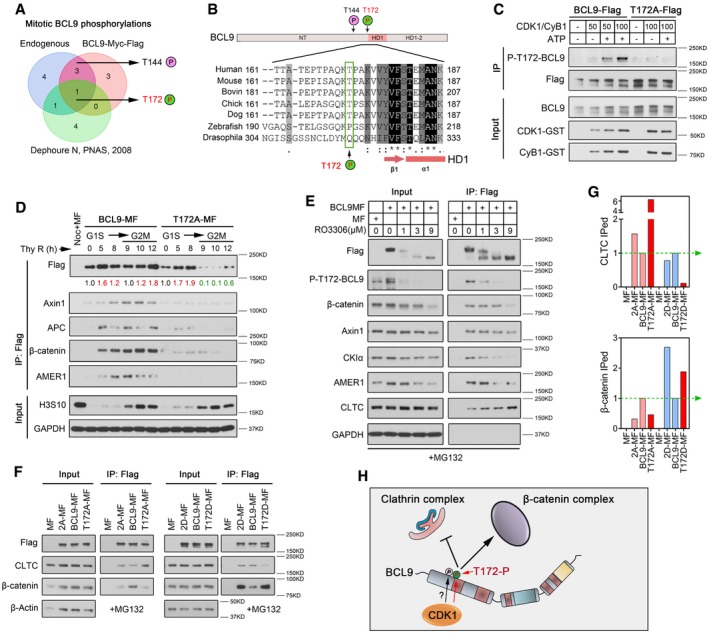
- Venn diagram analysis of BCL9 mitotic phosphorylation sites in HeLa cells by mass spectrometry.
- Schematic diagram of the position of two phosphorylation sites on the BCL9 N‐terminal domain, and protein alignment analysis of the sequence around T172 in different species from http://www.uniprot.org.
- In vitro CDK1/cyclin B1 kinase assay for immunoprecipitation‐purified BCL9‐Flag or T172A‐Flag proteins.
- Immunoprecipitation analysis of transiently expressed BCL9‐MF or T172A‐MF interactors in synchronised cells, from G1S to G2M phase. The relative level of BCL9‐MF or T172A‐MF immunoprecipitation is determined; red numbers indicate up‐regulation; and green numbers indicate down‐regulation compared to the starting time point.
- Immunoprecipitation analysis of the transiently expressed BCL9‐MF interaction in mitotic cells treated with RO3306.
- Immunoprecipitation analysis of the interaction of the T172 inactive mutant T172A, the activation mutant T172D, and the double inactive mutant T172/T144 (2A) or activation mutant (2D) transiently expressed in mitotic cells.
- Quantification for the inactive or activation mutant interactions indicated in Fig 6F. The wild‐type BCL9 interaction is normalised to 1, indicated by a green dashed line with an arrow.
- A schematic diagram of BCL9 phosphorylation regulation of its interactions.
Figure EV6. CDK1‐driven BCL9 phosphorylation at T172 regulates BCL9 interaction with clathrin in mitosis (related to Fig 6).
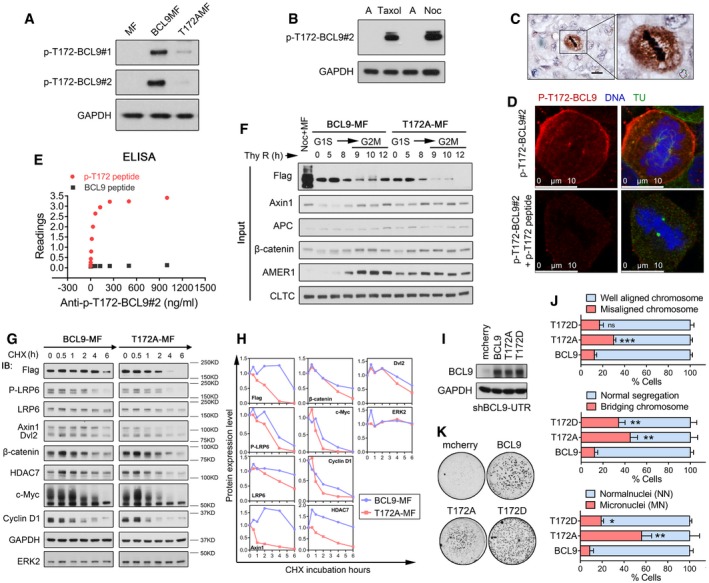
- Immunoblotting analysis of BCL9 T172 phosphorylation in mitotic cells with BCL9MF or T172AMF expression by two anti‐P‐T172‐BCL9 rabbit polyclonal antibodies.
- Immunoblotting analysis of endogenous BCL9 T172 phosphorylation in mitotic cells by a selected anti‐P‐T172‐BCL9 rabbit polyclonal antibody.
- Immunohistochemical staining of P‐T172‐BCL9 in a human HCC tissue. Scale bars represent 50 μm.
- Immunofluorescence analysis of P‐T172‐BCL9 subcellular localisation in mitotic cells treated with or without P‐T172 peptide blocking. Scale bars represent 10 μm.
- ELISA analysis to confirm anti‐P‐T172‐BCL9 antibody recognising T172 phosphorylation peptide specifically.
- Immunoblotting analysis of the input for Fig 6D.
- CHX chase analysis of the stability of overexpressed or endogenous proteins in mitotic cells with BCL9 stable knockdown by shBCL9‐UTR.
- Quantification of protein stability in the CHX chase assay mentioned above.
- Immunoblotting analysis of the BCL9 and its mutants expression in shBCL9‐UTR knockdown cells.
- Live‐cell imaging analysis of cell division problem by stable expression of BCL9, T172A or T172D. Twenty‐three H2BGFP‐mitotic stable cells were monitored for each time experiment; data are shown from three independent experiments. Significance was measured by two‐tailed unpaired t‐test, *P < 0.05, **P < 0.01, ***P < 0.001. All data are the mean ± SD.
- Oncogenic soft agar colony formation assay of cells stably expressing BCL9, T172A or T172D.
Source data are available online for this figure.
To investigate the effect of T172 phosphorylation on BCL9 interaction, we analysed wild‐type BCL9 or T172A mutant interaction in synchronised cells from G1S to G2M phase (Figs 6D and EV6F). T172A appeared unstable in mitosis and completely abolished β‐catenin complex binding (Figs 6D and EV6F), indicating a potential effect of T172 phosphorylation on BCL9 stability and interaction. To confirm these results, we employed a CHX chase assay in BCL9 or T172A transiently expressing cells in which endogenous BCL9 was knocked down simultaneously by a shRNA sequence targeting the 3′‐untranslated region (3′‐UTR) (also named shBCL9‐UTR). Our data further confirmed that T172A BCL9 was unstable compared to WT‐BCL9 in mitotic cells (Fig EV6G and H). Moreover, LRP6 signalosome and mitotic Wnt targets also appeared to be more stable in cells with WT‐BCL9 than with T172A (Fig EV6G and H), indicating that p‐T172‐BCL9 was involved in stabilising signalosome components and mitotic Wnt targets.
To confirm that CDK1 is required for BCL9 mitotic interaction, we incubated mitotic cells expressing BCL9‐myc‐Flag (BCL9MF) with RO3306, which dose dependently inhibited mitotic BCL9 phosphorylation, including p‐T172‐BCL9 (Fig 6E). Moreover, CDK1 inhibition suppressed BCL9 and β‐catenin complex interaction but notably increased BCL9 and CLTC binding (Fig 6E), demonstrating that BCL9 phosphorylation inhibits clathrin interaction. To further confirm these results, we cloned a T172D mutant mimicking T172 phosphorylation and a T144/T172 double mutant, and the transfection was manually optimised for similar bait protein level (Fig 6F and G). The IP result further confirmed that T172D inhibited clathrin binding and promoted β‐catenin complex binding, while T172A gave the opposite effect (Fig 6F and G).
To evaluate the biological function of T172 phosphorylation, T172A and T172D stably expressing cells were established in shBCL9‐UTR stable knockdown cells (Fig EV6I) and cell division and oncogenesis were further evaluated. To our surprise, both T172A and T172D expression resulted in abnormal cell division phenotype with MC, BC and MN (Fig EV6J), indicating p‐T172‐BCL9 controlled cell division. Moreover, oncogenic colony formation assays revealed that T172A‐expressing cells showed weak colony formation potential compared to WT‐BCL9‐expressing cells, while T172D‐expressing cells showed stronger colony formation ability (Fig EV6K).
Our data indicated that CDK1‐driven BCL9 phosphorylation promotes BCL9 oncogenic activity by regulating its stability and clathrin interaction to maintain normal mitotic Wnt signalling activity (Fig 6H).
P‐T172‐BCL9 is selectively expressed in human cancer tissues, and its expression correlates with cancer patient prognosis
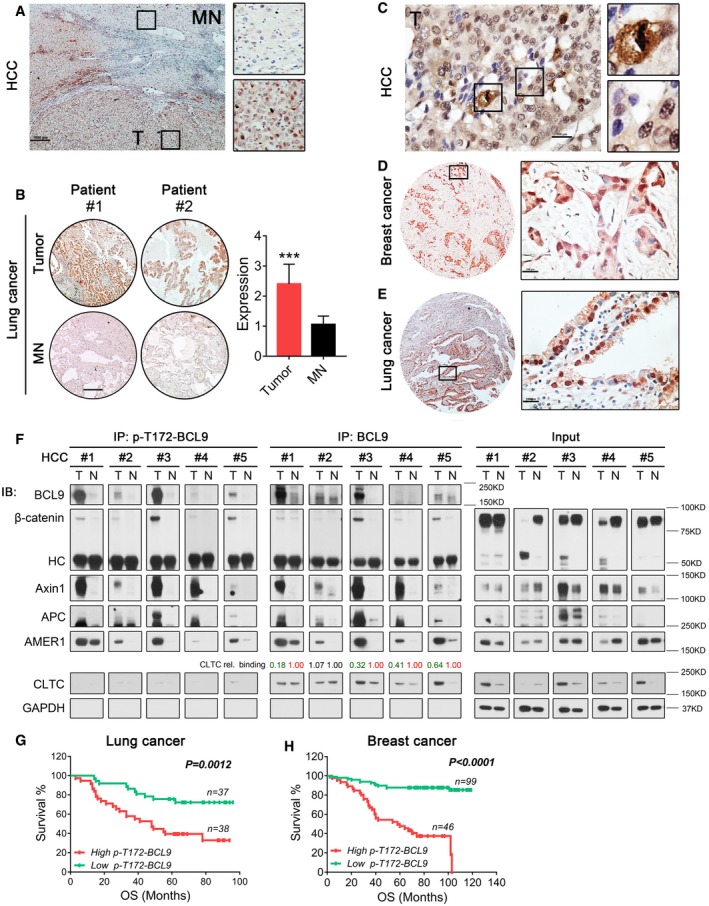
To examine the association of BCL9 phosphorylation with human cancer, we evaluated the expression of p‐T172‐BCL9 by IHC staining in the tissues of human hepatocellular carcinoma (HCC), breast cancer and lung cancer (Table EV5). T172 phosphorylation was significantly enriched in HCC and lung cancer tumours compared to matched normal tissues (Fig 7A and B). Interestingly, p‐T172‐BCL9 was also detected in interphase cells in addition to mitotic cells in human HCC, breast and lung cancers (Fig 7C–E). We further confirmed that p‐T172‐BCL9 was co‐expressed with CDK1 in HCC tissues (Fig EV7A).
Figure 7. T172 phosphorylation is selectively enriched in tumours, and its expression is correlated with cancer patient prognosis.
-
AImmunohistochemical staining of p‐T172‐BCL9 expression in a human HCC tissue. MN: matched normal tissues, T: tumour tissues. Scale bar represents 1,000 μm.
-
BIHC staining and the expression scored in a tissue array of 75 pairs of human lung cancer patient tissues. Scale bar represents 1,000 μm. Significance was measured by two‐tailed unpaired t‐test, ***P < 0.001, n = 75, all data are the mean ± SD.
-
CA representative figure of p‐T172‐BCL9 IHC staining in HCC to indicate the expression in mitotic or in interphase cells. Scale bar represents 100 μm.
-
D, ERepresentative figures of p‐T172‐BCL9 IHC staining in interphase cells of breast cancer (D) or lung cancer (E) patient tissue. Scale bars represent 100 μm.
-
FImmunoprecipitation analysis of the interaction in five pairs of human HCC patient tissues by a p‐T172‐BCL9 antibody or a BCL9 antibody. CLTC relative binding with BCL9 was determined.
-
G, HLog‐rank (Mantel–Cox) survival analysis based on high and low p‐T172‐BCL9 expression scored by immunohistochemical staining in two tissue arrays with (G) 75 lung cancer samples or (H) 145 breast cancer samples.
Figure EV7. T172 phosphorylation of BCL9 is selected enriched and interacted in tumours, and its expression is correlated with cancer patients' prognosis (related to Fig 7).
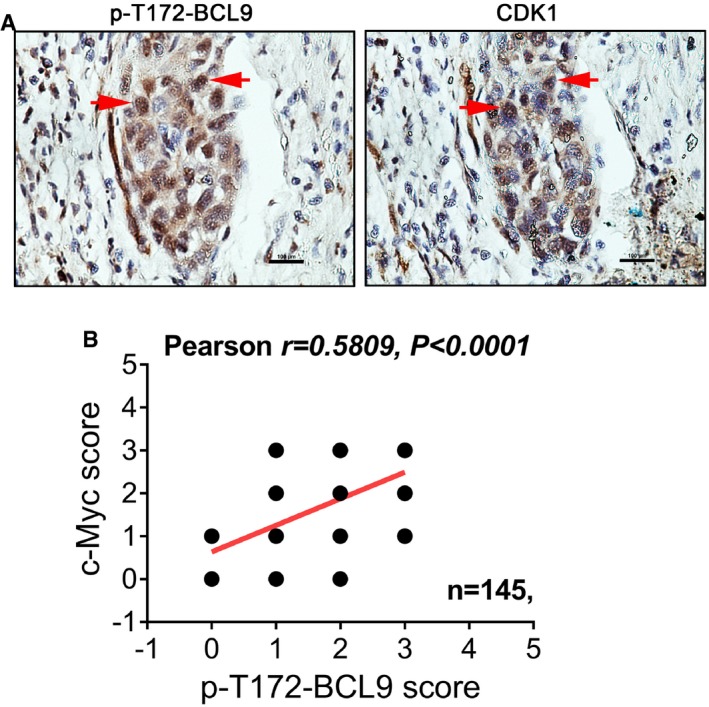
- Immunohistochemical staining of P‐T172‐BCL9 or CDK1 on two sequential HCC slides from one patient. The red arrows indicate the protein expression in the same tumour cells. Scale bars represent 100 μm.
- Pearson correlation analysis of the expression of P‐T172‐BCL9 and c‐Myc in 145 breast cancer patients by immunohistochemical staining.
To examine the BCL9 interaction in cancer tissues, we performed a tissue‐based IP with a T172 phosphorylation antibody or a BCL9 antibody in five pairs of human HCC tissues (Fig 7F and Table EV5). Both BCL9 and p‐T172‐BCL9 were highly enriched in HCC tumours compared to matched normal tissues. Moreover, p‐T172‐BCL9 was found to interact with the β‐catenin destruction complex primarily in tumour tissues (Fig 7F). P‐T172‐BCL9 barely interacted with clathrin in HCC tissues, while relatively strong binding was detected for the BCL9 and clathrin interaction. Interestingly, relatively weaker binding of BCL9 with CLTC was detected in tumour tissues than in matched normal tissues (Fig 7F), indicating that BCL9 phosphorylation may also inhibit clathrin in cancer tissues to activate mitotic Wnt signalling.
To evaluate the association of p‐T172‐BCL9 expression and the mitotic Wnt target, c‐Myc, we performed IHC staining of human c‐Myc protein with p‐T172‐BCL9 staining in a tissue array of 145 breast cancer patient samples. The p‐T172‐BCL9 expression score was significantly correlated with the c‐Myc expression score in breast cancer patients (Fig EV7B), indicating an association of BCL9 phosphorylation and c‐Myc in cancer. To examine whether p‐T172‐BCL9 is correlated with cancer patient survival, we evaluated the tissue arrays of 75 lung cancer patients and 145 breast cancer patients used above for p‐T172‐BCL9 expression, which were assessed for survival correlation (Fig 7G and H). Interestingly, high p‐T172‐BCL9 expression was significantly correlated with poor overall survival (OS) of patients with lung cancer (Fig 7G) or breast cancer (Fig 7H).
Overall, our data revealed an unexpected role of BCL9 in modulating mitotic Wnt signalling in oncogenic processes (Fig 8). When cancer cells enter mitosis, BCL9 is phosphorylated by CDK1 at T172 and possibly also at T144 and/or other random sites in N‐terminal. In mitosis, due to CDK14 activation by cyclin Y up‐regulation, the LRP6 co‐receptor is phosphorylated at PPPSP sites to promote active LRP6 signalosome formation (Davidson et al, 2009), and then, mitotic Wnt signalling can promote protein stabilisation. Meanwhile, N‐terminal phosphorylation of BCL9 further stabilises itself, LRP6 signalosome and mitotic Wnt targets by inhibiting clathrin binding with the signalosome and subsequent endocytosis and degradation of the signalosome. This helps to modulate precise cell division to preserve the growth potential of cancer cells (Fig 8).
Figure 8. A hypothetical model for BCL9 and its phosphorylation in regulating mitotic Wnt signalling to promote cancer cell growth.
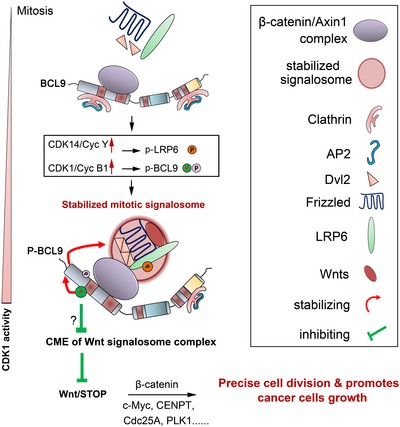
When cancer cells enter mitosis, BCL9 will bind to the β‐catenin/Axin1 complex via its N‐terminal, which includes HD2. As CDK14 and CDK1 kinases are activated by cyclin Y/Y‐L and cyclin B1, LRP6 is phosphorylated by CDK14, and BCL9 is phosphorylated by CDK1 on its N‐terminal. These changes will stabilise BCL9 and LRP6 signalosome components and prevent clathrin from binding and subsequent clathrin‐mediated‐endocytosis with mechanisms not fully explored, which further maintains mitotic Wnt/STOP signalling, modulates efficient cell division and promotes cancer cell growth.
Discussion
Here, we present evidence indicating that BCL9 phosphorylation modulates mitotic Wnt signalling in mitosis to promote cancer cell division and growth. The key discoveries of this study are that (i) BCL9 acts as a positive regulator in mitotic Wnt signalling; (ii) BCL9 regulates the mitotic spindle and cell division; (iii) BCL9 is localised in and regulates the mitotic β‐catenin destruction complex and clathrin complex; (iv) BCL9 stabilises the mitotic LRP6 signalosome by inhibiting the clathrin interaction and CME; (v) CDK1‐driven BCL9 phosphorylation at T172 inhibits the interaction of the BCL9 and clathrin complex to stabilise the LRP6 signalosome, and p‐T172‐BCL9 is correlated with cancer patient prognosis and is highly enriched in tumour tissues. Our discovery suggests a new potential model for the role of the oncoprotein BCL9 in mitotic Wnt signalling and linked the oncogenic kinase CDK1 to mitotic Wnt signalling in cancers.
The physiological role of Wnt/stabilisation of proteins (STOP) signalling has been reported in early Xenopus embryogenesis and in epididymal sperm maturation (Huang et al, 2015; Koch et al, 2015). Recently, the potential cancer‐associated roles of this signalling have also been suggested (Chen et al, 2017; Katoh & Katoh, 2017). One implicated oncogenic role of mitotic Wnt signalling is to stabilise proteins, some of which are prominent oncogenic drivers, such as c‐Myc, cyclin D1 or β‐catenin (Acebron et al, 2014). Moreover, mitotic Wnt signalling negative regulators that were confirmed here, including APC, Axin1 or CK Iα, are frequently mutated in human cancers (Polakis, 2012). These changes would lead to aberrant activation of Wnt signalling, stabilisation of targeted oncoproteins and chromosome instability caused by abnormal cell division. Here, we confirmed that increased BCL9 phosphorylation by aberrant CDK1 activation in cancer also contributes to mitotic Wnt signalling deregulation. In addition to phosphorylation, BCL9 gene expression has been shown to be highly enriched in many cancers, predominantly due to gene amplification (TCGA cbioportal, http://www.cbioportal.org/) and/or post‐transcriptional deregulation (Jia et al, 2011; Luna et al, 2017). In addition, the deregulation of mitotic progression regulators in cancer, such as spindle regulators (Chen et al, 2015, 2016), might induce abnormal mitotic distribution and lead to aberrant activation of mitotic Wnt signalling, which eventually leads to cancer progression and metastasis.
Mitotic cell division is a dynamic process controlled by CDK1 and other signalling cofactors (Malumbres & Barbacid, 2009). The phosphorylation of many CDK1 substrates, such as BCL9, shows a compatible dynamic change with cyclin B1 or securin expression level, increasing from prophase to metaphase and decreasing from metaphase to cytokinesis. Interestingly, mitotic Wnt signalling activity in cell lines is controlled in the same manner as CDK1 activity: activity is high in metaphase and low after anaphase. Thus, in addition to the regulation by CDK14/cyclin Y, another potential mechanism for regulating mitotic Wnt signalling by CDK1‐driven BCL9 phosphorylation might exist to further ensure the fidelity of chromosome segregation for cancer cell proliferation. Due to the cancer‐specific aberrant activation of CDK1 and its activators, BCL9 and other substrates could be phosphorylated in the interphase cells of cancer tissues and cancer xenograft, as demonstrated in our study. These cancer‐specific changes would trigger deregulation of the signalling network and/or stabilise CDK1 substrates including BCL9, to promote its oncogenic activity. Thus, BCL9 mitotic Wnt function may be present in interphase cancer cells of cancer tissues, but not normal tissues, to promote stabilisation of oncogenic drivers. Our discovery of BCL9 phosphorylation may be an example of a link between mitotic Wnt signalling and CDK1 in cancers. As the T172 phosphorylation site is conserved only in higher animals, this mechanism may offer an alternative programme to ensure genome integrity by modulating mitotic Wnt signalling activity modestly in response to external stimuli or environmental changes.
BCL9 has been reported as a nuclear regulator of β‐catenin and forms an enhanceosome driving Wnt target expression in Drosophila (Kramps et al, 2002; van Tienen et al, 2017). The mitotic interactors of BCL9 identified in this study are different from previous reported interactors in asychronised cells, such as Pygo, β‐catenin and Groucho/TLE co‐repressor (Kramps et al, 2002; van Tienen et al, 2017). This might be because the interactors reported here were identified specifically in mitosis HeLa cells. In mitosis, BCL9 is hyper‐phosphorylated on multiple sites, which mainly contributes to the formation of new complexes, the stabilisation of BCL9 and the mitotic novel function of BCL9 signalling. Moreover, it seems that BCL9 regulates its function and transcriptional target genes in a cell‐type‐ or tissue‐dependent manner. For cancer‐associated studies of BCL9, there are several studies reported that BCL9 had little effect on Wnt target expression and TCF reporter activity (Adachi et al, 2004; Brembeck et al, 2011). Here, we showed that in HeLa cells BCL9 also has little effect on Wnt target gene transcription, indicating that BCL9 has functions in Wnt signalling beyond transcription. However, in MM1S and Colo320 cells, BCL9 knockdown inhibited the gene expression of several Wnt targets, such as c‐Myc, cyclin D1 and VEGF (Mani et al, 2009). This might be due to the differences in the tissue sources, the differential expression of physiological interactors or cofactors with similar transcriptional functions, the different subcellular distribution, and the mutations, modification or variants of BCL9 in different cancer cell lines. A BCL9 cytoplasmic role has been indicated recently in tooth enamel formation via unknown regulation on exocytosis and vesicular trafficking (Cantu et al, 2017). This partially supports our discovery of the potential role of BCL9 in clathrin‐mediated trafficking, which still requires further verification. Although there is increasing evidence to support the mechanism of endocytic Wnt signalling in interphase cells, the detailed regulation in mitotic Wnt signalling is still unclear due to the controversy over mitotic endocytosis. In this study, we performed endocytosis‐related studies, primarily in unperturbed synchronised mitotic cells which have been validated with unaltered and functional endocytosis (Tacheva‐Grigorova et al, 2013), and we further verified the endocytosis effect on Wnt signalling.
The spindle regulation of BCL9 is possibly through its indirect effect on mitotic Wnt signalling activity. Some of the established mitotic Wnt signalling targets has been reported for spindle regulation, such as β‐catenin (Kaplan et al, 2004; Bahmanyar et al, 2008; Acebron et al, 2014), c‐Myc (O'Donovan et al, 2010), CENPT (Schleiffer et al, 2012), Cdc25A (Lindqvist et al, 2005) and PLK1 (Golsteyn et al, 1995). Thus, BCL9 may regulate spindle orientation via regulating these proteins. Moreover, BCL9 may also have a direct effect on spindle via interacting with spindle factors such as CHICA or HMMR, which has not been fully elucidated.
Here, we report a novel mechanistic role of BCL9 in regulating mitotic Wnt signalling in cancer where BCL9 is highly expressed and CDK1 is selectively activated. Therefore, CDK1/BCL9/mitotic Wnt signalling axis may act as a cancer‐associated oncogenic pathway, rather than a common regulatory pathway activated in normal tissues. In summary, CDK1/BCL9/mitotic Wnt signalling axis plays important roles in promoting the proliferation of cancer cell and could provide a valuable therapeutic target for cancer treatment.
Materials and Methods
Cell lines
Cell lines details were listed in key resources table in Appendix Supplementary Methods. Briefly, HeLa, H2BGFP‐HeLa, HCT116, MDA‐MB‐231, Calu‐1, 293Ta and HCCLM3 cells were cultured in Dulbecco's modified Eagle's medium (DMEM) with 10% foetal bovine serum (FBS) and 1% penicillin/streptomycin (PS). hTERT RPE1 cells cultured in DMEM:F12 medium with 10% foetal bovine serum (FBS) and 1% penicillin/streptomycin (PS).
Immunoprecipitation
The cells were harvested in ice‐cold PBS and put on ice for further experiments. For anti‐Flag M2 affinity gel based IP of BCL9‐MF, MF or T172A‐MF in Figs 3C, 4E and G, and 6C–F, cells were lysed by IP lysis buffer (Thermo Fisher Scientific) containing 25 mM Tris–HCl pH 7.4, 150 mM NaCl, 1% NP‐40, 1 mM EDTA, 5% glycerol, 2% protease inhibitor cocktail (for SILAC and MS identification, 5% protease inhibitor cocktail was used), 2% phosphatase inhibitor cocktail (for SILAC and MS identification, 5% phosphatase inhibitor cocktail was used), 0.1% benzonase nuclease. The lysate was clarified by centrifugation for 15 min 20,817 g, at 4°C. Protein concentration was determined by Pierce BCA Protein Assay Kit. Protein lysate was diluted to the concentration ≈ 1 mg/ml, and beads pre‐clean was further performed to remove the nonspecific binding to affinity gel for 1 h at 4°C by rotating on a roller. During this time, thoroughly suspend the anti‐Flag M2 affinity gel in the vial, in order to make a uniform suspension of the resin. For 1 mg total lysate, 20 μl affinity gel was employed, washed by three times with IP lysis buffer and spinned down by centrifugation for 30 s 5,000 g, at 4°C for each time. The lysate after beads pre‐clean was used for input and rest lysate was further mix with balanced Flag M2 affinity gel for IP for 3–4 h at 4°C by rotating on a roller. After that, centrifuge the resin for 30 s at 5,000 g and remove the supernatants with a narrow‐end pipette tip. The resin was further washed once by adding 1 ml IP lysis buffer with 0.5% protease inhibitor cocktail and 0.5% phosphatase inhibitor cocktail and transferred to a new tube for four more times wash. The supernatant was removed carefully by a narrow‐end pipette tip to fully remove the buffer. Then, the resin was added with 30 μl 1.5× NuPAGE LDS sample buffer with 0.1% DTT for elution by for 10 min heating at 95°C. The mix was further spinned down for 1 min 14,000 rpm at room temperature and carefully transferred to a clean tube for immunoblotting assay.
For endogenous IP by IgG, BCL9, Axin1, APC, CLTC antibody in Figs 3D, EV4A and C, and 4H, protein lysate with concentration 1 mg/ml was used. One day before IP process, the protein G and A Dynabeads was mixed 1:1 by volume and washed three times by IP lysis buffer, for 1 mg protein lysate, 5 μg antibody was mixed and incubated with 20 μl beads in IP lysis buffer with 5% BSA overnight. On the second day, cell lysate was prepared by IP lysis buffer as mentioned above and protein concentration was determined by BCA assay. 1 mg protein lysate was prepared and pre‐cleaned by 20 μl Dynabeads for 1 h, and then, the input was taken and the rest lysate was further mixed with prepared IP antibody beads for IP process. The IP was performed by rotating on a roller for 4 h at 4°C. After IP, the beads were washed for 4–5 times by 1 ml IP lysis buffer with MG132 on a DynaMag™‐2 Magnet (ThermoFisher Scientific). 40 μl 1× LDS sample buffer was used for elution by 10 min heating at 95°C.
For tissue‐based IP by BCL9 or P‐T172‐BCL9 antibody in Fig 7F, protein lysate with concentration 1.0 mg/ml was used. One day before IP process, the protein G and A Dynabeads were mixed 1:1 by volume and washed three times by IP lysis buffer, for 2 mg protein lysate, 8 μg antibody was mixed and incubated with 30 μl prepared beads in IP lysis buffer with 5% BSA overnight. On the second day, HCC tissues were cut into small pieces and further homogenised on ice by two hard surfaces slides in IP lysis buffer with 5% protease inhibitor cocktail, 5% phosphatase inhibitor cocktail, 0.2% benzonase nuclease, 20 μM MG‐132. The supernatant was transferred to a clean tube and spinned down by centrifugation for 10 min 14,000 rpm, at 4°C. After this, the supernatant was transferred to another clean tube and prepared for another centrifugation, these processes would be repeated for 4–5 times until the supernatant was clean without any fat or oil on top. The final protein lysate concentration was determined by BCA assay and 1.5 mg total protein was used for IP process. The IP was performed by rotating on a roller for overnight at 4°C. After IP, the beads were washed for five times by 1 ml IP lysis buffer with MG132 on a DynaMag™‐2 Magnet (ThermoFisher Scientific). 50 μl 1× LDS sample buffer was used for elution by 10 min heating at 95°C.
SILAC study of BCL9 interactome
HeLa cells were cultured in DMEM for SILAC (Thermo Fisher Scientific) supplemented with 10% dialysed FBS either containing normal isotopes of l‐lysine‐(12C614N2) (K0) and l‐arginine‐(12C614N4) (R0) (K0R0‐“light”, HeLa‐L) or stable isotope l‐lysine‐(13C615N2) (K8) and l‐arginine‐(13C615N4) (R10) (R10K8‐“heavy”, HeLa‐H) for at least six doublings to ensure efficient incorporation of labelled amino acids. The whole process of SILAC was briefly indicated in Fig EV3A. Briefly, around 4 μg MF (plus 12 μg pcDNA3.1) or 16 μg BCL9‐MF, which has been optimised to expresses same amount of Flag tag (data not shown), was transfected by TransIT‐LT1 transfection reagent (1:3) into HeLa‐L or HeLa‐H cells with density around 85% in 150 mm dishes. Eight dishes were used for transfection in both groups, and cells per dish were digested by trypsin and passage into three dishes to reach the density around 30%. When cells attached 6 h post‐plating, the cells were blocked by SILAC H or L medium with 2.5 mM thymidine for 24 h. The cells were further released into 50 ng/ml nocodazole for 14 h to enrich the mitotic cells. The mitotic cells were collected by shake‐off method and prepared for IP assay. The cells were lysed by IP lysate buffer, protein was quantified, and equal amount of protein in H or L was prepared for individual Flag IP assay as mentioned above. For IP, 35 mg total mitotic H (BCL9‐MF) or L (MF) lysate was employed, and 60 μl Flag‐M2 affinity gel was used for each IP assay. After IP and first wash, the beads from H and L cells' IP were mixed and washed for four more times. 30 μl 1× LDS sample buffer was used for elution by 10 min heating at 95°C. The IP elution was separated by a one‐dimensional NuPAGE™ 4–15% Bis‐Tris protein gels, stained with colloidal blue (Invitrogen). Protein bands were cut and digested with trypsin described previously (Chakraborty et al, 2015). The samples were further analysed on an Orbitrap Classic (Thermo Fisher Scientific). Identification and quantification were performed using MaxQuant version 1.5.0.30.
Additional and more detailed Materials and Methods are provided in the Appendix.
Author contributions
JC, MR, HX, SNK, AD and HG performed the experiments, and JC, MR, AD, WH, TX, LLO and KMH analysed the data. HLFS and JG performed the SILAC and MS/MS studies. KS performed the bioinformatics analysis. JC, WH and KMH designed the project, and JC and KMH wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Table EV1
Table EV2
Table EV3
Table EV4
Table EV5
Movie EV1
Movie EV2
Movie EV3
Source Data for Expanded View
Review Process File
Source Data for Figure 1
{kind=link}
Source Data for Figure 2
{kind=link}
Source Data for Figure 3
{kind=link}
Source Data for Figure 4
{kind=link}
Source Data for Figure 5
{kind=link}
Source Data for Figure 6
{kind=link}
Source Data for Figure 7
{kind=link}
Acknowledgements
We thank Dr. Philipp Kaldis, Institute of Molecular and Cell Biology, for critically reading and comments on the manuscript. We thank the SingHealth Tissue Repository (STR) for providing some of the human tissue samples, IHC staining and scoring. We thank the SingHealth Advanced Bioimaging Core for assisting in confocal imaging. We thank Prof. Erich Nigg for sharing the CHICA (FAM83D) antibody. This work was supported by grants from the National Medical Research Council of Singapore (NRNMRMH16101 to KMH and LLO), from Key projects of National Natural Science Foundation of China (81730108); Key Project of Zhejiang province Ministry of Science and Technology (2015C03055); Key Project of Hangzhou Ministry of Science and Technology (20162013A07, 20142013A63; to TX) and from the SingHealth Foundation, Start‐up Grant of HZNU, National Natural Science Foundation of China (81802338) (to JC).
The EMBO Journal (2018) 37: e99395
Contributor Information
Jianxiang Chen, Email: ncmcjx@yeah.net.
Kam Man Hui, Email: cmrhkm@nccs.com.sg.
References
- Acebron SP, Karaulanov E, Berger BS, Huang YL, Niehrs C (2014) Mitotic wnt signaling promotes protein stabilization and regulates cell size. Mol Cell 54: 663–674 [DOI] [PubMed] [Google Scholar]
- Adachi S, Jigami T, Yasui T, Nakano T, Ohwada S, Omori Y, Sugano S, Ohkawara B, Shibuya H, Nakamura T, Akiyama T (2004) Role of a BCL9‐related beta‐catenin‐binding protein, B9L, in tumorigenesis induced by aberrant activation of Wnt signaling. Cancer Res 64: 8496–8501 [DOI] [PubMed] [Google Scholar]
- Bahmanyar S, Kaplan DD, Deluca JG, Giddings TH Jr, O'Toole ET, Winey M, Salmon ED, Casey PJ, Nelson WJ, Barth AI (2008) beta‐Catenin is a Nek2 substrate involved in centrosome separation. Genes Dev 22: 91–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilic J, Huang YL, Davidson G, Zimmermann T, Cruciat CM, Bienz M, Niehrs C (2007) Wnt induces LRP6 signalosomes and promotes dishevelled‐dependent LRP6 phosphorylation. Science 316: 1619–1622 [DOI] [PubMed] [Google Scholar]
- Boucrot E, Kirchhausen T (2007) Endosomal recycling controls plasma membrane area during mitosis. Proc Natl Acad Sci USA 104: 7939–7944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brembeck FH, Wiese M, Zatula N, Grigoryan T, Dai Y, Fritzmann J, Birchmeier W (2011) BCL9‐2 promotes early stages of intestinal tumor progression. Gastroenterology 141: 1359–1370 [DOI] [PubMed] [Google Scholar]
- Cantu C, Pagella P, Shajiei TD, Zimmerli D, Valenta T, Hausmann G, Basler K, Mitsiadis TA (2017) A cytoplasmic role of Wnt/beta‐catenin transcriptional cofactors Bcl9, Bcl9 l, and Pygopus in tooth enamel formation. Sci Signal 10: eaah4598 [DOI] [PubMed] [Google Scholar]
- Chakraborty S, Lakshmanan M, Swa HL, Chen J, Zhang X, Ong YS, Loo LS, Akincilar SC, Gunaratne J, Tergaonkar V, Hui KM, Hong W (2015) An oncogenic role of Agrin in regulating focal adhesion integrity in hepatocellular carcinoma. Nat Commun 6: 6184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Xia H, Zhang X, Karthik S, Pratap SV, Ooi LL, Hong W, Hui KM (2015) ECT2 regulates the Rho/ERK signalling axis to promote early recurrence in human hepatocellular carcinoma. J Hepatol 62: 1287–1295 [DOI] [PubMed] [Google Scholar]
- Chen J, Rajasekaran M, Xia H, Zhang X, Kong SN, Sekar K, Seshachalam VP, Deivasigamani A, Goh BK, Ooi LL, Hong W, Hui KM (2016) The microtubule‐associated protein PRC1 promotes early recurrence of hepatocellular carcinoma in association with the Wnt/beta‐catenin signalling pathway. Gut 65: 1522–1534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Rajasekaran M, Hui KM (2017) Atypical regulators of Wnt/beta‐catenin signaling as potential therapeutic targets in Hepatocellular Carcinoma. Exp Biol Med (Maywood) 242: 1142–1149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clevers H, Nusse R (2012) Wnt/beta‐catenin signaling and disease. Cell 149: 1192–1205 [DOI] [PubMed] [Google Scholar]
- Davidson G, Shen J, Huang YL, Su Y, Karaulanov E, Bartscherer K, Hassler C, Stannek P, Boutros M, Niehrs C (2009) Cell cycle control of wnt receptor activation. Dev Cell 17: 788–799 [DOI] [PubMed] [Google Scholar]
- Davidson G, Niehrs C (2010) Emerging links between CDK cell cycle regulators and Wnt signaling. Trends Cell Biol 20: 453–460 [DOI] [PubMed] [Google Scholar]
- Dephoure N, Zhou C, Villen J, Beausoleil SA, Bakalarski CE, Elledge SJ, Gygi SP (2008) A quantitative atlas of mitotic phosphorylation. Proc Natl Acad Sci USA 105: 10762–10767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominguez‐Brauer C, Thu KL, Mason JM, Blaser H, Bray MR, Mak TW (2015) Targeting mitosis in cancer: emerging strategies. Mol Cell 60: 524–536 [DOI] [PubMed] [Google Scholar]
- Funk LC, Zasadil LM, Weaver BA (2016) Living in CIN: mitotic infidelity and its consequences for tumor promotion and suppression. Dev Cell 39: 638–652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gammons MV, Renko M, Johnson CM, Rutherford TJ, Bienz M (2016) Wnt signalosome assembly by DEP domain swapping of dishevelled. Mol Cell 64: 92–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golsteyn RM, Mundt KE, Fry AM, Nigg EA (1995) Cell cycle regulation of the activity and subcellular localization of Plk1, a human protein kinase implicated in mitotic spindle function. J Cell Biol 129: 1617–1628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habib SJ, Chen BC, Tsai FC, Anastassiadis K, Meyer T, Betzig E, Nusse R (2013) A localized Wnt signal orients asymmetric stem cell division in vitro . Science 339: 1445–1448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadjihannas MV, Bruckner M, Jerchow B, Birchmeier W, Dietmaier W, Behrens J (2006) Aberrant Wnt/beta‐catenin signaling can induce chromosomal instability in colon cancer. Proc Natl Acad Sci USA 103: 10747–10752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt LJ, Tuch BB, Villen J, Johnson AD, Gygi SP, Morgan DO (2009) Global analysis of Cdk1 substrate phosphorylation sites provides insights into evolution. Science 325: 1682–1686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang YL, Anvarian Z, Doderlein G, Acebron SP, Niehrs C (2015) Maternal Wnt/STOP signaling promotes cell division during early Xenopus embryogenesis. Proc Natl Acad Sci USA 112: 5732–5737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jallepalli PV, Lengauer C (2001) Chromosome segregation and cancer: cutting through the mystery. Nat Rev Cancer 1: 109–117 [DOI] [PubMed] [Google Scholar]
- Jia W, Eneh JO, Ratnaparkhe S, Altman MK, Murph MM (2011) MicroRNA‐30c‐2* expressed in ovarian cancer cells suppresses growth factor‐induced cellular proliferation and downregulates the oncogene BCL9. Mol Cancer Res 9: 1732–1745 [DOI] [PubMed] [Google Scholar]
- Johnson DG, Dent SY (2013) Chromatin: receiver and quarterback for cellular signals. Cell 152: 685–689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan KB, Burds AA, Swedlow JR, Bekir SS, Sorger PK, Nathke IS (2001) A role for the Adenomatous Polyposis Coli protein in chromosome segregation. Nat Cell Biol 3: 429–432 [DOI] [PubMed] [Google Scholar]
- Kaplan DD, Meigs TE, Kelly P, Casey PJ (2004) Identification of a role for beta‐catenin in the establishment of a bipolar mitotic spindle. J Biol Chem 279: 10829–10832 [DOI] [PubMed] [Google Scholar]
- Katoh M, Katoh M (2017) Molecular genetics and targeted therapy of WNT‐related human diseases (Review). Int J Mol Med 40: 587–606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kikuchi K, Niikura Y, Kitagawa K, Kikuchi A (2010) Dishevelled, a Wnt signalling component, is involved in mitotic progression in cooperation with Plk1. EMBO J 29: 3470–3483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch S, Acebron SP, Herbst J, Hatiboglu G, Niehrs C (2015) Post‐transcriptional Wnt signaling governs epididymal sperm maturation. Cell 163: 1225–1236 [DOI] [PubMed] [Google Scholar]
- Kramps T, Peter O, Brunner E, Nellen D, Froesch B, Chatterjee S, Murone M, Zullig S, Basler K (2002) Wnt/wingless signaling requires BCL9/legless‐mediated recruitment of pygopus to the nuclear beta‐catenin‐TCF complex. Cell 109: 47–60 [DOI] [PubMed] [Google Scholar]
- Lindqvist A, Kallstrom H, Lundgren A, Barsoum E, Rosenthal CK (2005) Cdc25B cooperates with Cdc25A to induce mitosis but has a unique role in activating cyclin B1‐Cdk1 at the centrosome. J Cell Biol 171: 35–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luna JM, Barajas JM, Teng KY, Sun HL, Moore MJ, Rice CM, Darnell RB, Ghoshal K (2017) Argonaute CLIP defines a deregulated miR‐122‐bound transcriptome that correlates with patient survival in human liver cancer. Mol Cell 67: 400–410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDonald BT, Tamai K, He X (2009) Wnt/beta‐catenin signaling: components, mechanisms, and diseases. Dev Cell 17: 9–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malumbres M, Barbacid M (2009) Cell cycle, CDKs and cancer: a changing paradigm. Nat Rev Cancer 9: 153–166 [DOI] [PubMed] [Google Scholar]
- Mani M, Carrasco DE, Zhang Y, Takada K, Gatt ME, Dutta‐Simmons J, Ikeda H, Diaz‐Griffero F, Pena‐Cruz V, Bertagnolli M, Myeroff LL, Markowitz SD, Anderson KC, Carrasco DR (2009) BCL9 promotes tumor progression by conferring enhanced proliferative, metastatic, and angiogenic properties to cancer cells. Cancer Res 69: 7577–7586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morin X, Bellaiche Y (2011) Mitotic spindle orientation in asymmetric and symmetric cell divisions during animal development. Dev Cell 21: 102–119 [DOI] [PubMed] [Google Scholar]
- Niehrs C, Acebron SP (2012) Mitotic and mitogenic Wnt signalling. EMBO J 31: 2705–2713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nusse R, Clevers H (2017) Wnt/beta‐catenin signaling, disease, and emerging therapeutic modalities. Cell 169: 985–999 [DOI] [PubMed] [Google Scholar]
- O'Donovan KJ, Diedler J, Couture GC, Fak JJ, Darnell RB (2010) The onconeural antigen cdr2 is a novel APC/C target that acts in mitosis to regulate c‐myc target genes in mammalian tumor cells. PLoS One 5: e10045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ong SE, Blagoev B, Kratchmarova I, Kristensen DB, Steen H, Pandey A, Mann M (2002) Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol Cell Proteomics 1: 376–386 [DOI] [PubMed] [Google Scholar]
- Ong TS, Dayanandan B, Fournier AE, Mandato CA (2010) GSK3 regulates mitotic chromosomal alignment through CRMP4. PLoS One 5: e14345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polakis P (2012) Wnt signaling in cancer. Cold Spring Harb Perspect Biol 4: a008052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulton JS, Mu FW, Roberts DM, Peifer M (2013) APC2 and Axin promote mitotic fidelity by facilitating centrosome separation and cytoskeletal regulation. Development 140: 4226–4236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Roche M, Rutherford TJ, Gupta D, Veprintsev DB, Saxty B, Freund SM, Bienz M (2012) An intrinsically labile alpha‐helix abutting the BCL9‐binding site of beta‐catenin is required for its inhibition by carnosic acid. Nat Commun 3: 680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Roche M, Worm J, Bienz M (2008) The function of BCL9 in Wnt/beta‐catenin signaling and colorectal cancer cells. BMC Cancer 8: 199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sampietro J, Dahlberg CL, Cho US, Hinds TR, Kimelman D, Xu W (2006) Crystal structure of a beta‐catenin/BCL9/Tcf4 complex. Mol Cell 24: 293–300 [DOI] [PubMed] [Google Scholar]
- Schleiffer A, Maier M, Litos G, Lampert F, Hornung P, Mechtler K, Westermann S (2012) CENP‐T proteins are conserved centromere receptors of the Ndc80 complex. Nat Cell Biol 14: 604–613 [DOI] [PubMed] [Google Scholar]
- Schlesinger A, Shelton CA, Maloof JN, Meneghini M, Bowerman B (1999) Wnt pathway components orient a mitotic spindle in the early Caenorhabditis elegans embryo without requiring gene transcription in the responding cell. Genes Dev 13: 2028–2038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segalen M, Johnston CA, Martin CA, Dumortier JG, Prehoda KE, David NB, Doe CQ, Bellaiche Y (2010) The Fz‐Dsh planar cell polarity pathway induces oriented cell division via Mud/NuMA in Drosophila and zebrafish. Dev Cell 19: 740–752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenov MV, Zhang X, He X (2008) DKK1 antagonizes Wnt signaling without promotion of LRP6 internalization and degradation. J Biol Chem 283: 21427–21432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stolz A, Neufeld K, Ertych N, Bastians H (2015) Wnt‐mediated protein stabilization ensures proper mitotic microtubule assembly and chromosome segregation. EMBO Rep 16: 490–499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tacheva‐Grigorova SK, Santos AJ, Boucrot E, Kirchhausen T (2013) Clathrin‐mediated endocytosis persists during unperturbed mitosis. Cell Rep 4: 659–668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taelman VF, Dobrowolski R, Plouhinec JL, Fuentealba LC, Vorwald PP, Gumper I, Sabatini DD, De Robertis EM (2010) Wnt signaling requires sequestration of glycogen synthase kinase 3 inside multivesicular endosomes. Cell 143: 1136–1148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takada K, Zhu D, Bird GH, Sukhdeo K, Zhao JJ, Mani M, Lemieux M, Carrasco DE, Ryan J, Horst D, Fulciniti M, Munshi NC, Xu W, Kung AL, Shivdasani RA, Walensky LD, Carrasco DR (2012) Targeted disruption of the BCL9/beta‐catenin complex inhibits oncogenic Wnt signaling. Sci Transl Med 4: 148ra117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Tienen LM, Mieszczanek J, Fiedler M, Rutherford TJ, Bienz M (2017) Constitutive scaffolding of multiple Wnt enhanceosome components by Legless/BCL9. Elife 6: e20882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto H, Sakane H, Yamamoto H, Michiue T, Kikuchi A (2008) Wnt3a and Dkk1 regulate distinct internalization pathways of LRP6 to tune the activation of beta‐catenin signaling. Dev Cell 15: 37–48 [DOI] [PubMed] [Google Scholar]
- Zhan T, Rindtorff N, Boutros M (2017) Wnt signaling in cancer. Oncogene 36: 1461–1473 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Table EV1
Table EV2
Table EV3
Table EV4
Table EV5
Movie EV1
Movie EV2
Movie EV3
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7