

Abstract
The synapse transmits, processes, and stores data within its tiny space. Effective and specific signaling requires precise alignment of the relevant components. This review examines current insights into mechanisms of AMPAR and NMDAR localization by PSD‐95 and their spatial distribution at postsynaptic sites to illuminate the structural and functional framework of postsynaptic signaling. It subsequently delineates how β2 adrenergic receptor (β2 AR) signaling via adenylyl cyclase and the cAMP‐dependent protein kinase PKA is organized within nanodomains. Here, we discuss targeting of β2 AR, adenylyl cyclase, and PKA to defined signaling complexes at postsynaptic sites, i.e., AMPARs and the L‐type Ca2+ channel Cav1.2, and other subcellular surface localizations, the role of A kinase anchor proteins, the physiological relevance of the spatial restriction of corresponding signaling, and their interplay with signal transduction by the Ca2+‐ and calmodulin‐dependent kinase CaMKII. How localized and specific signaling by cAMP occurs is a central cellular question. The dendritic spine constitutes an ideal paradigm for elucidating the dimensions of spatially restricted signaling because of their small size and defined protein composition.
Keywords: AKAP, cAMP, NMDA receptors, norepinephrine, PSD‐95
Subject Categories: Neuroscience, Signal Transduction
Glossary
- AC
adenylate cyclase
- AKAP
A kinase anchor protein
- AMPAR
α‐amino‐3‐hydroxy‐5‐methyl‐4‐isoxazolepropionic acid receptor; AMPA‐type glutamate receptor
- CaMKII
Ca2+/calmodulin‐dependent protein kinase II
- Cav1.2
class C L‐type Ca2+ channel
- Cav1.3
class D L‐type Ca2+ channel
- CI‐AMPAR
Ca2+‐impermeable AMPAR
- CNIH2/3
cornichon homologues 2 and 3
- CP‐AMPAR
Ca2+‐permeable AMPAR
- EPSP
excitatory postsynaptic potential
- GABA
γ‐aminobutyric acid
- GKAP
guanylate kinase domain‐associated protein (also known as SAPAP)
- GK
guanylate kinase
- GluA
AMPA‐type glutamate receptor subunit
- GluN
NMDA‐type glutamate receptor subunit
- GRK
G protein‐coupled receptor kinase
- GsPCR
Gs protein‐coupled receptor
- LTD
long‐term depression
- LTP
long‐term potentiation
- MAP2B
microtubule‐associated protein type 2B
- mEPSC
miniature excitatory postsynaptic current
- mGluR
metabotropic glutamate receptor
- NFκB
nuclear factor kappa‐light‐chain‐enhancer of activated B cells
- NMDAR
N‐methyl‐D‐aspartate receptor; NMDA‐type glutamate receptor
- PDZ
PSD‐95–discs large–zonula occludens homology domain
- PFC
prefrontal cortex
- PKA
cAMP‐dependent protein kinase; protein kinase A
- PKC
protein kinase C
- PP1
serine/threonine protein phosphatase 1
- PP2B
protein phosphatase 2B/calcineurin
- PSD‐93
postsynaptic density protein of 93 kDa
- PSD‐95
postsynaptic density protein of 95 kDa
- PSD
postsynaptic density
- PTT‐LTP
prolonged theta tetanus‐long‐term potentiation
- Pyk2
protein tyrosine kinase 2; Ca‐dependent tyrosine kinase (CADTK)
- SAP102
synapse‐associated protein of 102 kDa
- SAP97
synapse‐associated protein of 97 kDa
- SAPAP
synapse‐associated protein–associated protein (also known as GKAP)
- SH3
Src homology domain 3
- Src
proto‐oncogene tyrosine‐protein kinase
- STEP
striatal‐enriched protein tyrosine phosphatase
- Stg
stargazing (γ2)
- SynDIG1
synapse differentiation‐induced gene 1
- TARP
transmembrane AMPAR regulatory protein
- TNF
tumor necrosis factor
- γ8
transmembrane AMPAR regulatory protein (TARP) γ8
Introduction
The mind‐staggering capabilities of the brain depend on the complexity of neuronal connections formed by ~1015 synapses. Synapses not only transmit and process signals; they also constitute the basic unit for information storage such as episodic memory or motor skills utilizing different forms of synaptic plasticity such as long‐term potentiation (LTP—see Glossary) and long‐term depression (LTD). LTP is a permanent increase in the otherwise remarkably stable synaptic strength following brief synaptic stimulations at high frequency (1 s, 50–200 Hz), and LTD is a decrease induced by longer synaptic stimulations at modest frequency (Collingridge et al, 2004; Lisman & Hell et al, 2008; Sudhof & Malenka, 2008; Kessels & Malinow, 2009; Huganir & Nicoll, 2013; Morris, 2013). Furthermore, synapses decode and process signals in a dynamically regulated manner. For instance, spine Na+ channels amplify (Araya et al, 2007) and K+ channels dampen (Kim et al, 2007) postsynaptic excitation. Thus, the synapse integrates in its minute space a large number of signaling mechanisms, constituting a unique system for studying localized signaling.
Synaptic functions are modulated by various signaling pathways such as the prominent β2 AR–PKA cascade (Lee et al, 2000; Oh et al, 2006; Hu et al, 2007; Lu et al, 2007; Joiner et al, 2010; Havekes et al, 2012; Qian et al, 2012, 2017; Murphy et al, 2014a; Patriarchi et al, 2016). The widely studied PKA is exemplary for localized signaling via formation of signaling complexes. It resides in stable complexes with AMPARs (Rosenmund et al, 1994; Tavalin et al, 2002; Joiner et al, 2010) and the L‐type Ca2+ channel Cav1.2 (Davare et al, 1999; Balijepalli et al, 2006). Remarkably, these AMPAR and Cav1.2 complexes also contain the β2 AR, trimeric Gs protein, and adenylyl cyclase (AC), for highly localized regulation via cAMP (Davare et al, 2001; Joiner et al, 2010; Wang et al, 2010). PKA is important for many forms of learning and memory in the broadest sense, which are as diverse as declarative and spatial memory (Lee et al, 2003), fear conditioning (Hu et al, 2007) and its reversal (Clem & Huganir, 2010; particularly relevant for posttraumatic stress disorder), drug addiction (Wolf & Tseng, 2012), and plasticity of sensory maps (Fischer et al, 2004). The β2 AR is the main postsynaptic mediator of signaling by norepinephrine (Joiner et al, 2010; Qian et al, 2012, 2017; Patriarchi et al, 2016). Norepinephrine is important for arousal, acuity of behavioral tasks, and learning in novel and emotionally charged situations (Cahill et al, 1994; Berman & Dudai, 2001; Hu et al, 2007; Minzenberg et al, 2008; Carter et al, 2010; He et al, 2015).
After a thorough overview of the overall organization of the postsynaptic site of glutamatergic synapses, this review will assess our knowledge of the molecular details and spatial and functional aspects of the classical postsynaptic signaling by β2 AR–AC–PKA pathways. The review will then contrast those pre‐assembled pathways with signaling by CaMKII, which is recruited to postsynaptic sites that experience heightened synaptic activity and Ca2+ influx through the NMDAR. This Ca2+ influx and the ensuing activation of CaMKII are absolutely critical for various forms of learning and LTP (Collingridge et al, 2004; Lisman & Hell et al, 2008; Sudhof & Malenka, 2008; Kessels & Malinow, 2009; Huganir & Nicoll, 2013; Morris, 2013). We propose that the diffuse signaling by NE throughout large brain regions by volume release sensitizes a broad population of synapses to induction of synaptic plasticity during alert states via preformed β2 AR complexes with AMPARs and Cav1.2, whereas the activity‐driven and highly specific recruitment of CaMKII to a small number of individual synapses that experience Ca2+ influx serves as the defining key step in the induction of LTP at those selected synapses for storage of specific information such as environmental maps or fear conditioning. Whether assembly of signaling complexes is constitutive or induced, target association is essential to minimize off target effects by cAMP, PKA, or CaMKII.
AMPARs and NMDARs
More than 80% of synapses in the cortex are glutamatergic (Micheva et al, 2010). The AMPAR mediates most of the basal postsynaptic response with the slower NMDAR contributing a smaller fraction especially during the later part of an EPSP. AMPARs consist of four homologous subunits (GluA1‐4) (Traynelis et al, 2010). Diheterotetrameric GluA1/2 receptors account for ~80% and GluA2/3 receptors for most of the rest of postsynaptic AMPARs in forebrain under basal conditions (Wenthold et al, 1996; Lu et al, 2009; Traynelis et al, 2010; Fig 1). A minority of AMPARs consist of GluA1 only, which plays a role during the early phase of LTP at certain ages as well as LTD (see below). These AMPARs lack GluA2, which acquires via RNA editing an Arg at a position within its pore that is otherwise occupied by Gln in GluA1, GluA3, GluA4, and unedited GluA2. The positive charge reduces single‐channel conductance and makes GluA2‐containing AMPAR Ca2+‐impermeable (CI‐AMPARs). GluA2‐lacking AMPARs are Ca2+‐permeable (CP‐AMPARs).
Figure 1. Postsynaptic AMPARs, NMDARs, and PSD‐95.
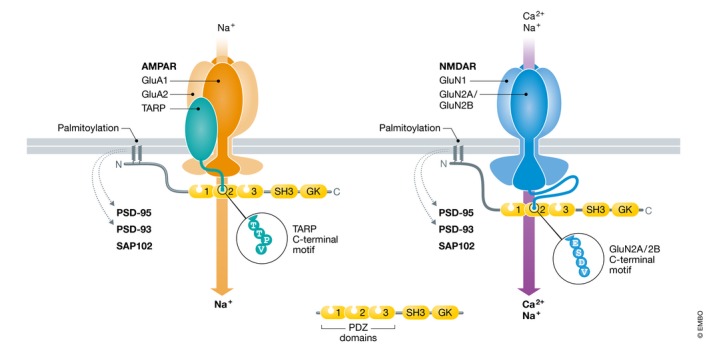
AMPARs mostly consist of two GluA1 and two GluA2 subunits (blue) plus one or more TARP subunits (green). TARPs mediate postsynaptic localization by binding to PSD‐95 (yellow) and its homologues, PSD‐93, and SAP102, which contain three PDZ domains followed by an SH3 and a GK domain. NMDARs mostly consist of two GluN1 and two GluN2A/2B subunits (purple); GluN2A/2B directly bind to PSD‐93/PSD‐95/SAP102. AMPARs are mainly Na+‐ and K+‐permeable, whereas NMDARs also conduct Ca2+. Postsynaptic anchoring of PSD‐95 and some PSD‐93 isoforms but not of SAP102 requires palmitoylation of their N‐termini (depicted in zigzag), which fosters their interactions with AMPARs and NMDARs. The C‐terminal ESDV motif of GluN2A/2B has a higher affinity for PDZ1 and PDZ2 than the TTPV motif of TARPs.
Like AMPARs, the homologous NMDARs are tetramers, which are formed by two GluN1 and two GluN2 subunits. One GluN1 and four GluN2 (A‐D) genes exist, with GluN1/2A and GluN1/2B being the predominant isoforms in forebrain (Traynelis et al, 2010; Gray et al, 2011). Whereas most AMPARs are mainly permeable for Na+ (and K+), NMDARs also conduct Ca2+ (Fig 1). AMPARs and NMDARs are clustered at the postsynaptic density (PSD), a protein‐dense meshwork, and precisely juxtaposed to the presynaptic active zone (Tang et al, 2016; Biederer et al, 2017) for fast and efficient postsynaptic responses (Clements et al, 1992).
Postsynaptic Distribution of AMPARs and NMDARs
A typical PSD is 300–400 nm in diameter (Harris & Stevens, 1989; Shepherd & Harris, 1998; Dani et al, 2010). It contains 30–150 AMPARs and 20–30 NMDARs according to immunogold EM (Nusser et al, 1998; Tanaka et al, 2005; Fukazawa & Shigemoto, 2012), EM tomography (Chen et al, 2008, 2015), electrophysiology (Bekkers & Stevens, 1989; Spruston et al, 1995; Smith et al, 2003), glutamate uncaging (Matsuzaki et al, 2001), and proteomic analysis of Triton X‐100‐treated PSDs (Sheng & Hoogenraad, 2007). PSD size is linearly correlated with AMPAR (Takumi et al, 1999; Shinohara et al, 2008; Fukazawa & Shigemoto, 2012; Chen et al, 2015) but not NMDAR content, which is fairly invariant between synapses of quite different sizes (Takumi et al, 1999; Racca et al, 2000; Shinohara et al, 2008; Chen et al, 2015). Because of the disproportional content of Ca2+‐permeable NMDARs, small spines have larger Ca2+ transients than large spines (Nimchinsky et al, 2004).
AMPARs are ~24–27 nm and NMDARs ~30‐32 nm apart when measured center to center (Chen et al, 2008, 2015; Fig 2). NMDARs tend to localize toward the PSD center when AMPAR density seems to be skewed toward the periphery (Matsubara et al, 1996; Kharazia & Weinberg, 1997; Petralia et al, 1998; Somogyi et al, 1998; Chen et al, 2008, 2015) although some other work suggests a more uniform distribution of AMPARs within the PSD (Nusser et al, 1994). AMPARs are concentrated in clusters of ~100 nm diameter, together with PSD‐95 and its binding partners GKAP, Shank, and Homer (Fukata et al, 2013; MacGillavry et al, 2013; Nair et al, 2013; Sinnen et al, 2017). Localization of AMPARs in those nanoclusters is thought to be functionally important because affinity of AMPARs for glutamate is fairly low and only AMPARs that are exactly juxtaposed to presynaptic release sites might be effectively activated (Franks et al, 2003; Lisman & Raghavachari, 2006). In fact, those nanoclusters appear to be closely aligned with presynaptic release sites for fast and efficient synaptic transmission (Tang et al, 2016; Biederer et al, 2017; Hruska et al, 2018). New functional evidence for this hypothesis has recently been provided by optogenetically induced binding of GluA1 to postsynaptic proteins (Sinnen et al, 2017). Accordingly, optogenetic AMPAR recruitment to spines did not augment the mEPSC amplitude in synapses that already contained functional AMPAR (although it did induce AMPAR responses in so‐called silent synapses in which no AMPAR activity was detected before light exposure). Glutamate uncaging, which stimulates AMPARs over the whole surface of dendritic spines, demonstrated that AMPARs were clearly increased on the spine surface of synapses that did contain AMPAR before light application, yet responses to presynaptic glutamate release were not. Furthermore, stochastic optical reconstruction microscopy (STORM) imaging showed that light‐induced recruitment occurred into the postsynaptic density, the postsynaptic site of glutamatergic synapses. These results indicate that augmenting AMPAR content on the spine surface is not sufficient for augmenting postsynaptic response, thereby supporting the hypothesis that AMPARs must be present near the presynaptic glutamate release sites for their effective activation (MacGillavry et al, 2013; Nair et al, 2013; Tang et al, 2016; Hruska et al, 2018). However, it still remains unclear whether only AMPARs in those nanoclusters or AMPARs inside the PSD in general are activated during regular synaptic transmission.
Figure 2. AMPAR and NMDAR dimensions and distribution at the postsynaptic site.
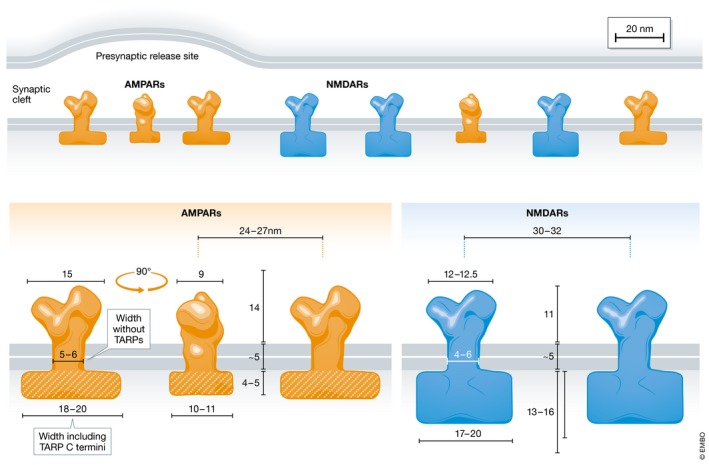
The extracellular N‐termini of inactive AMPARs are V‐shaped dimers of dimers and 14 nm high and 9 nm × 15 nm wide at the tip (Nakagawa et al, 2005; Sobolevsky et al, 2009). The transmembrane segment is ~5 nm long and 5–6 nm in diameter without TARPs (Nakagawa et al, 2006; Sobolevsky et al, 2009) and significantly wider with TARPs (Nakagawa et al, 2005). The intracellular AMPAR C‐termini in conjunction with C‐termini of associated TARPs are 18–20 nm × 10–11 nm wide and 4–5 nm high (Chen et al, 2008, 2015). The extracellular N‐termini of NMDARs are 11 nm high and 12 nm × 12.5 nm wide at the tip (Lee et al, 2014). The transmembrane segment is ~5 nm long and 4–6 nm wide (Lee et al, 2014). The C‐termini of NMDARs are 17–20 nm × 13–14 nm wide and 13–16 nm high (Chen et al, 2008, 2015). The center‐to‐center distance is on average 24 nm for AMPARs and 32 nm for NMDARs (Chen et al, 2008, 2015). The models are based on Nakagawa et al (2006).
However, the PSD is not static. Some AMPARs in PSDs are basically immobile on a minute timescale and apparently fairly stably anchored within PSDs (Tardin et al, 2003) especially when PSD‐95 is overexpressed (Kerr & Blanpied, 2012). Other AMPARs show modest mobility within postsynaptic sites (Tardin et al, 2003). Extrasynaptic AMPARs are highly mobile and can diffuse right through synaptic sites (Tardin et al, 2003), but PDZ interactions between PSD‐95 and AMPAR complexes (see below) can trap AMPARs at postsynaptic sites (Bats et al, 2007). More recent superresolution microscopy shows that AMPARs are either confined to the nanodomains described in the preceding paragraph with very low diffusion (presumably because they are anchored through protein interactions with PSD‐95 and its homologues) or diffuse seemingly freely in and out of the nanodomains, apparently untethered (Nair et al, 2013). Some AMPARs can diffuse in and out of a nanodomain but become temporarily confined inside the nanodomain (Nair et al, 2013). Nevertheless, the majority of postsynaptic AMPARs can turn over within minutes (Ashby et al, 2006) if not faster in hippocampal cultures (Groc et al, 2004) as well as acute slices (Heine et al, 2008) by lateral diffusion (Makino & Malinow, 2009), endocytosis, and insertion into the plasma membrane via exocytosis (Lissin et al, 1999; Ehlers, 2000; Passafaro et al, 2001; Petrini et al, 2009). In fact, in 40% of spines imaged by Nair et al (2013) showed nanodomains that were not stable for more than 5 min.
Postsynaptic NMDARs turn over much more slowly, on the hour‐to‐day timescale (Lissin et al, 1999; Tovar & Westbrook, 2002; Groc et al, 2004). NMDAR clusters are 100–200 nm in diameter (Chen et al, 2015; Hanamura et al, 2017; Ladepeche et al, 2018).
With respect to the overall dimensions of a synapse, it is noteworthy that spine volume and PSD size are strongly correlated with each other (Harris & Stevens, 1989), with presynaptic volume, vesicle content, and active zone size (Harris & Stevens, 1989; Schikorski & Stevens, 1997), and with AMPAR content (Nusser et al, 1998; Kharazia & Weinberg, 1999; Takumi et al, 1999). Spine size is also well correlated with postsynaptic response strength as determined by glutamate uncaging (Matsuzaki et al, 2001; Asrican et al, 2007; Araya et al, 2014). During LTP, AMPAR content increases within 1–2 min in parallel with spine size (Matsuzaki et al, 2004; Steiner et al, 2008; Zhang et al, 2008; Bosch et al, 2014) (see also Yang et al, 2008b) and F‐actin (Fukazawa et al, 2003; Honkura et al, 2008; Bosch et al, 2014) mediating a lasting increase in synaptic strength (Collingridge et al, 2004; Lisman & Hell et al, 2008; Sudhof & Malenka, 2008; Kessels & Malinow, 2009; Huganir & Nicoll, 2013). Similarly, NMDAR‐dependent LTD correlates with a decrease in spine size and F‐actin (Okamoto et al, 2004; Zhou et al, 2004) and requires actin depolymerization (Wang et al, 2007).
Over time, F‐actin defines not only spine size but also PSD size and AMPAR content although it is unclear how F‐actin does so, a critical question for understanding synaptic strength and plasticity. However, LTD but not spine shrinkage depends on the protein phosphatase PP1 and spine shrinkage but not LTD on slingshot or a related phosphatase (Zhou et al, 2004; Wang et al, 2007), separating the two events and thereby potentially uncoupling synaptic strength and spine size though this uncoupling lasts likely only for a limited time.
Postsynaptic AMPAR and NMDAR targeting by PSD‐95
PSD‐95 and its homologues PSD‐93 and SAP102 are important for postsynaptic AMPAR targeting (El‐Husseini et al, 2000; Schnell et al, 2002; Beique et al, 2006; Elias et al, 2006; Schluter et al, 2006; Bats et al, 2007). There are roughly 300 PSD‐95, 60 PSD‐93, and 40 SAP102 in an average PSD (Sheng & Hoogenraad, 2007). The abundance of SAP97, a fourth PSD‐95 homologue linking PKA to GluA1 (see below), in PSD fractions is less certain as it is easily extracted with Triton X‐100 (Leonard et al, 1998), which is required for purifying PSDs. SAP97 is likely not more abundant than PSD‐93 or SAP102 given its relatively low apparent synaptic enrichment by immunofluorescence microscopy (Valtschanoff et al, 2000). PSD‐95 in turn is localized to the synapse by α‐actinin binding to the N‐terminus of PSD‐95 (Matt et al, 2018a). Postsynaptic targeting of PSD‐95 also requires its palmitoylation of Cys3 and Cys5 near its N‐terminus (Craven et al, 1999; El‐Husseini Ael et al, 2002). α‐Actinin binding to PSD‐95 is not only important for the localization of PSD‐95 but also of a substantial fraction (~40%) of AMPARs. Postsynaptic localization of most of the remaining AMPARs depends on PSD‐93 and SAP102 (Elias et al, 2006, 2008), which do not bind to α‐actinin and are anchored at postsynaptic sites independent of α‐actinin (Matt et al, 2018a). The estimated presence of ~400 molecules PSD‐95 and its homologues fits remarkably well with affinity determinations for PDZ interactions specifically for the interaction of the first two PDZ domains of PSD‐95 with GluN2 subunits and TARPs. The average spine volume is 0.062 ± 0.08 μm3 (Harris & Stevens, 1989). If 400 PSD‐95 homologues are even distributed throughout the spine, the concentration would be ~10 μM. Consistently, K d values for the higher affinity GluN2 subunits are ~1 μM, whereas those for TARPs are in the range of 3–12 μM (Lim et al, 2002; Dakoji et al, 2003; Hafner et al, 2015; Pedersen et al, 2017).
Based on EM tomography, PSD‐95 family members adopt an extended conformation to form vertical filaments (~5 nm diameter) that protrude from the PSD into the cytosol with their C‐termini being 20–30 nm away from the PSD (Chen et al, 2008, 2011, 2015; Jeyifous et al, 2016) in agreement with immunogold EM localization of GKAP (~25 nm) and Shank (~30 nm) (Valtschanoff & Weinberg, 2001), with GKAP binding to the C‐terminal GK domain of PSD‐95 and Shank to GKAP. Notably, isolated PSD‐95 family members show a C‐shaped structure suggesting that its conformation can be regulated to expand (Nakagawa et al, 2004). In fact, palmitoylation induces an elongated shape of PSD‐95, which is required for its binding to AMPARs and NMDARs (Jeyifous et al, 2016). The PSD‐95 filaments are 13 nm apart (Chen et al, 2008, 2011) when distances between AMPARs are ~27 nm and NMDARs ~30 nm (Chen et al, 2015). Accordingly, the density of PSD‐95 is higher than that of AMPARs and NMDARs. This finding is in agreement with the estimated PSD content of ~400 PSD‐95 family members. If one assumes that an average PSD contains 100 glutamate receptors and one receptor associates with 2–4 PSD‐95 family members, there would likely be some surplus of PSD‐95. Accordingly, PSD‐95 might associate with a limited number of postsynaptic protein complexes that do not contain AMPARs or NMDARs. One prominent binding partner is neuroligin, which augments synaptogenesis and functional availability of postsynaptic AMPARs, although its PDZ interaction does not appear relevant for this function (Shipman et al, 2011).
Postsynaptic targeting of AMPARs depends on auxiliary subunits known as TARPs with γ2 (stargazin), γ3, γ4, and γ8 being the main isoforms (Chen et al, 2000; Jackson & Nicoll, 2011). CNIH2/3 (Schwenk et al, 2009; Herring et al, 2013), SynDIG1 (Kalashnikova et al, 2010; Chenaux et al, 2016), and SynDIG4 (Prrt1) (Matt et al, 2018b) also promote synaptic AMPAR targeting, but their function is not as well defined and could be limited to augmenting AMPAR availability at extrasynaptic sites in the case of SynDIG4 (Matt et al, 2018b). TARPs bind with their very C‐terminal ends to the first two PDZ domains of PSD‐95 and its homologues PSD‐93 and SAP102 for postsynaptic localization of AMPARs (Fig 1; El‐Husseini et al, 2000; Schnell et al, 2002; Beique et al, 2006; Elias et al, 2006; Schluter et al, 2006; Bats et al, 2007). GluN2A and 2B bind directly with their C‐termini to these PDZ domains (Kornau et al, 1995) and removal of PSD‐95 and its homologues reduces excitatory postsynaptic currents (EPSCs) by NMDARs, suggesting that these scaffolds contribute channel functional availability (Elias et al, 2006, 2008; Ehrlich et al, 2007; Fig 1). However, PDZ interactions appear less critical for postsynaptic availability of NMDARs than of AMPARs as NMDAR EPSCs are less sensitive to a reduction in PSD‐93/95 (Elias et al, 2006, 2008; Schluter et al, 2006; Ehrlich et al, 2007; Matt et al, 2018a) or mutations that affect PDZ binding (Schnell et al, 2002; Prybylowski et al, 2005). Nevertheless, acute disruption of PDZ binding does strongly increase lateral mobility of synaptic NMDARs (Bard et al, 2010), as it does increase diffusion of AMPARs (Sainlos et al, 2011). Furthermore, under conditions under which the PSD‐93/PSD‐95/SAP102 levels are more strongly reduced than by combined knockdown of PSD‐93/PSD‐95, a decrease in postsynaptic NMDAR function becomes obvious (Elias et al, 2006; Levy et al, 2015). Notably, GluN2A and 2B have much higher affinities for PDZ1 and PDZ2 than TARPs (Lim et al, 2002). The ESDV sequence at the C‐termini of GluN2A and 2B subunits constitutes a nearly optimal binding motif for those PDZ domains with Q or E being desirable at the −3 position and D or E the −1 position in addition to the previously established requirement of S/T at the −2 and V the 0 position (Lim et al, 2002; Zhu et al, 2016). The C‐terminal TTPV sequence of γ2/3/4/8 is much less optimal; peptides mimicking the last 11 residues of GluN2A and 2B, which contain the C‐terminal ESDV sequence, possess a Kd of ~1 μM for PDZ2 of PSD‐95, whereas peptides mimicking the last 10 residues of γ2, which contain the C‐terminal TTPV sequence, have a Kd of ~12 μM (Lim et al, 2002; Dakoji et al, 2003; Pedersen et al, 2017) although extending the length of the γ2 peptide decreases the K d to 3.4 μM (Hafner et al, 2015). Thus, NMDARs will tie up available PSD‐93/PSD‐95/SAP102 before AMPARs have access. In other words, when the concentration of PSD‐93/PSD‐95/SAP102 is limited, a loss of postsynaptic AMPAR function will first be observed before a loss of NMDAR function.
In addition to potentially contributing to the direct anchoring of NMDARs at postsynaptic sites, PSD‐95 might more indirectly foster postsynaptic content or functional availability of NMDARs by recruiting regulatory proteins, as abrogating PSD‐95 binding of GluN2A decreases current density in HEK293 cells by more than 10‐fold without significantly affecting surface expression (Lin et al, 2004). For instance, Pyk2 binds to PSD‐95 to augment postsynaptic NMDAR function via Src during LTP (Huang et al, 2001; Bartos et al, 2010; Fig 3). Pyk2/Src might act in part by promoting phosphorylation of GluN2B on Y1472 (Yang et al, 2013), which impairs AP2 binding to this site and thereby NMDAR endocytosis (Roche et al, 2001; Prybylowski et al, 2005). As GluN2A does not share this endocytic motif, its regulation by PSD‐95 must utilize a different mechanism that could potentially directly affect the channel activity of the NMDAR (Lin et al, 2004). A role of PSD‐95 in promoting AMPAR trafficking to the postsynaptic site via β2 AR–PKA signaling in addition to AMPAR anchoring is discussed below.
Figure 3. Role of PSD‐95‐anchored Pyk2 in NMDAR function.
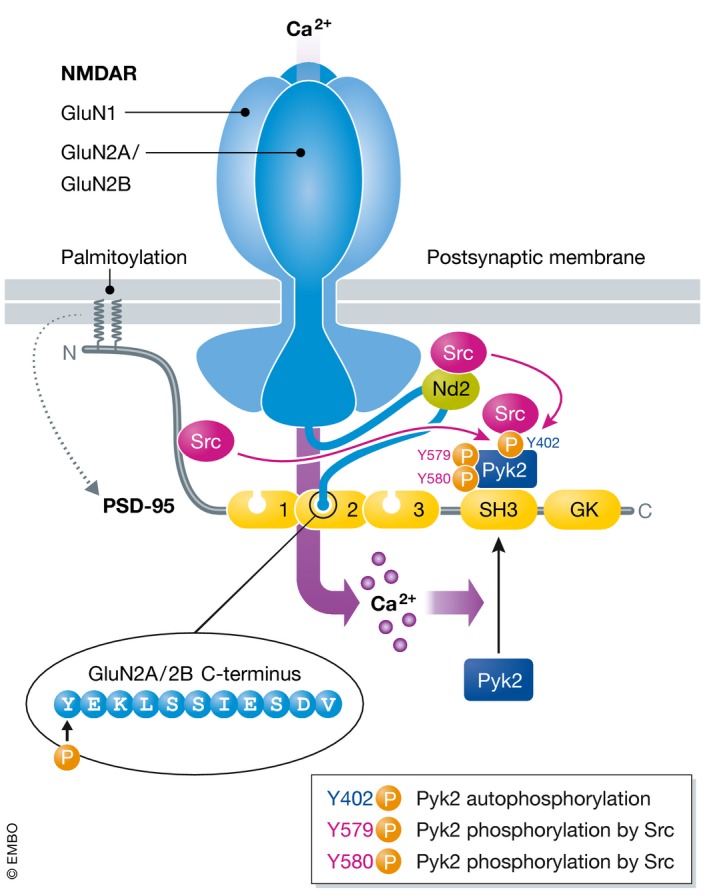
Ca2+ influx induces Pyk2 relocation to postsynaptic sites via Ca2+/CaM‐stimulated binding to the SH3 domain of PSD‐95 (Bartos et al, 2010). At the same time, Pyk2 trans‐autophosphorylates on Y402 (Bartos et al, 2010), which creates a binding site for the SH2 domain of Src. Src might be recruited to pY402 from the N‐terminus of PSD‐95, which binds Src and modestly suppresses Src activity (Kalia et al, 2006), or from NADH dehydrogenase 2 (ND2), a Src‐anchoring protein in the NMDAR complex in addition to PSD‐95 (Gingrich et al, 2004). Binding to pY402 activates Src, which in turn phosphorylates Pyk2 on Y579 and Y580 located in its activation loop to strongly augment Pyk2 activity (Park et al, 2004; Yang et al, 2013). Src binding obviously protects pY402 from dephosphorylation, which is mediated by STEP (Xu et al, 2012). In addition, Y402 phosphorylation can likely be more effectively renewed by Pyk2 when phosphorylated by Src on Y579/Y580 for self‐perpetuating activation of this Pyk2/Src complex as a form of molecular memory (Bartos et al, 2010).
Induction of LTP by multiple glutamate uncaging pulses, which activate Ca2+ influx through NMDARs, increases spine size and spine content of GluA1, F‐actin (Matsuzaki et al, 2004; Honkura et al, 2008), and especially its regulator cofilin within 1 min, but total PSD‐95 content of spines and PSD size do not start to significantly increase before 1 h postinduction (Bosch et al, 2014; Meyer et al, 2014). Thus, PSD expansion and recruitment of PSD‐95 trail far behind the recruitment of AMPARs. Either PSD‐95 is not mediating the increase in postsynaptic AMPARs in the early phases of LTP, or spare PSD‐95 in the PSD (see above) does so by an increase in its affinity for TARPs. Such an increase could translate into more pre‐existing PSD‐95 molecules binding incoming AMPARs. In fact, phosphorylation of stargazin/γ2 at its C‐terminus by CaMKII at multiple sites, which had been implicated earlier by Tomita et al (2005) in LTP, detaches the otherwise positively charged C‐terminus from the plasma membrane for increased binding to PSD‐95 and postsynaptic AMPAR localization (Opazo et al, 2010; Sumioka et al, 2010; Hafner et al, 2015). However, more recent work by Tomita and co‐workers suggests a role of CaMKII‐mediated phosphorylation of γ8 and not of γ2 in LTP, leaving open the question of whether and which TARPs are truly the relevant CaMKII targets (Sumioka et al, 2011; Park et al, 2016; Sheng et al, 2018).
Why does glutamate uncaging not increase postsynaptic PSD‐95 content when spine size does go up? It appears that the glutamate‐induced Ca2+ influx triggers two antagonistic events. One leads to actual net loss of PSD‐95 from spines and the other to its accumulation in spines. The net loss of PSD‐95 upon Ca2+ influx is well established; it is augmented by phosphorylation of PSD‐95 on S73 within its first PDZ domain by CaMKII (Steiner et al, 2008) and on T19 by GSK3β (Nelson et al, 2013). In addition, the loss depends on Ca2+ influx‐induced binding of Ca2+/CaM to the very N‐terminus of PSD‐95 (Fig 4; Zhang et al, 2014). This interaction antagonizes palmitoylation of PSD‐95 on Cys3 and Cys5, which is required for its postsynaptic localization and reversed upon NMDAR activation (El‐Husseini Ael et al, 2002; Zhang et al, 2014). Ca2+/CaM recruitment to the PSD‐95 N‐terminus also displaces α‐actinin (Matt et al, 2018a), which anchors PSD‐95 at the postsynaptic site (Matt et al, 2018a). Mutating Tyr12 near the N‐terminus of PSD‐95 to Glu not only abrogates Ca2+/CaM binding and with it loss of PSD‐95(Y12E) from spines but actually causes a substantial increase in postsynaptic accumulation of this PSD‐95 mutant in spines upon Ca2+ influx. This latter finding is remarkable as it shows that Ca2+ influx can engage a mechanism that augments postsynaptic localization of PSD‐95, but for endogenous PSD‐95, this mechanism is overridden by the Ca2+/CaM‐driven displacement of PSD‐95 from spines. We propose that the absence of postsynaptic PSD‐95 accumulation during the first hour following glutamate‐induced spine LTP is at least in part due to the Ca2+/CaM‐mediated loss of PSD‐95 from spines, which curbs and effectively antagonizes the mechanism that would otherwise lead to immediate increase in postsynaptic PSD‐95. Ca2+/CaM‐induced displacement of PSD‐95 from spines is also relevant in homeostatic scaling down of the strength of all synapses in a neuron in response to a chronic increase in its excitatory input (Chowdhury et al, 2018).
Figure 4. Displacement of PSD‐95 from postsynaptic sites by Ca2+/CaM.
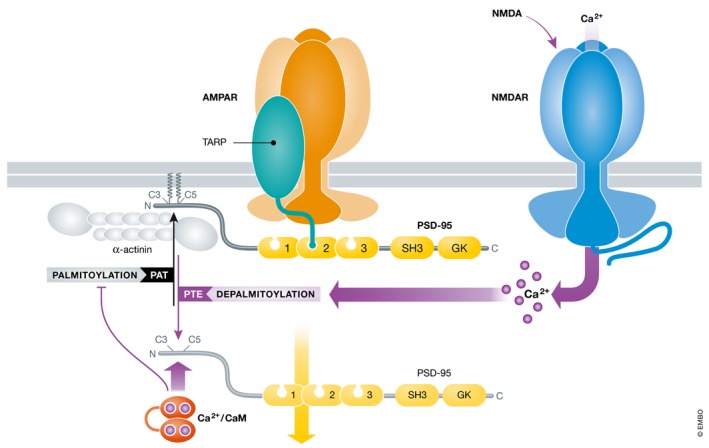
Ca2+ influx via NMDARs likely stimulates depalmitoylation of PSD‐95 by a hypothetical palmitoyl thioesterase (PTE). Binding of Ca2+/CaM to depalmitoylated PSD‐95 will prevent re‐palmitoylation by a palmitoyl transferase (PAT). Lack of palmitoylation will reduce postsynaptic PSD‐95 anchoring, leading to a loss of PSD‐95 from postsynaptic sites (Zhang et al, 2014). Ca2+/CaM also disrupts binding of PSD‐95 to α‐actinin, which anchors otherwise PSD‐95 at postsynaptic sites (Matt et al, 2018a).
Although the reason for the delay in the increased size of PSDs and their PSD‐95 content following LTP is unclear, this delay does correlate with the finding that 1–2 h after saturating LTP by multiple trains of stimuli, further LTP can be induced by additional stimuli (e.g., Cao & Harris, 2014, and ref. therein). Perhaps delayed PSD‐95 recruitment sets the stage for further LTP.
Complex signaling within spines
PSDs harbor at least 50 kinases (Collins et al, 2006), and hundreds of signaling mechanisms likely regulate postsynaptic functions (Coba et al, 2009). For instance, at least 15 different Rho family GEFs have been implicated in regulating spine size and morphology by controlling F‐actin (Penzes et al, 2008; Kiraly et al, 2010; Kim et al, 2011). Such complexity enhances reliability of signaling as paradigmatically shown for TNF–NFκB signaling in fibroblasts (Cheong et al, 2011). It can explain why manipulations often lead to effects that are small or difficult to reproduce as alternative mechanisms can compensate. The best studied and perhaps most prevalent postsynaptic kinase signaling is by PKA and CaMKII, the focus of the remainder of this article.
PKA: structure, regulation, and localization by AKAPs
PKA is a tetramer formed by two regulatory (R) and two catalytic (C) subunits (Fig 5). Four genes encode RIα,β and RIIα,β and three genes Cα,β,γ (Taylor et al, 2012). R subunits homo‐dimerize via their N‐terminal dimerization domains forming a four‐helix crossing bundle (Beene & Scott, 2007). C subunit activity is suppressed by a pseudosubstrate segment on R, which binds to the catalytic site on C and is released by cAMP (Brandon et al, 1997), although C does not have to be fully released from R in order to catalyze phosphorylation as indicated by correlating full dissociation of C from R or better a lack thereof with full enzymatic activity (Johnson et al, 1993; Yang et al, 1995). A recent publication seems to confirm these earlier studies as both R and C co‐immunoprecipitate with the A kinase anchor protein AKAP5 from cell lysate prepared after strong β‐adrenergic stimulation of the cells (Smith et al, 2017). Yet, follow‐up work suggests that the co‐immunoprecipitation is due to re‐association of C with R upon cell lysis as cAMP becomes diluted (Walker‐Gray et al, 2017).
Figure 5. Overall structure and regulation of PKA .
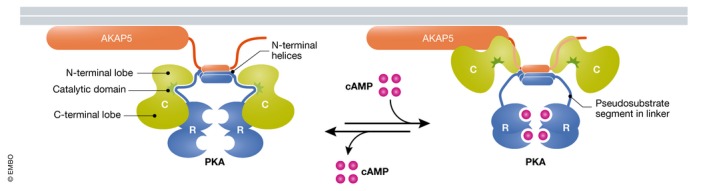
The holoenzyme consists of two R and two C subunits. The catalytic center of the C subunit resides in the cleft between its N and C lobe. The N‐termini of the R subunits homo‐dimerize by forming a four‐helix bundle, which are followed by flexible linkers. The pseudosubstrate segment in each linker binds to the catalytic site of the C subunit to suppress activity. cAMP binds with high cooperativity to two sites, CNBA and CNBB, on each R subunit to release the pseudosubstrate segment from the catalytic site. AKAP5 binds with an amphipathic α‐helix to the four‐helix bundle formed by the R subunits.
Phosphotransfer and substrate release is modestly fast for most kinases (~500/s) (Zhou & Adams, 1997; Shaffer & Adams, 1999). However, ADP release is very slow for PKA (~20/s) (Zhou & Adams, 1997). The consequent low turnover number (<20/s) confers special importance to association of PKA with substrate proteins for effective phosphorylation of key targets and at the same time helps avoid phosphorylation of unintended targets. RII and to a lesser degree RI dimers are recruited to a number of substrates by AKAPs (Beene & Scott, 2007; Dai et al, 2009). AKAPs are a group of diverse and typically multifunctional scaffolding proteins, which share an amphipathic helix that interacts with the four‐helix bundle formed by the N‐terminal dimerization regions of two R subunits (Fig 5; Newlon et al, 2001).
Regulation of AMPARs by AKAP5‐anchored PKA and PP2B during various forms of synaptic plasticity
AKAP5 is the most prevalent AKAP at postsynaptic sites (Smith et al, 2006; Lu et al, 2007; Tunquist et al, 2008; Weisenhaus et al, 2010). AKAP5 refers to human AKAP79 and rodent AKAP150, which is much larger than AKAP79 due to the insert of 36 imperfect octapeptide repeats of unknown function. A segment near the very C‐terminus of AKAP5 is the docking site for PKA RII subunits (Fig 5; Carr et al, 1992; Murphy et al, 2014b). AKAP5 also anchors the Ca2+/CaM‐activated phosphatase calcineurin (PP2B). PP2B binds to the PIAIIIT motif in the central AKAP5 region, a modification of the PXIXIT motif used by other proteins to bind PP2B (Li et al, 2012). The three N‐terminal polybasic segments of AKAP5 can bind PKC, Ca2+/CaM, F‐actin, cadherin, and PIP2 and augment targeting of AKAP5 to spines (Klauck et al, 1996; Dell'Acqua et al, 1998; Gomez et al, 2002; Gorski et al, 2005; Tavalin, 2008; Patel et al, 2017; Woolfrey et al, 2018). Further postsynaptic targeting occurs via binding of the middle region of AKAP5 to the SH3 and GK domains of PSD‐95 and SAP97 (Colledge et al, 2000; Bhattacharyya et al, 2009; Nikandrova et al, 2010; Fig 6).
Figure 6. Schematic structure of AKAP5 and its complexes with AMPARs and Cav1.2.
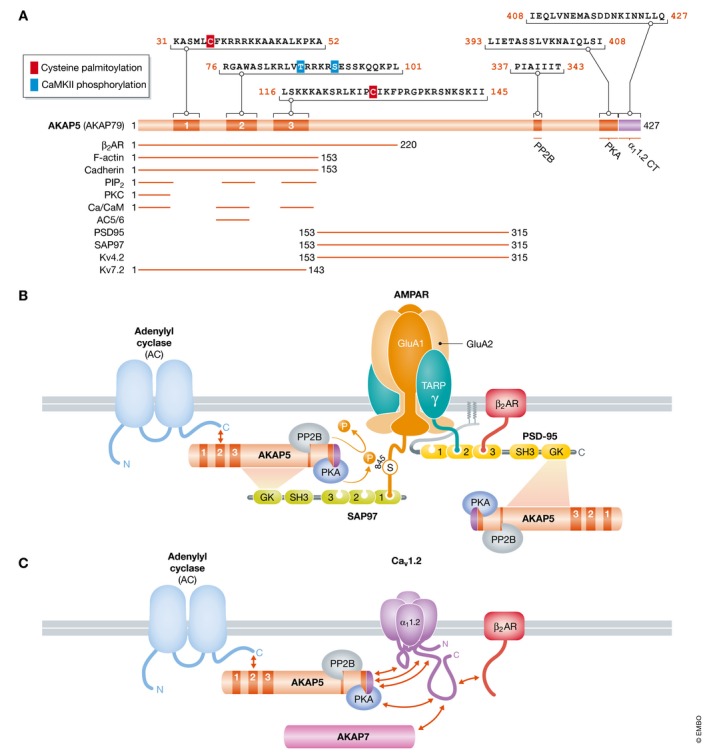
(A) Overview of AKAP5 binding partners and their binding sites. Residue numbering refers to human AKAP79. The N‐terminus is formed by three segments designated A, B, and C. These segments are polybasic regions, each of which can bind to Ca2+/CaM and PIP2. In addition, PKC binds to A and adenylyl cyclases 5 and 6 (AC5/6) bind via their N‐termini to B. PP2B binds to a PIXIT‐like motif near the center of AKAP5 and PKA to a motif that is about 20 residues upstream of the very C‐terminus. Immediately downstream of the PKA site is a leucine zipper‐like motif that binds to a leucine zipper‐like motif near the C‐terminus of the Cav1.2 α11.2 subunit. PSD‐95 and SAP97 interact through their SH3 and GK domains with a broad region in the center of AKAP5, which also binds to KV4.2. Other binding sites are less clearly defined. The two palmitoylation sites are identified by red squares and the two known CaMKII phosphorylation sites by blue squares. (B) The β2 AR–AMPAR complex. AC binds to the N‐terminus, PP2B to the middle region, and PKA to the C‐terminus of AKAP5, which is connected with AMPARs via SAP97, which binds to the very C‐terminus of GluA1 (Leonard et al, 1998; Tavalin et al, 2002; Zhang et al, 2013). The β2 AR binds to the third PDZ domain of PSD‐95, which is linked to AMPARs via TARPs (γ), which bind with their very C‐termini to the second and, with lower affinity, also first and third PDZ domains of PSD‐95 (Hafner et al, 2015). PSD‐95 might recruit a second AKAP5/PKA/PP2B complex. PKA and PP2B mediate phosphorylation and dephosphorylation of S845 on GluA1, respectively. (C) The β2 AR directly binds to the C‐terminus of α11.2. AKAP5 binds to three different regions as depicted (red arrows); the leucine zipper‐like segment near C‐terminus of α11.2 also binds alternatively AKAP7. AKAP5 recruits PKA, PP2B, and likely ACs to the Cav1.2 complex.
Like PSD‐95 (see above; El‐Husseini et al, 2000), AKAP5 is a positive regulator of spine size and AMPAR content (Robertson et al, 2009); this effect depends on AKAP5's central domain, which binds to PSD‐95 (Robertson et al, 2009). Perhaps AKAP5 works in conjunction with PSD‐95 as part of a structural framework that determines synaptic size and strength, possibly including GKAP, Shank, and Homer (Sala et al, 2001; Baron et al, 2006). Consistent with this notion, the increase in postsynaptic strengthen by PSD‐95 expression requires its SH3‐GK region (Xu et al, 2008) perhaps for AKAP5 association (Robertson et al, 2009) although the SH3‐GK region binds other proteins such as GKAP and Pyk2, which could be involved in spine enlargement (Sala et al, 2001; Bartos et al, 2010).
AKAP5 is linked to GluA1 via SAP97 (Colledge et al, 2000; Tavalin et al, 2002; Bhattacharyya et al, 2009), which binds with its first or second PDZ domain to the C‐terminus of GluA1 (Fig 6; Leonard et al, 1998; Cai et al, 2002). Additional, more indirect association of AKAP5 with GluA1 could be via TARP‐associated PSD‐95 and via the β2 AR (Dai et al, 2009), which directly binds to AKAP5 and indirectly to GluA1 via PSD‐95/TARP (6; Joiner et al, 2010).
SAP97‐anchored AKAP5 recruits PKA and the antagonistic PP2B to GluA1 for dynamic phosphorylation and dephosphorylation of S845 in the cytosolic C‐terminus of GluA1 (Tavalin et al, 2002; Hoshi et al, 2005; Sanderson et al, 2012; Diering et al, 2014). S845 phosphorylation augments channel opening (Banke et al, 2000) and surface expression of GluA1 (Fig 7) (Sun et al, 2005; Gao et al, 2006; Oh et al, 2006; Man et al, 2007; Joiner et al, 2010) especially in the perisynaptic space (Oh et al, 2006; Yang et al, 2008a,b, 2010; He et al, 2009; Diering et al, 2014). The perisynaptic space is functionally defined as containing AMPARs that are activated upon presynaptic stimulation when glutamate reuptake is inhibited so that a higher concentration of glutamate can reach the space surrounding the postsynaptic site upon presynaptic glutamate release. Recruitment of AMPARs to this space provides a readily available reserve pool of AMPARs for postsynaptic insertion during LTP and constitutes a form of synaptic metaplasticity that fosters LTP (Esteban et al, 2003; Sun et al, 2005; Oh et al, 2006; Man et al, 2007; Yang et al, 2008a; Qian et al, 2012; Diering et al, 2014). In fact, an extrasynaptic surface pool of AMPARs is the only specific requirement for LTP in replacement experiments in which endogenous GluA1, GluA2, and GluA3 are all knocked out in individual neurons (Granger et al, 2013). Accordingly, the abundance of receptors in this reserve pool will determine, which receptors become incorporated into the PSD during LTP. When all subunits are present, S845 phosphorylation appears to afford an advantage that might become a necessity under certain conditions for activity‐driven insertion of GluA1 homomeric AMPARs as formed when GFP‐GluA1 is ectopically expressed (Esteban et al, 2003). However, LTP is normal in S845A knock‐in mice (Lee et al, 2010). The discrepancy between these two papers can be explained by recent findings that implicate the N‐terminal domain of GluA1 as important for its postsynaptic targeting and show that adding a tag such as GFP as in Esteban et al (2003) impairs this targeting (Diaz‐Alonso et al, 2017; Watson et al, 2017). Accordingly, GFP‐GluA1 requires S845 to augment postsynaptic targeting when this targeting is impaired, while S845 is less important for postsynaptic targeting of endogenous untagged GluA1.
Figure 7. Two‐step model of AMPAR surface trafficking and lateral diffusion to the postsynaptic site.
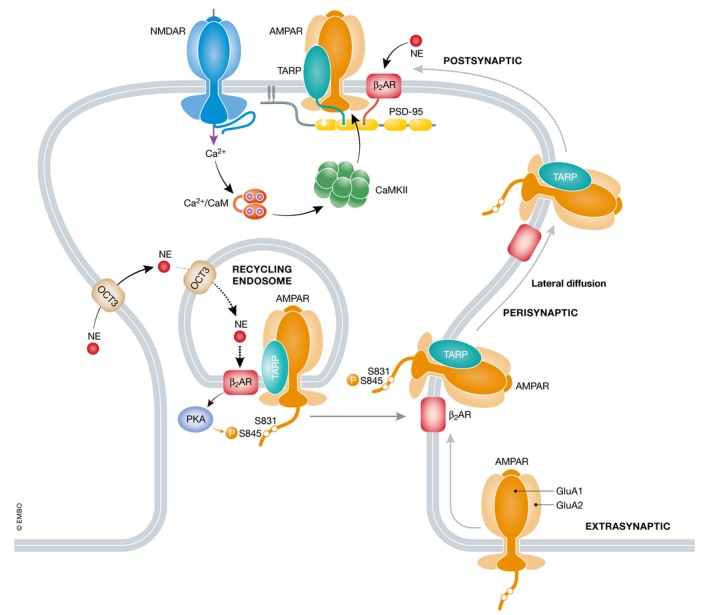
A significant portion of AMPARs are synthesized in the endoplasmic reticulum (ER) in dendrites possibly without TARPs, which might associate with the AMPAR core in a later secretory compartment (e.g., recycling endosomes (REs)) or the plasma membrane (Bowen et al, 2017). Surface insertion via REs is promoted by intracellular norepinephrine (NE) signaling, which is transported from the extracellular space via the OCT3 transporter first into the cytosol and then, as proposed here, into the lumen of REs, analogous to NE transport into the Golgi apparatus (Irannejad et al, 2017). This NE transport enables intracellular stimulation of β2 ARs that are associated with GluA1 in RE analogous stimulation of β2 ARs in endosomes (Tsvetanova & von Zastrow, 2014). The resulting S845 phosphorylation by PKA induces insertion of AMPARs into the perisynaptic membrane via unknown mechanisms. From there, AMPARs laterally diffuse into the postsynaptic density, where they are trapped especially upon activation of CaMKII by Ca2+ influx via NMDARs.
In 2‐week‐old mice, GluA2‐lacking CP‐AMPARs (presumably GluA1 homomers) are transiently postsynaptically inserted right after induction of LTP (Plant et al, 2006) in a PKA‐ and AKAP5‐dependent manner (Lu et al, 2007; Sanderson et al, 2016). This process is necessary for stabilization of LTP at 2 weeks of age (Plant et al, 2006) especially when induced by minimal stimulation (Lu et al, 2007; Sanderson et al, 2016). This process is not necessary for LTP in 3‐ to 4‐week‐old mice (Lu et al, 2007; Sanderson et al, 2016) but becomes necessary again at 8 weeks and older (Lu et al, 2007). The presence of four versus two S845 residues in GluA1 homomers versus GluA1/A2 diheteromers might be important for the temporary postsynaptic insertion of GluA1 homomers. In support of this notion, the increase in postsynaptic AMPARs during homeostatic synaptic plasticity upon chronic inhibition of neuronal activity or specifically L‐type Ca2+ channels or NMDARs is often (but not always; see below) due to postsynaptic insertion of GluA1 homomeric CP‐AMPARs (Thiagarajan et al, 2005; Sutton et al, 2006; Soares et al, 2013; Kim & Ziff, 2014; Sanderson et al, 2018) and requires S845 phosphorylation (Diering et al, 2014; Kim & Ziff, 2014) and anchoring of PKA by AKAP5, which is important for phosphorylation of S845 (Sanderson et al, 2018). Because GluA2‐lacking CP‐AMPARs including GluA1 homomers have a higher ion channel conductance than GluA2‐containing CI‐AMPARs, their insertion will immediately augment postsynaptic strength and also mediate Ca2+ influx, which promotes synaptic homeostasis (Kim & Ziff, 2014) and LTP under certain conditions (Plant et al, 2006; Lu et al, 2007; Sanderson et al, 2016). In fact, recent work indicates that blocking CP‐AMPAR during the silencing phase also prevents upscaling suggesting that this Ca2+ influx during this phase is important in this process (Sanderson et al, 2018). However, other work indicates that homeostatic synaptic upscaling depends on GluA2 rather than GluA1 (Gainey et al, 2009, 2015; Goold & Nicoll, 2010; Tan et al, 2015; Ancona Esselmann et al, 2017) or does not specifically require either GluA1 or GluA2 (Altimimi & Stellwagen, 2013), possibly reflecting differences in the precise neuronal systems and signaling states within neurons between different laboratories.
In contrast to Plant et al (2006), who described postsynaptic insertion of CP‐AMPARs right after LTP induction and their need for LTP maintenance, Adesnik and Nicoll (2007) did not find such evidence. Although both groups state that they used 2‐ to 3‐week‐old mice, this discrepancy could be explained if the mice used by Plant et al were actually closer to 2 weeks and those by Adesnik et al closer to 3 weeks in their development as systematic comparisons of 12‐ to 14‐day and 20‐ to 22‐day old mice find that in the younger but not older mice, CP‐AMPARs are required for single tetanus LTP (Lu et al, 2007; Sanderson et al, 2016).
Long‐term depression, the flip side of LTP, can be induced by two different mechanisms, prolonged Ca2+ influx via NMDARs (Dudek & Bear, 1992) and activation of mGluR1/5 receptors (Bolshakov & Siegelbaum, 1994; Oliet et al, 1997). NMDAR‐ but not mGluR1/5‐dependent LTD requires anchoring of PP2B by SAP97/AKAP5 (Jurado et al, 2010; Sanderson et al, 2012, 2016). Elimination of PP2B binding to AKAP5 or of AKAP5 to the SH3 domain of PSD‐95 abrogates removal of AMPARs and PSD‐95 from spines during chemical LTD (Jurado et al, 2010; Sanderson et al, 2012). Furthermore, AKAP5‐anchored PKA is required for transient recruitment of CP‐AMPARs during LTD and AKAP5‐anchored PP2B for subsequent removal of those CP‐AMPARs (Sanderson et al, 2016). Abrogating PKA binding to AKAP5 by deletion of the PKA binding site on AKAP5 prevents the transient recruitment of CP‐AMPAR during the 1‐Hz/15‐min stimulation (Sanderson et al, 2016), and little to no change in synaptic strength occurs upon this 1‐Hz/15‐min stimulation when the PKA binding site is deleted from AKAP5 (Lu et al, 2008; Sanderson et al, 2016). In fact, PKA has to be active during induction for LTD to occur (Lu et al, 2008). Abrogating PP2B binding to AKAP5 by deletion of the PP2B binding site on AKAP5 does not affect the initial recruitment of CP‐AMPARs to the postsynaptic site but prevents the subsequent removal of these CP‐AMPARs presumably by impairing dephosphorylation of S845 in GluA1, which is important for LTD (Lee et al, 2010). Due to the recruitment of CP‐AMPARs during the early phases of LTD induction and the failure of their removal, LTP rather than LTD is observed in mice that lack the PP2B binding site in AKAP5 (Sanderson et al, 2016). Divergent findings that suggest PKA anchoring by AKAP5 is not necessary for LTD (Jurado et al, 2010) are likely explained by differences in the developmental stages of the two systems with LTD requiring PKA anchoring by AKAP5 at one stage as in Lu et al (2008) but not the other stage as in Jurado et al (2010), analogous to the age dependence of LTP on AKAP5‐anchored PKA (Lu et al, 2007; Sanderson et al, 2016). Finally, although deletion of the PKA binding site in AKAP5 abrogated LTD, a full knockout of AKAP5 did not overtly affect LTD at 2 weeks of age (Weisenhaus et al, 2010). Perhaps complete removal of AKAP5 by knockout allowed compensation by another AKAP such as AKAP12 (see below) though the situation appears different at other ages where AKAP5 KD and KO did abolish LTD (Tunquist et al, 2008; Jurado et al, 2010).
Role of AKAP12 in postsynaptic signaling
AKAP12 (gravin, AKAP250, SSeCKS) is also required for several forms of LTP (Havekes et al, 2012). These forms include theta burst stimulation, which the authors find to depend on β2 AR signaling, and LTP induced by a prolonged theta tetanus (PTT‐LTP induced by a 3‐min‐long 5‐Hz tetanus, with a β AR agonist present; Havekes et al, 2012), which also requires β2 AR signaling (Qian et al, 2012), anchoring of AC and PKA by AKAP5 (Zhang et al, 2013), and S845 phosphorylation (Qian et al, 2012). The overall structural elements of AKAP12 exhibit remarkable similarities to AKAP5: It binds with N‐terminal motifs to PKC and negatively charged phospholipids, which is inhibited by CaM binding to this very region, with central motifs to the β2 AR and PP2B, and with a C‐terminal motif PKA (Nauert et al, 1996; Shih et al, 1999; Tao et al, 2003; Dai et al, 2009; Havekes et al, 2012). Why both AKAP12 and AKAP5 are required for PTT‐LTP (Havekes et al, 2012; Zhang et al, 2013) and what kinds of non‐redundant roles these two AKAPs play in PTT‐LTP are unclear.
Loss of AKAP12 impairs several forms of learning including spatial learning (Morris Water Maze), in contrast to the minimal effect on the Morris Water Maze task from loss of AKAP5 (Weisenhaus et al, 2010), and fear conditioning (M. Zhang & J.W. Hell, unpublished results). Tunquist et al (2008) report impaired Morris Water Maze learning in a different strain of AKAP5 KO mice. However, the AKAP5 KO mice still showed a tendency toward an increase in the time spent in the target quadrant during test runs of their memory. Although this increase was statistically not significantly at the P = 0.05 level compared to other areas in the Morris Water Maze, it is unclear whether the difference in the time spent in the target quadrant was actually statistically different from the time WT mice spent in the target quadrant. Whether memory retention is affected in one of the two AKAP5 KO mouse strains in this test thus remains unclear. Perhaps AKAP12 can compensate for loss of AKAP5 but not vice versa in these tests pointing toward a unique function of AKAP12 that remains to be revealed.
Binding of AKAP12 to the β2 AR is strongly increased upon PKA‐mediated phosphorylation of AKAP12 within its central, β2 AR binding motif (Tao et al, 2003), which is important for de‐ and re‐sensitization of the β2 AR (Lin et al, 2000). This finding suggests that recruitment of PKA by AKAP12 to the β2 AR could be activity driven rather than a more static anchoring mechanism as seen for other AKAPs. However, PKA binding to AKAP12 is required for phosphorylation of AKAP12 itself and the consequent increase in β2 AR. Perhaps AKAP12 is an AKAP to stably anchor PKA for its own phosphorylation and PKA‐driven binding of AKAP12 to the β2 AR serves to recruit other binding partners such as PKC for β2 AR de‐ and re‐sensitization.
Potential role of the AKAP MAP2B in postsynaptic signaling
The microtubule binding protein MAP2B also anchors PKA, constituting a bona fide AKAP. Work with a mutant mouse in which the N‐terminus of MAP2B, which anchors PKA, was deleted to prevent PKA anchoring suggested that PKA anchored at microtubules in the dendritic shaft is required for LTP (Zhong et al, 2009). Accordingly, during LTP induction the catalytic subunit is released from MAP2B to relocate into spines to support LTP, which is impaired in these mice. However, it is possible that effects other than the loss of PKA anchoring at microtubules are responsible for the loss of LTP as the MAP2B deletion mouse has a dramatically altered cytoarchitecture especially for apical dendritic arborization (Khuchua et al, 2003). Thus, this mutation has a widespread pleiotropic effect that will likely affect numerous neuronal functions, and conclusions about any specific molecular effect or mechanism cannot readily be drawn.
Localized signaling from the β2 AR to AMPARs
Although cAMP is a readily diffusible second messenger, signaling downstream of GsPCRs, which act by stimulating adenylate cyclase (AC) through Gs, such as β ARs, can have different effects for different GsPCR even within the same cell (Steinberg & Brunton, 2001; Dai et al, 2009). Such differential effects require spatially restricted signaling by cAMP (Steinberg & Brunton, 2001; Dai et al, 2009). The discoveries of signaling complexes formed between the β2 AR and two of its most prominent targets in the brain, the L‐type Ca2+ channel Cav1.2 (Davare et al, 2001) and GluA1 subunit (Joiner et al, 2010) (see also Wang et al, 2010), that also contain Gs, AC, and PKA thus constitute true milestones in defining cAMP signaling. In detail, AKAP5 is linked together with AC, PKA, and the antagonistic phosphatase PP2B via SAP97 to GluA1 (Fig 6B; Leonard et al, 1998; Tavalin et al, 2002; Sanderson & Dell'Acqua, 2011; Zhang et al, 2013). The β2 AR is recruited to the AMPAR complex via its binding to the third PDZ domain of PSD‐95 (Joiner et al, 2010), which is linked to AMPARs by binding with its second and potentially also first and, possibly in AMPAR complexes not containing the β2 AR, third PDZ domains to TARPs (Schnell et al, 2002; Hafner et al, 2015). The formation of this remarkable protein complex allows for highly localized cAMP signaling within nanodomains, as illustrated by the finding that only those GluA1 subunits that are part of β2 AR‐associated AMPARs become phosphorylated upon β AR stimulation, with β2 AR‐associated AMPARs accounting for likely much less than 20% of all AMPAR complexes (Joiner et al, 2010). Furthermore, the increase in surface expression in cultured hippocampal neurons and in postsynaptic AMPAR response in pyramidal neurons in the prefrontal cortex upon β2 AR stimulation is blocked by disrupting the β2 AR–AMPAR complex by two different peptides that interfere with the β2 AR–PSD‐95 and PSD‐95–TARP interactions (Joiner et al, 2010). Accordingly, the β2 AR has to be associated with AMPARs for their regulation.
A remarkable flip side to the localized stimulatory signaling from the β2 AR to GluA1 is the localized inhibitory signaling at individual synapses by the α2 AR and GABA B receptor, which are coupled to the AC‐inhibitory Gi protein (Lur & Higley, 2015). In layer 5 pyramidal neurons in prefrontal cortex (PFC), activation of the α2 AR and GABA B receptor specifically reduces synaptic transmission by AMPARs and NMDARs, respectively, but not vice versa. This selectivity within single postsynaptic sites is likely due to co‐localization of the α2 AR and GABA B receptor with the AMPAR and NMDAR, respectively, such that Gi activated by the α2 AR or GABA B receptor has only immediate access to the respective glutamate receptor complexes. However, the cellular and molecular basis of this selectivity is unclear except it requires the regulator of G protein signaling RGS4, which limits the duration of Gi activation by promoting their GTPase activity (Lur & Higley, 2015).
Regulation of synaptic AMPAR trafficking by β2 AR signaling
As detailed above, PKA drives AMPARs into the plasma membrane and especially into the perisynaptic space through phosphorylation of GluA1 on S845 to foster LTP (Fig 7) (Ehlers, 2000; Esteban et al, 2003; Sun et al, 2005; Gao et al, 2006; Oh et al, 2006; Man et al, 2007; Yang et al, 2008a,b, 2010; He et al, 2009; Joiner et al, 2010; Diering et al, 2014). Consistently, several forms of LTP, and especially those mediated by weak stimulation paradigms, require cAMP signaling and PKA for induction (Blitzer et al, 1995; Thomas et al, 1996; Gelinas & Nguyen, 2005; Lu et al, 2007; Qian et al, 2012, 2017). For instance, PTT‐LTP is induced by a prolonged theta tetanus (5 Hz, 3 min) but only when the β2 AR is stimulated (Qian et al, 2012). Furthermore, PTT‐LTP also requires anchoring of AC and PKA by AKAP5 (Zhang et al, 2013), and S845 phosphorylation (Qian et al, 2012).
Precisely how PKA‐mediated S845 phosphorylation augments surface trafficking of AMPARs is still unclear. One important question is how norepinephrine (NE), the main endogenous β AR agonist in the brain, can control trafficking of AMPAR–β2 AR complexes from inside neurons to the surface. Recent work shows that NE is transported into the cell and from the cytosol into the lumen of intracellular vesicles via the transporter OCT3, which allows stimulation of β ARs inside cells (Tsvetanova & von Zastrow, 2014; Irannejad et al, 2017). We propose that NE accesses the lumen of recycling endosomes (REs), which contain recycled as well as newly synthesized AMPARs (Bowen et al, 2017), where it stimulates the β2 ARs associated with AMPARs to trigger S845 phosphorylation (Fig 7). This phosphorylation then enhances insertion of AMPARs into the cell surface through mechanisms that are currently unknown. Consistent with this model, AKAP5 is targeted to RE via its palmitoylation on Cys36 and Cys129 and disruption of this palmitoylation interferes with trafficking of REs and AMPARs (Keith et al, 2012; Woolfrey et al, 2015).
Analogous considerations apply to signaling by dopamine via the Gs‐coupled D1 and D5 receptors (D1/5R), which increase GluA1 surface expression in cultured PFC and hippocampal neurons (Sun et al, 2005; Gao et al, 2006). Induction of chemical LTP with a subthreshold concentration of glycine leads to synaptic AMPAR incorporation if D1/5R activation with SKF81297 precedes the glycine treatment (Sun et al, 2005; Gao et al, 2006). In the hippocampal CA1 region, D1/5R stimulation also converts the induction of spike timing‐dependent synaptic depression into potentiation at certain time points (Brzosko et al, 2015).
Localized and dynamic signaling from the β2 AR to Cav1.2
Like AMPARs, Cav1.2 forms a complex that contains the β2 AR, Gs, AC, and PKA for highly localized regulation via cAMP (Davare et al, 1999, 2001; Balijepalli et al, 2006) as well as the antagonistic phosphatases PP2A (Davare et al, 2000; Hall et al, 2006; Xu et al, 2010) and PP2B, the latter anchored via AKAP5 (Oliveria et al, 2007; Fig 6C). In cell‐attached single‐channel recordings, application of the β2 AR‐selective agonist albuterol results in a remarkably strong, more than twofold increase in channel open probability when applied inside the patch electrode but no increase at all when applied to the outside of the electrode. Although in the latter case > 99% of the cell surface and thereby the β2 ARs are agonist accessible, β2 AR stimulation does not allow the resulting cAMP, which is produced throughout the cell except the small patch that is physically occluded by the electrode, to effectively reach the channels under the patch (Chen‐Izu et al, 2000; Davare et al, 2001). These results suggest that cAMP signaling is limited to less than 200 nm.
The C‐terminus of β2 AR directly binds to a small region of the α11.2 C‐terminus encompassing S1928 (Patriarchi et al, 2016; Fig 6C). S1928 is the main phosphorylation site for PKA (De Jongh et al, 1996), and its phosphorylation is increased in the brain in vivo upon β AR stimulation in WT but not AKAP5 KO mice (Hall et al, 2007). Nevertheless, S1928A KI mice have perfectly normal β AR regulation of Cav1.2 in the heart (Lemke et al, 2008). As it turns out, phosphorylation of S1928 serves two different functions: It increases channel activity in neurons (Patriarchi et al, 2016) and vascular smooth muscle cells (Lemke et al, 2008; Patriarchi et al, 2016; Nystoriak et al, 2017); at the same time, it displaces the β2 AR from Cav1.2, which creates a refractory period of about 5 min during which Cav1.2 cannot be re‐phosphorylated upon its dephosphorylation and also not re‐stimulated by β AR agonist application (Patriarchi et al, 2016). A peptide that mimics the interaction site and disrupts the β2 AR–Cav1.2 interaction prevents β AR stimulation of Cav1.2 (Patriarchi et al, 2016), which is further evidence not only for the requirement of β2 AR binding to Cav1.2 for channel regulation but also for localized signaling by this cAMP‐mediated mechanism.
Stimulation of the β2 AR induces its phosphorylation by GPCR kinases (GRKs), leading to recruitment of β‐arrestin and endocytosis of the β2 AR (Shenoy & Lefkowitz, 2011; Staus et al, 2018). Does the Cav1.2‐associated β2 AR also undergo endocytosis? Obviously, displacement of the β2 AR from Cav1.2 would create a situation during which the β2 AR could easily be endocytosed although endocytosis does not contribute to the refractory period of Cav1.2 regulation by β2 AR stimulation (Patriarchi et al, 2016). Furthermore, most recent work now shows that GRKs phosphorylate only β2 AR monomers while the β2 ARs in Cav1.2 complexes are dimers and those dimers are only phosphorylated by PKA (Shen et al, 2018). The PKA phosphorylation sites are different from the GRK sites. Only GRK‐ but not PKA‐phosphorylated β2 ARs undergo endocytosis (Shen et al, 2018) (see also Staus et al, 2018), suggesting that Cav1.2‐associated β2 ARs are not destined for endocytosis. Finally, phosphorylation of not only Cav1.2 itself on S1928 but also the β2 ARs on its PKA sites S261/S262 is required for upregulation of Cav1.2 activity by β2 AR signaling (Shen et al, 2018).
Role of regulation of Cav1.2 by β2 AR signaling in PTT‐LTP
As described above, LTP induced by a 3‐min‐long 5‐Hz tetanus requires the β2 AR and phosphorylation of the AMPAR subunit GluA1 on S845 by PKA (Qian et al, 2012). That Cav1.2 also forms a complex with β2 AR, which makes it a prime target for NE signaling, raises the possibility that Cav1.2 stimulation by the β2 AR is also required for PTT‐LTP especially as Cav1.2 is co‐localized with the β2 AR at postsynaptic sites (Davare et al, 2001). In fact, PTT‐LTP was only mildly if at all affected by NMDAR antagonists but completely blocked by dihydropyridines (DHPs) that inhibit L‐type channel (Qian et al, 2017). Hippocampal slices from KI mice with a point mutation that renders Cav1.2 insensitive to DHPs showed PTT‐LTP that was not affected by DHPs (Qian et al, 2017). Accordingly, Cav1.2 is absolutely required for PTT‐LTP. In fact, PTT‐LTP was completely absent in slices from S1928A KI mice (but not affected in slices from S1700A KI mice) (Qian et al, 2017) and blocked by the peptide that displaces the β2 AR from Cav1.2 (Patriarchi et al, 2016). These results indicate that upregulation of both AMPAR and Cav1.2 activity by β2 AR–PKA signaling is essential for PTT‐LTP (Fig 8). PKA augments not only postsynaptic AMPAR recruitment (see above) but also open probability of AMPARs (Banke et al, 2000), which will amplify depolarization of postsynaptic sites. PKA will not only increase open probability and thereby activity of Cav1.2 (Patriarchi et al, 2016) but also make Cav1.2 more sensitive to depolarization; i.e., Cav1.2 will open more readily for the same level of depolarization upon its stimulation by PKA (Gray & Johnston, 1987; Sculptoreanu et al, 1993). These multiple effects will most likely act in a highly synergistic manner to drive the Ca2+ influx that is necessary for PTT‐LTP (Fig 8).
Figure 8. Role of upregulation of AMPAR and Cav1.2 activity by β2 AR–PKA signaling during PTT‐LTP .
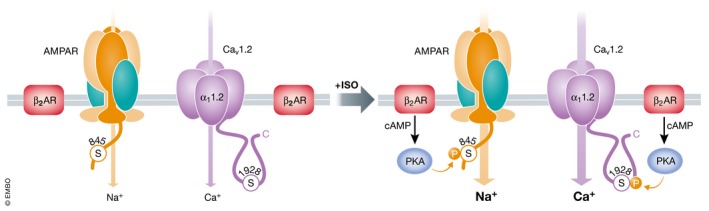
Basal phosphorylation of GluA1 on S845 and of S1928 in the Cav1.2 α11.2 subunit by PKA is low. β2 AR stimulation activates PKA via Gs, AC (not depicted), and cAMP. The consequent phosphorylation of S845 increases Po of the AMPAR and AMPAR postsynaptic accumulation and thereby Na+ influx and depolarization during synaptic transmission. S1928 phosphorylation renders Cav1.2 more sensitive to depolarization and increases Po of Cav1.2. The resulting increase in Ca2+ influx triggers via yet‐to‐be‐defined signaling pathways the potentiation in PTT‐LTP.
Role of CaMKII in regulation of postsynaptic AMPAR localization
Activation of PKA and the phosphorylation of GluA1 on its PKA site S845 is not always required for LTP (Lee et al, 2003, 2010; Lu et al, 2007; Granger et al, 2013) nor is it sufficient by itself to increase postsynaptic AMPAR content in the hippocampal CA1 region (Esteban et al, 2003; Oh et al, 2006; Joiner et al, 2010) although PKA activation does increase postsynaptic AMPAR strength in cultured hippocampal neurons (Diering et al, 2014) and in PFC slices (Joiner et al, 2010). LTP also requires Ca2+ influx and activation and signaling by the Ca2+‐ and calmodulin‐dependent protein kinase CaMKII (Malenka et al, 1989; Malinow et al, 1989; Hayashi et al, 2000; Esteban et al, 2003; Gao et al, 2006; Halt et al, 2012; Huganir & Nicoll, 2013; Hell, 2014; Herring & Nicoll, 2016). The role of PKA in promoting surface expression of AMPARs paired with the requirement of CaMKII for LTP is best explained by a two‐step model (Fig 7) (Penn et al, 2017) (see also Opazo & Choquet, 2011, for a similar three‐step model): AMPARs are first targeted to perisynaptic sites, and then, CaMKII induces trapping of AMPARs at the PSD proper. Accordingly, postsynaptic AMPAR accumulation during LTP requires exocytosis and subsequent lateral diffusion (Penn et al, 2017). Cross‐linking of biotin‐tagged AMPAR subunits at the cell surface impaired LTP indicating that lateral diffusion of AMPARs in the plasma membrane is required for LTP. A slowly developing potentiation that was not blocked by cross‐linking was prevented by inhibition of Ca2+‐triggered exocytosis. The central role of CaMKII in governing postsynaptic AMPAR activity, presumably mostly through promoting postsynaptic AMPAR accumulation, is also illustrated by the surprising recent finding that single‐cell KO of CaMKIIα, the most prevalent CaMKII in forebrain, or double KO of CaMKIIα plus CaMKIIβ reduces AMPAR EPSCs under otherwise basal conditions by 50% and also NMDAR EPSCs by 30% and abrogated pairing‐induced LTP (Incontro et al, 2018). Remarkably, rescue of AMPAR EPSCs as well as LTP required not only CaMKIIα that is catalytically active but also its binding to the NMDAR GluN2B subunit (see the next paragraph).
A key difference between signaling by NE–β2 ARs–cAMP–PKA and by CaMKII is that the former is based on predetermined co‐localization and co‐assembly of all components except for NE, which is widely and diffusely released whereas CaMKII is evenly distributed throughout the dendritic shaft (Strack & Hell et al, 2008). However, stimulation of Ca2+ influx through postsynaptic NMDARs will lead to recruitment of CaMKII to the stimulated spines as seen with ectopically expressed GFP‐CaMKII (Shen & Meyer, 1999; Otmakhov et al, 2004; Rose et al, 2009) as well as endogenous CaMKII (Merrill et al, 2005; Strack & Hell et al, 2008). This recruitment depends on CaMKII binding to residues 1290‐1310 in the C‐terminus of GluN2B (Strack & Colbran, 1998; Leonard et al, 1999; Bayer et al, 2001; Halt et al, 2012).
LTP induced by multiple 1‐s/100‐Hz tetani does not require PKA. It is possible that strong Ca2+ influx triggers AMPAR surface insertion via synaptotagmin‐1‐ and synaptotagmin‐7‐mediated acute exocytosis (Wu et al, 2017). Alternatively, strong LTP induction paradigms could activate CaMKII more so than weaker ones (e.g., two versus one 100‐Hz tetanus in 8‐week‐old mice; Lu et al, 2007). In this way, CaMKII could compensate for a lack of PKA signaling by phosphorylating S831 in the C‐terminus of GluA1. S831 is close to S845 and is a prominent phosphorylation site for CaMKII (Roche et al, 1996). Consistent with this hypothesis, LTP is lost in GluA1 S831A/S845A double KI mice (Lee et al, 2003) while it is normal in single S831A and S845A KI mice (Lee et al, 2010). Hence, one site is both sufficient and required for LTP. Perhaps trafficking of GluA1 to the perisynaptic space can be stimulated by phosphorylation of GluA1 on either S831 by CaMKII or S845 by PKA. In fact, CaMKII can foster surface delivery of GluA1 (Gao et al, 2006).
Phosphorylation of TARPs by CaMKII
At the postsynaptic site, NMDAR‐associated CaMKII acts locally to phosphorylate AMPARs on S831 (Halt et al, 2012), which will increase single‐channel conductance of AMPARs (Kristensen et al, 2011) and might contribute to an increase in postsynaptic response during LTP as LTP has been associated with an increase in single‐channel conductance (Benke et al, 1998) (Fig 9). Alternatively, this increase could also be due to recruitment of GluA1 homomeric AMPARs (Plant et al, 2006; Sanderson et al, 2016), which have a higher conductance than GluA2‐containing AMPARs (Traynelis et al, 2010). However, no deficit in LTP has been found so far in S831A KI mice indicating that S831 phosphorylation is not strictly required (Lee et al, 2010). Rather, the main mechanism of LTP is a persistent increase in postsynaptic AMPAR number, which could be mediated by phosphorylation of the cytosolic C‐termini of the AMPAR auxiliary TARP subunits γ2 (Tomita et al, 2005; Sumioka et al, 2010; Hafner et al, 2015) or γ8 (Park et al, 2016). These phosphorylations strengthen binding of γ2 and γ8 to PSD‐95, thereby trapping of AMPARs at postsynaptic sites (Chen et al, 2000; Schnell et al, 2002; Elias et al, 2006, 2008; Schluter et al, 2006; Opazo et al, 2010; Hafner et al, 2015) (Fig 9). However, the apparent contradictions between findings that specifically implicate CaMKII‐mediated phosphorylation of γ2 (Tomita et al, 2005) versus γ8 (Park et al, 2016) in LTP have to be addressed. There is also a disagreement between recent studies that suggest either phosphorylation of γ8 on S277 and S281 but not PDZ anchoring (Park et al, 2016) or PDZ anchoring of γ8 but not S277 and S281 phosphorylation (Sheng et al, 2018) is critical for LTP. The work by Park et al (2016) is based on KI mice in which the CaMKII phosphorylation sites had been eliminated and the work by Sheng et al (2018) on replacement of all endogenous AMPARs with a GluA1–γ8 fusion protein. Perhaps fusing γ8 to GluA1 leads to a conformation of the γ8 C‐terminus that augments PSD‐95 binding similar to the conformation that is induced by CaMKII‐mediated phosphorylation as shown for γ2 (Sumioka et al, 2010; Hafner et al, 2015).
Figure 9. Hypothetical postsynaptic AMPAR regulation and trapping by CaMKII .
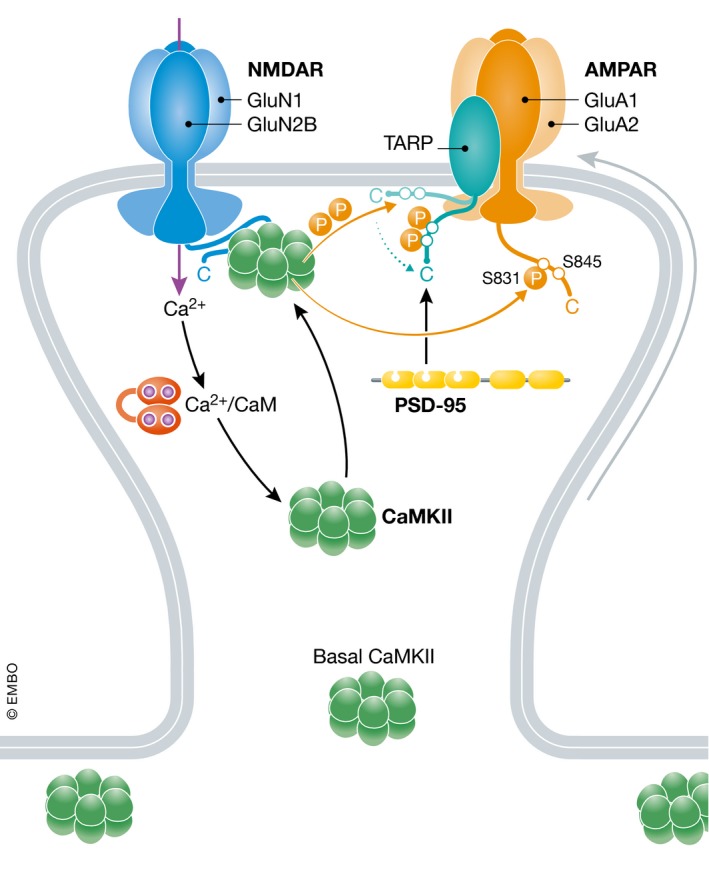
AMPARs reach the postsynaptic density by lateral diffusion. Ca2+ influx via the NMDA receptor will lead to activation of CaMKII by Ca2+/CaM. The immediate autophosphorylation on T286 causes binding of CaMKII to the GluN2B C‐terminus, which is important for phosphorylation of postsynaptic proteins (Halt et al, 2012). Phosphorylation of GluA1 on S831 increases channel activity. The Ca2+ influx also augments detachment of the TARP C‐termini from the cytosolic face of the plasma membrane, which are then phosphorylated by CaMKII. Phosphorylation of either γ2 or γ8 will increase their binding to PSD‐95 to trap AMPARs. Recruitment of CaMKII to postsynaptic sites that are activated during LTP is likely part of the mechanism that ensures synapse specificity of LTP (Hell, 2014).
Phosphorylation of Kalirin and Trio by CaMKII
A second, increasingly prominent CaMKII target that is relevant for LTP is Kalirin 7, a splice isoform of Kalirin (also called Duo). Kalirin 7 is a guanine nucleotide exchange factor (GEF) for the small G protein Rac (Penzes et al, 2008). Activation of Rac by Kalirin 7 augments formation of F‐actin via p21‐activated kinase PAK and thereby spine enragement as well as postsynaptic AMPAR accumulation (Penzes et al, 2008). Ca2+ influx through NMDARs induces phosphorylation of Kalirin 7 on S95 by CaMKII, which leads to activation of PAK, spine enlargement, and an increase in postsynaptic AMPAR content (Xie et al, 2007). More recent work shows that Kalirin 7 as well as the closely related Trio fulfills overlapping functions with respect to synaptic maturation and LTP (Herring & Nicoll, 2016). The increase in postsynaptic AMPAR responses upon overexpression of either Kalirin 7 or Trio depended on basal spontaneous synaptic activity; it was prevented by concurrent inhibition of AMPARs and NMDARs. Knockdown (KD) of both proteins resulted in an ~80% loss of spines and of synaptic transmission by both AMPARs and NMDARs. These and earlier results indicate that these two proteins together mediate spine and synapse maturation under basal conditions (Xie et al, 2007; Herring & Nicoll, 2016). However, KD of Kalirin was insufficient to impair LTP and KD of Trio had only a modest effect. KD of both proteins abrogated synapse formation, but synapse formation can be rescued by KD‐resistant Kalirin 7, which also rescued LTP. Importantly, rescue of LTP was not achieved if the CaMKII phosphorylation site S95 had been mutated to alanine. An analogous phosphorylation site is present in Trio, and LTP was rescued by KD‐resistant WT Trio but not phosphorylation‐deficient T66A Trio (Herring & Nicoll, 2016). Thus, phosphorylation of one of these two Rac GEFs by CaMKII is a critical step in LTP. Finally, mutations in the Rac GEF domain of Trio that have been linked to autism spectrum disorders affect AMPAR function (Sadybekov et al, 2017).
Emerging CaMKII targets in LTP: SynGAP and neuroligin‐1
SynGAP is a Ras GTPase‐activating protein; i.e., it terminates Ras signaling by stimulating its hydrolysis of GTP to GDP (Carlisle et al, 2008). Ras in turn promotes postsynaptic delivery of AMPARs (Zhu et al, 2002). Recent work now finds that CaMKII phosphorylation of SynGAP leads to displacement of SynGAP from the postsynaptic site, thereby fostering LTP (Araki et al, 2015; Walkup et al, 2016).
Finally, the postsynaptic cell adhesion protein neuroligin‐1 is important for synapse formation by interacting with presynaptic neurexin (Scheiffele et al, 2000). More recent work indicates that phosphorylation of neuroligin‐1 on T739 promotes synapse stabilization and strength (Bemben et al, 2014).
Where PKA and CaMKII intersect
Given the prominence of PKA and CaMKII signaling at the postsynaptic site, it can be expected that signaling pathways by these two kinases interconnect. In fact, PKA‐mediated phosphorylation of the NMDAR subunit GluN2B on S1166, which can be induced by β2 AR stimulation, selectively increases Ca2+ permeability of NMDARs (Skeberdis et al, 2006; Murphy et al, 2014a). This phosphorylation is important for induction of CaMKII‐dependent LTP by augmenting Ca2+ influx, which in turn is required for CaMKII activation.
On the other hand, CaMKII might antagonize PKA signaling in spines by phosphorylating AKAP5 on multiple serine and threonine residues in its polybasic regions, including T87 and S92 in region B (Fig 6A; Woolfrey et al, 2018). Interestingly, this phosphorylation is inhibited by Ca2+/CaM binding to the polybasic regions and could only proceed after CaMKII becomes constitutively active through its autophosphorylation of T286 (Woolfrey et al, 2018). T286 phosphorylation results in Ca2+/CaM‐independent so‐called autonomous CaMKII activity due to impaired rebinding of the autoinhibitory domain to the catalytic domain upon removal of Ca2+/CaM from this autoinhibitory domain (Hell, 2014). In addition, Ca2+/CaM had to be removed from the polybasic AKAP5 regions before autonomously active CaMKII could phosphorylate this region. Functionally, phosphorylation of AKAP5 by CaMKII proved to be important for removal of AKAP5 from spines (Woolfrey et al, 2018). This removal required in addition depalmitoylation of AKAP5, which in turn needed CaMKII activity. This complex mechanism is critical for LTD induction during which cytosolic Ca2+ levels remain elevated for some time before falling to resting levels. It is presumably during this phase of Ca2+ removal and with it of Ca2+/CaM from AKAP5 that autonomously active CaMKII phosphorylates AKAP5. Although the molecular consequences remain to be determined, the loss of AKAP5 from spines upon this CaMKII‐dependent phosphorylation and depalmitoylation likely translates into a reduction of PKA and thereby of PKA‐mediated signaling in spines. The situation might be more complicated as loss of AKAP5 would also translate into loss of the phosphatase PP2B, which antagonizes postsynaptic PKA signaling.
Conclusion
The intricacy of signaling mechanisms at the postsynaptic site and of the underlying protein interactions we have unveiled so far is truly mind‐boggling. One wonders how much more complex the whole postsynaptic signaling network will turn out to be. Postsynaptic signaling as it regulates postsynaptic AMPAR content and thereby synaptic strength is functionally highly relevant because changes in synaptic strength underly many forms of physiological as well as pathological learning. If we are to truly understand postsynaptic signaling and how it relates to synaptic strength in health and disease, an important long‐term goal, we need to keep digging for years to come.
Conflict of interest
All authors declare that they have no conflict of interest.
Acknowledgements
We thank Dr. T. Nakagawa, Vanderbilt University, Nashville, TN, for discussion and help with modeling of glutamate receptor structure and Dr. Lucas Matt, University of Tübingen, Germany, for help with figures.
The EMBO Journal (2018) 37: e99771
See the Glossary for abbreviations used in this article.
References
- Adesnik H, Nicoll RA (2007) Conservation of glutamate receptor 2‐containing AMPA receptors during long‐term potentiation. J Neurosci 27: 4598–4602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altimimi HF, Stellwagen D (2013) Persistent synaptic scaling independent of AMPA receptor subunit composition. J Neurosci 33: 11763–11767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ancona Esselmann SG, Diaz‐Alonso J, Levy JM, Bemben MA, Nicoll RA (2017) Synaptic homeostasis requires the membrane‐proximal carboxy tail of GluA2. Proc Natl Acad Sci USA 114: 13266–13271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araki Y, Zeng M, Zhang M, Huganir RL (2015) Rapid dispersion of SynGAP from synaptic spines triggers AMPA receptor insertion and spine enlargement during LTP. Neuron 85: 173–189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araya R, Nikolenko V, Eisenthal KB, Yuste R (2007) Sodium channels amplify spine potentials. Proc Natl Acad Sic USA 104: 12347–12352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araya R, Vogels TP, Yuste R (2014) Activity‐dependent dendritic spine neck changes are correlated with synaptic strength. Proc Natl Acad Sci USA 111: E2895–E2904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashby MC, Maier SR, Nishimune A, Henley JM (2006) Lateral diffusion drives constitutive exchange of AMPA receptors at dendritic spines and is regulated by spine morphology. J Neurosci 26: 7046–7055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asrican B, Lisman J, Otmakhov N (2007) Synaptic strength of individual spines correlates with bound Ca2+‐calmodulin‐dependent kinase II. J Neurosci 27: 14007–14011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balijepalli RC, Foell JD, Hall DD, Hell JW, Kamp TJ (2006) From the Cover: Localization of cardiac L‐type Ca2+ channels to a caveolar macromolecular signaling complex is required for beta2‐adrenergic regulation. Proc Natl Acad Sci USA 103: 7500–7505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banke TG, Bowie D, Lee H, Huganir RL, Schousboe A, Traynelis SF (2000) Control of GluR1 AMPA receptor function by cAMP‐dependent protein kinase. J Neurosci 20: 89–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bard L, Sainlos M, Bouchet D, Cousins S, Mikasova L, Breillat C, Stephenson FA, Imperiali B, Choquet D, Groc L (2010) Dynamic and specific interaction between synaptic NR2‐NMDA receptor and PDZ proteins. Proc Natl Acad Sic USA 107: 19561–19566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baron MK, Boeckers TM, Vaida B, Faham S, Gingery M, Sawaya MR, Salyer D, Gundelfinger ED, Bowie JU (2006) An architectural framework that may lie at the core of the postsynaptic density. Science 311: 531–535 [DOI] [PubMed] [Google Scholar]
- Bartos JA, Ulrich JD, Li H, Beazely MA, Chen Y, Macdonald JF, Hell JW (2010) Postsynaptic clustering and activation of Pyk2 by PSD‐95. J Neurosci 30: 449–463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bats C, Groc L, Choquet D (2007) The interaction between Stargazin and PSD‐95 regulates AMPA receptor surface trafficking. Neuron 53: 719–734 [DOI] [PubMed] [Google Scholar]
- Bayer KU, De Koninck P, Leonard AS, Hell JW, Schulman H (2001) Interaction with the NMDA receptor locks CaMKII in an active conformation. Nature 411: 801–805 [DOI] [PubMed] [Google Scholar]
- Beene DL, Scott JD (2007) A‐kinase anchoring proteins take shape. Curr Opin Cell Biol 19: 192–198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beique JC, Lin DT, Kang MG, Aizawa H, Takamiya K, Huganir RL (2006) Synapse‐specific regulation of AMPA receptor function by PSD‐95. Proc Natl Acad Sci USA 103: 19535–19540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bekkers JM, Stevens CF (1989) NMDA and non‐NMDA receptors are co‐localized at individual excitatory synapses in cultured rat hippocampus. Nature 341: 230–233 [DOI] [PubMed] [Google Scholar]
- Bemben MA, Shipman SL, Hirai T, Herring BE, Li Y, Badger JD II, Nicoll RA, Diamond JS, Roche KW (2014) CaMKII phosphorylation of neuroligin‐1 regulates excitatory synapses. Nat Neurosci 17: 56–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benke TA, Luthi A, Isaac JT, Collingridge GL (1998) Modulation of AMPA receptor unitary conductance by synaptic activity. Nature 393: 793–797 [DOI] [PubMed] [Google Scholar]
- Berman DE, Dudai Y (2001) Memory extinction, learning anew, and learning the new: dissociations in the molecular machinery of learning in cortex. Science 291: 2417–2419 [DOI] [PubMed] [Google Scholar]
- Bhattacharyya S, Biou V, Xu W, Schluter O, Malenka RC (2009) A critical role for PSD‐95/AKAP interactions in endocytosis of synaptic AMPA receptors. Nat Neurosci 12: 172–181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biederer T, Kaeser PS, Blanpied TA (2017) Transcellular nanoalignment of synaptic function. Neuron 96: 680–696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blitzer RD, Wong T, Nouranifar R, Iyengar R, Landau EM (1995) Postsynaptic cAMP pathway gates early LTP in hippocampal CA1 region. Neuron 15: 1403–1414 [DOI] [PubMed] [Google Scholar]
- Bolshakov VY, Siegelbaum SA (1994) Postsynaptic induction and presynaptic expression of hippocampal long‐term depression. Science 264: 148–152 [DOI] [PubMed] [Google Scholar]
- Bosch M, Castro J, Saneyoshi T, Matsuno H, Sur M, Hayashi Y (2014) Structural and molecular remodeling of dendritic spine substructures during long‐term potentiation. Neuron 82: 444–459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowen AB, Bourke AM, Hiester BG, Hanus C, Kennedy MJ (2017) Golgi‐independent secretory trafficking through recycling endosomes in neuronal dendrites and spines. eLife 6: e27362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandon EP, Idzerda RL, McKnight GS (1997) PKA isoforms, neural pathways, and behaviour: making the connection. Curr Opin Neurobiol 7: 397–403 [DOI] [PubMed] [Google Scholar]
- Brzosko Z, Schultz W, Paulsen O (2015) Retroactive modulation of spike timing‐dependent plasticity by dopamine. eLife 4: e09685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahill L, Prins B, Weber M, McGaugh JL (1994) Beta‐adrenergic activation and memory for emotional events. Nature 371: 702–704 [DOI] [PubMed] [Google Scholar]
- Cai C, Coleman SK, Niemi K, Keinanen K (2002) Selective binding of synapse‐associated protein 97 to GluR‐A a‐amino‐5‐hydroxy‐3‐methyl‐4‐isoxazole‐4‐propionate receptor subunit is determined by a novel sequence motif. J Biol Chem 277: 31484–31490 [DOI] [PubMed] [Google Scholar]
- Cao G, Harris KM (2014) Augmenting saturated LTP by broadly spaced episodes of theta‐burst stimulation in hippocampal area CA1 of adult rats and mice. J Neurophysiol 112: 1916–1924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlisle HJ, Manzerra P, Marcora E, Kennedy MB (2008) SynGAP regulates steady‐state and activity‐dependent phosphorylation of cofilin. J Neurosci 28: 13673–13683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carr DW, Stofko‐Hahn RE, Fraser IDC, Cone RD, Scott JD (1992) Localization of the cAMP‐dependent protein kinase to the postsynaptic densities by A‐kinase anchoring proteins. J Biol Chem 267: 16816–16823 [PubMed] [Google Scholar]
- Carter ME, Yizhar O, Chikahisa S, Nguyen H, Adamantidis A, Nishino S, Deisseroth K, de Lecea L (2010) Tuning arousal with optogenetic modulation of locus coeruleus neurons. Nat Neurosci 13: 1526–1533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Chetkovich DM, Petralia RS, Sweeney NT, Kawasaki Y, Wenthold RJ, Bredt DS, Nicoll RA (2000) Stargazing regulates synaptic targeting of AMPA receptors by two distinct mechanisms. Nature 408: 936–943 [DOI] [PubMed] [Google Scholar]
- Chen X, Winters C, Azzam R, Li X, Galbraith JA, Leapman RD, Reese TS (2008) Organization of the core structure of the postsynaptic density. Proc Natl Acad Sci USA 105: 4453–4458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Nelson CD, Li X, Winters CA, Azzam R, Sousa AA, Leapman RD, Gainer H, Sheng M, Reese TS (2011) PSD‐95 Is Required to Sustain the Molecular Organization of the Postsynaptic Density. J Neurosci 31: 6329–6338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Levy JM, Hou A, Winters C, Azzam R, Sousa AA, Leapman RD, Nicoll RA, Reese TS (2015) PSD‐95 family MAGUKs are essential for anchoring AMPA and NMDA receptor complexes at the postsynaptic density. Proc Natl Acad Sci USA 112: E6983–E6992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chenaux G, Matt L, Hill TC, Kaur I, Liu XB, Kirk LM, Speca DJ, McMahon SA, Zito K, Hell JW, Diaz E (2016) Loss of SynDIG1 reduces excitatory synapse maturation but not formation in vivo. eNeuro 3: e0130‐16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen‐Izu Y, Xiao RP, Izu LT, Cheng H, Kuschel M, Spurgeon H, Lakatta EG (2000) G(i)‐dependent localization of beta(2)‐adrenergic receptor signaling to L‐type Ca(2+) channels. Biophys J 79: 2547–2556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheong R, Rhee A, Wang CJ, Nemenman I, Levchenko A (2011) Information transduction capacity of noisy biochemical signaling networks. Science 334: 354–358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chowdhury D, Turner M, Patriarchi T, Hergarden AC, Anderson D, Zhang Y, Sun J, Chen CY, Ames JB, Hell JW (2018) Ca(2+)/calmodulin binding to PSD‐95 mediates homeostatic synaptic scaling down. EMBO J 37: 122–138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clem RL, Huganir RL (2010) Calcium‐permeable AMPA receptor dynamics mediate fear memory erasure. Science 330: 1108–1112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clements JD, Lester RA, Tong G, Jahr CE, Westbrook GL (1992) The time course of glutamate in the synaptic cleft. Science 258: 1498–1501 [DOI] [PubMed] [Google Scholar]
- Coba MP, Pocklington AJ, Collins MO, Kopanitsa MV, Uren RT, Swamy S, Croning MD, Choudhary JS, Grant SG (2009) Neurotransmitters drive combinatorial multistate postsynaptic density networks. Sci Signal 2: ra19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colledge M, Dean RA, Scott GK, Langeberg LK, Huganir RL, Scott JD (2000) Targeting of PKA to glutamate receptors through a MAGUK‐AKAP complex. Neuron 27: 107–119 [DOI] [PubMed] [Google Scholar]
- Collingridge GL, Isaac JT, Wang YT (2004) Receptor trafficking and synaptic plasticity. Nat Rev Neurosci 5: 952–962 [DOI] [PubMed] [Google Scholar]
- Collins MO, Husi H, Yu L, Brandon JM, Anderson CN, Blackstock WP, Choudhary JS, Grant SG (2006) Molecular characterization and comparison of the components and multiprotein complexes in the postsynaptic proteome. J Neurochem 97(Suppl 1): 16–23 [DOI] [PubMed] [Google Scholar]
- Craven SE, El‐Husseini AE, Bredt DS (1999) Synaptic targeting of the postsynaptic density protein PSD‐95 mediated by lipid and protein motifs. Neuron 22: 497–509 [DOI] [PubMed] [Google Scholar]
- Dai S, Hall DD, Hell JW (2009) Supramolecular Assemblies and Localized Regulation of Voltage‐gated Ion Channels. Physiol Rev 89: 411–452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dakoji S, Tomita S, Karimzadegan S, Nicoll RA, Bredt DS (2003) Interaction of transmembrane AMPA receptor regulatory proteins with multiple membrane associated guanylate kinases. Neuropharmacology 45: 849–856 [DOI] [PubMed] [Google Scholar]
- Dani A, Huang B, Bergan J, Dulac C, Zhuang X (2010) Superresolution imaging of chemical synapses in the brain. Neuron 68: 843–856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davare MA, Dong F, Rubin CS, Hell JW (1999) The A‐kinase anchor protein MAP2B and cAMP‐dependent protein kinase are associated with class C L‐type calcium channels in neurons. J Biol Chem 274: 30280–30287 [DOI] [PubMed] [Google Scholar]
- Davare MA, Horne MC, Hell JW (2000) Protein Phosphatase 2A is associated with class C L‐type calcium channels (Cav1.2) and antagonizes channel phosphorylation by cAMP‐dependent protein kinase. J Biol Chem 275: 39710–39717 [DOI] [PubMed] [Google Scholar]
- Davare MA, Avdonin V, Hall DD, Peden EM, Burette A, Weinberg RJ, Horne MC, Hoshi T, Hell JW (2001) A beta2 adrenergic receptor signaling complex assembled with the Ca2+ channel Cav1.2. Science 293: 98–101 [DOI] [PubMed] [Google Scholar]
- Dell'Acqua ML, Faux MC, Thorburn J, Thorburn A, Scott JD (1998) Membrane‐targeting sequences on AKAP79 bind phosphatidylinositol‐4, 5‐ bisphosphate. EMBO J 17: 2246–2260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz‐Alonso J, Sun YJ, Granger AJ, Levy JM, Blankenship SM, Nicoll RA (2017) Subunit‐specific role for the amino‐terminal domain of AMPA receptors in synaptic targeting. Proc Natl Acad Sci USA 114: 7136–7141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diering GH, Gustina AS, Huganir RL (2014) PKA‐GluA1 coupling via AKAP5 controls AMPA receptor phosphorylation and cell surface targeting during bidirectional homeostatic plasticity. Neuron 84: 790–805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudek SM, Bear MF (1992) Homosynaptic long‐term depression in area CA1 of hippocampus and effects of N‐methyl‐D‐aspartate receptor blockade. Proc Natl Acad Sci USA 89: 4363–4367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehlers MD (2000) Reinsertion or degradation of AMPA receptors determined by activity‐dependent endocytic sorting. Neuron 28: 511–525 [DOI] [PubMed] [Google Scholar]
- Ehrlich I, Klein M, Rumpel S, Malinow R (2007) PSD‐95 is required for activity‐driven synapse stabilization. Proc Natl Acad Sci USA 104: 4176–4181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- El‐Husseini AE, Schnell E, Chetkovich DM, Nicoll RA, Bredt DS (2000) PSD‐95 involvement in maturation of excitatory synapses. Science 290: 1364–1368 [PubMed] [Google Scholar]
- El‐Husseini Ael D, Schnell E, Dakoji S, Sweeney N, Zhou Q, Prange O, Gauthier‐Campbell C, Aguilera‐Moreno A, Nicoll RA, Bredt DS (2002) Synaptic strength regulated by palmitate cycling on PSD‐95. Cell 108: 849–863 [DOI] [PubMed] [Google Scholar]
- Elias GM, Funke L, Stein V, Grant SG, Bredt DS, Nicoll RA (2006) Synapse‐Specific and Developmentally Regulated Targeting of AMPA Receptors by a Family of MAGUK Scaffolding Proteins. Neuron 52: 307–320 [DOI] [PubMed] [Google Scholar]
- Elias GM, Elias LA, Apostolides PF, Kriegstein AR, Nicoll RA (2008) Differential trafficking of AMPA and NMDA receptors by SAP102 and PSD‐95 underlies synapse development. Proc Natl Acad Sci USA 105: 20953–20958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esteban JA, Shi SH, Wilson C, Nuriya M, Huganir RL, Malinow R (2003) PKA phosphorylation of AMPA receptor subunits controls synaptic trafficking underlying plasticity. Nat Neurosci 6: 136–143 [DOI] [PubMed] [Google Scholar]
- Fischer QS, Beaver CJ, Yang Y, Rao Y, Jakobsdottir KB, Storm DR, McKnight GS, Daw NW (2004) Requirement for the RIIbeta isoform of PKA, but not calcium‐stimulated adenylyl cyclase, in visual cortical plasticity. J Neurosci 24: 9049–9058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franks KM, Stevens CF, Sejnowski TJ (2003) Independent sources of quantal variability at single glutamatergic synapses. J Neurosci 23: 3186–3195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukata Y, Dimitrov A, Boncompain G, Vielemeyer O, Perez F, Fukata M (2013) Local palmitoylation cycles define activity‐regulated postsynaptic subdomains. J Cell Biol 202: 145–161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukazawa Y, Saitoh Y, Ozawa F, Ohta Y, Mizuno K, Inokuchi K (2003) Hippocampal LTP is accompanied by enhanced F‐actin content within the dendritic spine that is essential for late LTP maintenance in vivo. Neuron 38: 447–460 [DOI] [PubMed] [Google Scholar]
- Fukazawa Y, Shigemoto R (2012) Intra‐synapse‐type and inter‐synapse‐type relationships between synaptic size and AMPAR expression. Curr Opin Neurobiol 22: 446–452 [DOI] [PubMed] [Google Scholar]
- Gainey MA, Hurvitz‐Wolff JR, Lambo ME, Turrigiano GG (2009) Synaptic scaling requires the GluR2 subunit of the AMPA receptor. J Neurosci 29: 6479–6489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gainey MA, Tatavarty V, Nahmani M, Lin H, Turrigiano GG (2015) Activity‐dependent synaptic GRIP1 accumulation drives synaptic scaling up in response to action potential blockade. Proc Natl Acad Sci USA 112: E3590–E3599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao C, Sun X, Wolf ME (2006) Activation of D1 dopamine receptors increases surface expression of AMPA receptors and facilitates their synaptic incorporation in cultured hippocampal neurons. J Neurochem 98: 1664–1677 [DOI] [PubMed] [Google Scholar]
- Gelinas JN, Nguyen PV (2005) Beta‐adrenergic receptor activation facilitates induction of a protein synthesis‐dependent late phase of long‐term potentiation. J Neurosci 25: 3294–3303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gingrich JR, Pelkey KA, Fam SR, Huang Y, Petralia RS, Wenthold RJ, Salter MW (2004) Unique domain anchoring of Src to synaptic NMDA receptors via the mitochondrial protein NADH dehydrogenase subunit 2. Proc Natl Acad Sci USA 101: 6237–6242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez LL, Alam S, Smith KE, Horne E, Dell'Acqua ML (2002) Regulation of A‐kinase anchoring protein 79/150‐cAMP‐dependent protein kinase postsynaptic targeting by NMDA receptor activation of calcineurin and remodeling of dendritic actin. J Neurosci 22: 7027–7044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goold CP, Nicoll RA (2010) Single‐cell optogenetic excitation drives homeostatic synaptic depression. Neuron 68: 512–528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorski JA, Gomez LL, Scott JD, Dell'Acqua ML (2005) Association of an A‐kinase‐anchoring protein signaling scaffold with cadherin adhesion molecules in neurons and epithelial cells. Mol Biol Cell 16: 3574–3590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granger AJ, Shi Y, Lu W, Cerpas M, Nicoll RA (2013) LTP requires a reserve pool of glutamate receptors independent of subunit type. Nature 493: 495–500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray R, Johnston D (1987) Noradrenaline and beta‐adrenoceptor agonists increase activity of voltage‐dependent calcium channels in hippocampal neurons. Nature 327: 620–622 [DOI] [PubMed] [Google Scholar]
- Gray JA, Shi Y, Usui H, During MJ, Sakimura K, Nicoll RA (2011) Distinct modes of AMPA receptor suppression at developing synapses by GluN2A and GluN2B: single‐cell NMDA receptor subunit deletion in vivo. Neuron 71: 1085–1101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groc L, Heine M, Cognet L, Brickley K, Stephenson FA, Lounis B, Choquet D (2004) Differential activity‐dependent regulation of the lateral mobilities of AMPA and NMDA receptors. Nat Neurosci 7: 695–696 [DOI] [PubMed] [Google Scholar]
- Hafner AS, Penn AC, Grillo‐Bosch D, Retailleau N, Poujol C, Philippat A, Coussen F, Sainlos M, Opazo P, Choquet D (2015) Lengthening of the Stargazin Cytoplasmic Tail Increases Synaptic Transmission by Promoting Interaction to Deeper Domains of PSD‐95. Neuron 86: 475–489 [DOI] [PubMed] [Google Scholar]
- Hall DD, Feekes JA, Arachchige Don AS, Shi M, Hamid J, Chen L, Strack S, Zamponi GW, Horne MC, Hell JW (2006) Binding of protein phosphatase 2A to the L‐type calcium channel Cav1.2 next to Ser 1928, its main PKA site, is critical for Ser1928 dephosphorylation. Biochemistry 45: 3448–3459 [DOI] [PubMed] [Google Scholar]
- Hall DD, Davare MA, Shi M, Allen ML, Weisenhaus M, McKnight GS, Hell JW (2007) Critical role of cAMP‐dependent protein kinase anchoring to the L‐type calcium channel Cav1.2 via A‐kinase anchor protein 150 in neurons. Biochemistry 46: 1635–1646 [DOI] [PubMed] [Google Scholar]
- Halt AR, Dallapiazza R, Yu H, Stein IS, Qian H, Junti S, Wojcik S, Brose N, Sliva A, Hell JW (2012) CaMKII binding to GluN2B is Critical During Memory Consolidation. EMBO J 31: 1203–1216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanamura K, Washburn HR, Sheffler‐Collins SI, Xia NL, Henderson N, Tillu DV, Hassler S, Spellman DS, Zhang G, Neubert TA, Price TJ, Dalva MB (2017) Extracellular phosphorylation of a receptor tyrosine kinase controls synaptic localization of NMDA receptors and regulates pathological pain. PLoS Biol 15: e2002457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris KM, Stevens JK (1989) Dendritic spines of CA 1 pyramidal cells in the rat hippocampus: serial electron microscopy with reference to their biophysical characteristics. J Neurosci 9: 2982–2997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Havekes R, Canton DA, Park AJ, Huang T, Nie T, Day JP, Guercio LA, Grimes Q, Luczak V, Gelman IH, Baillie GS, Scott JD, Abel T (2012) Gravin orchestrates protein kinase A and beta2‐adrenergic receptor signaling critical for synaptic plasticity and memory. J Neurosci 32: 18137–18149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi Y, Shi SH, Esteban JA, Piccini A, Poncer JC, Malinow R (2000) Driving AMPA receptors into synapses by LTP and CaMKII: requirement for GluR1 and PDZ domain interaction. Science 287: 2262–2267 [DOI] [PubMed] [Google Scholar]
- He K, Song L, Cummings LW, Goldman J, Huganir RL, Lee HK (2009) Stabilization of Ca2+‐permeable AMPA receptors at perisynaptic sites by GluR1‐S845 phosphorylation. Proc Natl Acad Sci USA 106: 20033–20038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He K, Huertas M, Hong SZ, Tie X, Hell JW, Shouval H, Kirkwood A (2015) Distinct eligibility traces for LTP and LTD in cortical synapses. Neuron 88: 528–538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heine M, Groc L, Frischknecht R, Beique JC, Lounis B, Rumbaugh G, Huganir RL, Cognet L, Choquet D (2008) Surface mobility of postsynaptic AMPARs tunes synaptic transmission. Science 320: 201–205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hell JW (2014) CaMKII: claiming center stage in postsynaptic function and organization. Neuron 81: 249–265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herring BE, Shi Y, Suh YH, Zheng CY, Blankenship SM, Roche KW, Nicoll RA (2013) Cornichon proteins determine the subunit composition of synaptic AMPA receptors. Neuron 77: 1083–1096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herring BE, Nicoll RA (2016) Kalirin and Trio proteins serve critical roles in excitatory synaptic transmission and LTP. Proc Natl Acad Sci USA 113: 2264–2269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honkura N, Matsuzaki M, Noguchi J, Ellis‐Davies GC, Kasai H (2008) The subspine organization of actin fibers regulates the structure and plasticity of dendritic spines. Neuron 57: 719–729 [DOI] [PubMed] [Google Scholar]
- Hoshi N, Langeberg LK, Scott JD (2005) Distinct enzyme combinations in AKAP signalling complexes permit functional diversity. Nat Cell Biol 7: 1066–1073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hruska M, Henderson N, Le Marchand SJ, Jafri H, Dalva MB (2018) Synaptic nanomodules underlie the organization and plasticity of spine synapses. Nat Neurosci 21: 671–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu H, Real E, Takamiya K, Kang MG, Ledoux J, Huganir RL, Malinow R (2007) Emotion enhances learning via norepinephrine regulation of AMPA‐receptor trafficking. Cell 131: 160–173 [DOI] [PubMed] [Google Scholar]
- Huang Y, Lu W, Ali DW, Pelkey KA, Pitcher GM, Lu YM, Aoto H, Roder JC, Sasaki T, Salter MW, MacDonald JF (2001) CAKbeta/Pyk2 kinase is a signaling link for induction of long‐term potentiation in CA1 hippocampus. Neuron 29: 485–496 [DOI] [PubMed] [Google Scholar]
- Huganir RL, Nicoll RA (2013) AMPARs and synaptic plasticity: the last 25 years. Neuron 80: 704–717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Incontro S, Diaz‐Alonso J, Iafrati J, Vieira M, Asensio CS, Sohal VS, Roche KW, Bender KJ, Nicoll RA (2018) The CaMKII/NMDA receptor complex controls hippocampal synaptic transmission by kinase‐dependent and independent mechanisms. Nat Commun 9: 2069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irannejad R, Pessino V, Mika D, Huang B, Wedegaertner PB, Conti M, von Zastrow M (2017) Functional selectivity of GPCR‐directed drug action through location bias. Nat Chem Biol 13: 799–806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson AC, Nicoll RA (2011) The expanding social network of ionotropic glutamate receptors: TARPs and other transmembrane auxiliary subunits. Neuron 70: 178–199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeyifous O, Lin EI, Chen X, Antinone SE, Mastro R, Drisdel R, Reese TS, Green WN (2016) Palmitoylation regulates glutamate receptor distributions in postsynaptic densities through control of PSD95 conformation and orientation. Proc Natl Acad Sci USA 113: E8482–E8491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson DA, Leathers VL, Martinez AM, Walsh DA, Fletcher WH (1993) Fluorescence resonance energy transfer within a heterochromatic cAMP‐dependent protein kinase holoenzyme under equilibrium conditions: new insights into the conformational changes that result in cAMP‐dependent activation. Biochemistry 32: 6402–6410 [DOI] [PubMed] [Google Scholar]
- Joiner ML, Lise MF, Yuen EY, Kam AY, Zhang M, Hall DD, Malik ZA, Qian H, Chen Y, Ulrich JD, Burette AC, Weinberg RJ, Law PY, El‐Husseini A, Yan Z, Hell JW (2010) Assembly of a beta(2)‐adrenergic receptor‐GluR1 signalling complex for localized cAMP signalling. EMBO J 29: 482–495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Jongh KS, Murphy BJ, Colvin AA, Hell JW, Takahashi M, Catterall WA (1996) Specific phosphorylation of a site in the full length form of the a1 subunit of the cardiac L‐type calcium channel by adenosine 3’,5’‐cyclic monophosphate‐dependent protein kinase. Biochemistry 35: 10392–10402 [DOI] [PubMed] [Google Scholar]
- Jurado S, Biou V, Malenka RC (2010) A calcineurin/AKAP complex is required for NMDA receptor‐dependent long‐term depression. Nat Neurosci 13: 1053–1055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalashnikova E, Lorca RA, Kaur I, Barisone GA, Li B, Ishimaru T, Trimmer JS, Mohapatra DP, Diaz E (2010) SynDIG1: an activity‐regulated, AMPA‐ receptor‐interacting transmembrane protein that regulates excitatory synapse development. Neuron 65: 80–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalia LV, Pitcher GM, Pelkey KA, Salter MW (2006) PSD‐95 is a negative regulator of the tyrosine kinase Src in the NMDA receptor complex. EMBO J 25: 4971–4982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keith DJ, Sanderson JL, Gibson ES, Woolfrey KM, Robertson HR, Olszewski K, Kang R, El‐Husseini A, Dell'Acqua ML (2012) Palmitoylation of A‐kinase anchoring protein 79/150 regulates dendritic endosomal targeting and synaptic plasticity mechanisms. J Neurosci 32: 7119–7136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr JM, Blanpied TA (2012) Subsynaptic AMPA receptor distribution is acutely regulated by actin‐driven reorganization of the postsynaptic density. J Neurosci 32: 658–673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessels HW, Malinow R (2009) Synaptic AMPA receptor plasticity and behavior. Neuron 61: 340–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kharazia VN, Weinberg RJ (1997) Tangential synaptic distribution of NMDA and AMPA receptors in rat neocortex. Neurosci Lett 238: 41–44 [DOI] [PubMed] [Google Scholar]
- Kharazia VN, Weinberg RJ (1999) Immunogold localization of AMPA and NMDA receptors in somatic sensory cortex of albino rat. J Comp Neurol 412: 292–302 [DOI] [PubMed] [Google Scholar]
- Khuchua Z, Wozniak DF, Bardgett ME, Yue Z, McDonald M, Boero J, Hartman RE, Sims H, Strauss AW (2003) Deletion of the N‐terminus of murine map2 by gene targeting disrupts hippocampal ca1 neuron architecture and alters contextual memory. Neuroscience 119: 101–111 [DOI] [PubMed] [Google Scholar]
- Kim J, Jung SC, Clemens AM, Petralia RS, Hoffman DA (2007) Regulation of dendritic excitability by activity‐dependent trafficking of the A‐type K+ channel subunit Kv4.2 in hippocampal neurons. Neuron 54: 933–947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JY, Oh MH, Bernard LP, Macara IG, Zhang H (2011) The RhoG/ELMO1/Dock180 signaling module is required for spine morphogenesis in hippocampal neurons. J Biol Chem 286: 37615–37624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Ziff EB (2014) Calcineurin mediates synaptic scaling via synaptic trafficking of Ca2+‐permeable AMPA receptors. PLoS Biol 12: e1001900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiraly DD, Eipper‐Mains JE, Mains RE, Eipper BA (2010) Synaptic plasticity, a symphony in GEF. ACS Chem Neurosci 1: 348–365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klauck TM, Faux MC, Labudda K, Langeberg LK, Jaken S, Scott JD (1996) Coordination of three signaling enzymes by AKAP79, a mammalian scaffold protein. Science 271: 1589–1592 [DOI] [PubMed] [Google Scholar]
- Kornau HC, Schenker LT, Kennedy MB, Seeburg PH (1995) Domain interaction between NMDA receptor subunits and the postsynaptic density protein PSD‐95. Science 269: 1737–1740 [DOI] [PubMed] [Google Scholar]
- Kristensen AS, Jenkins MA, Banke TG, Schousboe A, Makino Y, Johnson RC, Huganir R, Traynelis SF (2011) Mechanism of Ca(2)/calmodulin‐dependent kinase II regulation of AMPA receptor gating. Nat Neurosci 14: 727–735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ladepeche L, Planaguma J, Thakur S, Suarez I, Hara M, Borbely JS, Sandoval A, Laparra‐Cuervo L, Dalmau J, Lakadamyali M (2018) NMDA receptor autoantibodies in autoimmune encephalitis cause a subunit‐specific nanoscale redistribution of nmda receptors. Cell Rep 23: 3759–3768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HK, Barbarosie M, Kameyama K, Bear MF, Huganir RL (2000) Regulation of distinct AMPA receptor phosphorylation sites during bidirectional synaptic plasticity. Nature 405: 955–959 [DOI] [PubMed] [Google Scholar]
- Lee HK, Takamiya K, Han JS, Man H, Kim CH, Rumbaugh G, Yu S, Ding L, He C, Petralia RS, Wenthold RJ, Gallagher M, Huganir RL (2003) Phosphorylation of the AMPA receptor GluR1 subunit is required for synaptic plasticity and retention of spatial memory. Cell 112: 631–643 [DOI] [PubMed] [Google Scholar]
- Lee HK, Takamiya K, He K, Song L, Huganir RL (2010) Specific roles of AMPA receptor subunit GluR1 (GluA1) phosphorylation sites in regulating synaptic plasticity in the CA1 region of hippocampus. J Neurophysiol 103: 479–489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CH, Lu W, Michel JC, Goehring A, Du J, Song X, Gouaux E (2014) NMDA receptor structures reveal subunit arrangement and pore architecture. Nature 511: 191–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemke T, Welling A, Christel CJ, Blaich A, Bernhard D, Lenhardt P, Hofmann F, Moosmang S (2008) Unchanged beta‐adrenergic stimulation of cardiac L‐type calcium channels in Ca v 1.2 phosphorylation site S1928A mutant mice. J Biol Chem 283: 34738–34744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonard AS, Davare MA, Horne MC, Garner CC, Hell JW (1998) SAP97 is associated with the a‐amino‐3‐hydroxy‐5‐methylisoxazole‐4‐propionic acid receptor GluR1 subunit. J Biol Chem 273: 19518–19524 [DOI] [PubMed] [Google Scholar]
- Leonard AS, Lim IA, Hemsworth DE, Horne MC, Hell JW (1999) Calcium/calmodulin‐dependent protein kinase II is associated with the N‐methyl‐D‐aspartate receptor. Proc Natl Acad Sci USA 96: 3239–3244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy JM, Chen X, Reese TS, Nicoll RA (2015) Synaptic Consolidation Normalizes AMPAR Quantal Size following MAGUK Loss. Neuron 87: 534–548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Pink MD, Murphy JG, Stein A, Dell'Acqua ML, Hogan PG (2012) Balanced interactions of calcineurin with AKAP79 regulate Ca2+‐calcineurin‐NFAT signaling. Nat Struct Mol Biol 19: 337–345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim IA, Hall DD, Hell JW (2002) Selectivity and promiscuity of the first and second PDZ domains of PSD‐ 95 and synapse‐associated protein 102. J Biol Chem 277: 21697–21711 [DOI] [PubMed] [Google Scholar]
- Lin F, Wang H, Malbon CC (2000) Gravin‐mediated formation of signaling complexes in beta 2‐adrenergic receptor desensitization and resensitization. J Biol Chem 275: 19025–19034 [DOI] [PubMed] [Google Scholar]
- Lin Y, Skeberdis VA, Francesconi A, Bennett MV, Zukin RS (2004) Postsynaptic density protein‐95 regulates NMDA channel gating and surface expression. J Neurosci 24: 10138–10148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisman J, Raghavachari S (2006) A unified model of the presynaptic and postsynaptic changes during LTP at CA1 synapses. Sci STKE 2006: re11 [DOI] [PubMed] [Google Scholar]
- Lisman JE, Hell JW (2008) Long‐term Potentiation In Structural and Functional Organization of the Synapse, Hell JW, Ehlers MD. (eds), pp. 501–534. Heidelberg: Springer; [Google Scholar]
- Lissin DV, Carroll RC, Nicoll RA, Malenka RC, von Zastrow M (1999) Rapid, activation‐induced redistribution of ionotropic glutamate receptors in cultured hippocampal neurons. J Neurosci 19: 1263–1272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y, Allen M, Halt AR, Weisenhaus M, Dallapiazza RF, Hall DD, Usachev YM, McKnight GS, Hell JW (2007) Age‐dependent requirement of AKAP150‐anchored PKA and GluR2‐lacking AMPA receptors in LTP. EMBO J 26: 4879–4890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y, Zhang M, Lim IA, Hall DD, Allen ML, Medvedeva Y, McKnight GS, Usachev YM, Hell JW (2008) AKAP150‐anchored PKA activity is important for LTD during its induction phase. J Physiol 586: 4155–4164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu W, Shi Y, Jackson AC, Bjorgan K, During MJ, Sprengel R, Seeburg PH, Nicoll RA (2009) Subunit composition of synaptic AMPA receptors revealed by a single‐cell genetic approach. Neuron 62: 254–268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lur G, Higley MJ (2015) Glutamate receptor modulation is restricted to synaptic microdomains. Cell Rep 12: 326–334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacGillavry HD, Song Y, Raghavachari S, Blanpied TA (2013) Nanoscale scaffolding domains within the postsynaptic density concentrate synaptic AMPA receptors. Neuron 78: 615–622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makino H, Malinow R (2009) AMPA receptor incorporation into synapses during LTP: the role of lateral movement and exocytosis. Neuron 64: 381–390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malenka RC, Kauer JA, Perkel DJ, Mauk MD, Kelly PT, Nicoll RA, Waxham MN (1989) An essential role for postsynaptic calmodulin and protein kinase activity in long‐term potentiation. Nature 340: 554–557 [DOI] [PubMed] [Google Scholar]
- Malinow R, Schulman H, Tsien RW (1989) Inhibition of postsynaptic PKC or CaMKII blocks induction but not expression of LTP. Science 245: 862–866 [DOI] [PubMed] [Google Scholar]
- Man H‐Y, Sekine‐Aizawa Y, Huganir R (2007) Regulation of alpha‐amino‐3‐hydroxy‐5‐methyl‐4‐isoxazolepropionic acid receptor trafficking through PKA phosphorylation of the Glu receptor 1 subunit. Proc Natl Acad Sci USA 104: 3579–3584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsubara A, Laake JH, Davanger S, Usami S, Ottersen OP (1996) Organization of AMPA receptor subunits at a glutamate synapse: a quantitative immunogold analysis of hair cell synapses in the rat organ of Corti. J Neurosci 16: 4457–4467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuzaki M, Ellis‐Davies GC, Nemoto T, Miyashita Y, Iino M, Kasai H (2001) Dendritic spine geometry is critical for AMPA receptor expression in hippocampal CA1 pyramidal neurons. Nat Neurosci 4: 1086–1092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuzaki M, Honkura N, Ellis‐Davies GC, Kasai H (2004) Structural basis of long‐term potentiation in single dendritic spines. Nature 429: 761–766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matt L, Kim K, Hergarden AC, Patriarchi T, Malik ZA, Park DK, Chowdhury D, Buonarati OR, Henderson PB, Gokcek Sarac C, Zhang Y, Mohapatra D, Horne MC, Ames JB, Hell JW (2018a) alpha‐actinin anchors PSD‐95 at postsynaptic sites. Neuron 97: 1094–1109.e9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matt L, Kirk LM, Chenaux G, Speca DJ, Puhger KR, Pride MC, Qneibi M, Haham T, Plambeck KE, Stern‐Bach Y, Silverman JL, Crawley JN, Hell JW, Diaz E (2018b) SynDIG4/Prrt1 is required for excitatory synapse development and plasticity underlying cognitive function. Cell Rep 22: 2246–2253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merrill MA, Chen Y, Strack S, Hell JW (2005) Activity‐driven postsynaptic translocation of CaMKII. Trends Pharmacol Sci 26: 645–653 [DOI] [PubMed] [Google Scholar]
- Meyer D, Bonhoeffer T, Scheuss V (2014) Balance and stability of synaptic structures during synaptic plasticity. Neuron 82: 430–443 [DOI] [PubMed] [Google Scholar]
- Micheva KD, Busse B, Weiler NC, O'Rourke N, Smith SJ (2010) Single‐synapse analysis of a diverse synapse population: proteomic imaging methods and markers. Neuron 68: 639–653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minzenberg MJ, Watrous AJ, Yoon JH, Ursu S, Carter CS (2008) Modafinil shifts human locus coeruleus to low‐tonic, high‐phasic activity during functional MRI. Science 322: 1700–1702 [DOI] [PubMed] [Google Scholar]
- Morris RG (2013) NMDA receptors and memory encoding. Neuropharmacology 74: 32–40 [DOI] [PubMed] [Google Scholar]
- Murphy JA, Stein IS, Lau CG, Peixoto RT, Aman TK, Kaneko N, Aromolaran K, Saulnier JL, Popescu GK, Sabatini BL, Hell JW, Zukin RS (2014a) Phosphorylation of Ser1166 on GluN2B by PKA is critical to synaptic NMDA receptor function and Ca2+ signaling in spines. J Neurosci 34: 869–879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy JG, Sanderson JL, Gorski JA, Scott JD, Catterall WA, Sather WA, Dell'Acqua ML (2014b) AKAP‐anchored PKA maintains neuronal L‐type calcium channel activity and NFAT transcriptional signaling. Cell Rep 7: 1577–1588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nair D, Hosy E, Petersen JD, Constals A, Giannone G, Choquet D, Sibarita JB (2013) Super‐resolution imaging reveals that AMPA receptors inside synapses are dynamically organized in nanodomains regulated by PSD95. J Neurosci 33: 13204–13224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa T, Futai K, Lashuel HA, Lo I, Okamoto K, Walz T, Hayashi Y, Sheng M (2004) Quaternary structure, protein dynamics, and synaptic function of SAP97 controlled by L27 domain interactions. Neuron 44: 453–467 [DOI] [PubMed] [Google Scholar]
- Nakagawa T, Cheng Y, Ramm E, Sheng M, Walz T (2005) Structure and different conformational states of native AMPA receptor complexes. Nature 433: 545–549 [DOI] [PubMed] [Google Scholar]
- Nakagawa T, Cheng Y, Sheng M, Walz T (2006) Three‐dimensional structure of an AMPA receptor without associated stargazin/TARP proteins. Biol Chem 387: 179–187 [DOI] [PubMed] [Google Scholar]
- Nauert JB, Klauck TM, Langeberg LK, Scott JD (1996) Gravin, an autoantigen recognized by serum from myasthenia gravis patients, is a kinase scaffolding protein. Curr Biol 7: 52–62 [DOI] [PubMed] [Google Scholar]
- Nelson CD, Kim MJ, Hsin H, Chen Y, Sheng M (2013) Phosphorylation of threonine‐19 of PSD‐95 by GSK‐3beta is required for PSD‐95 mobilization and long‐term depression. J Neurosci 33: 12122–12135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newlon MG, Roy M, Morikis D, Carr DW, Westphal R, Scott JD, Jennings PA (2001) A novel mechanism of PKA anchoring revealed by solution structures of anchoring complexes. EMBO J 20: 1651–1662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikandrova YA, Jiao Y, Baucum AJ, Tavalin SJ, Colbran RJ (2010) Ca2+/calmodulin‐dependent protein kinase II binds to and phosphorylates a specific SAP97 splice variant to disrupt association with AKAP79/150 and modulate alpha‐amino‐3‐hydroxy‐5‐methyl‐4‐isoxazolepropionic acid‐type glutamate receptor (AMPAR) activity. J Biol Chem 285: 923–934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimchinsky EA, Yasuda R, Oertner TG, Svoboda K (2004) The number of glutamate receptors opened by synaptic stimulation in single hippocampal spines. J Neurosci 24: 2054–2064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nusser Z, Mulvihill E, Streit P, Somogyi P (1994) Subsynaptic segregation of metabotropic and ionotropic glutamate receptors as revealed by immunogold localization. Neuroscience 61: 421–427 [DOI] [PubMed] [Google Scholar]
- Nusser Z, Lujan R, Laube G, Roberts JD, Molnar E, Somogyi P (1998) Cell type and pathway dependence of synaptic AMPA receptor number and variability in the hippocampus. Neuron 21: 545–559 [DOI] [PubMed] [Google Scholar]
- Nystoriak MA, Nieves‐Cintron M, Patriarchi T, Buonarati OR, Prada MP, Morotti S, Grandi E, Fernandes JD, Forbush K, Hofmann F, Sasse KC, Scott JD, Ward SM, Hell JW, Navedo MF (2017) Ser1928 phosphorylation by PKA stimulates the L‐type Ca2+ channel CaV1.2 and vasoconstriction during acute hyperglycemia and diabetes. Sci Signal 10: eaaf9647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh MC, Derkach VA, Guire ES, Soderling TR (2006) Extrasynaptic membrane trafficking regulated by GluR1 Serine 845 phosphorylation primes AMPA receptors for long‐term potentiation. J Biol Chem 281: 752–758 [DOI] [PubMed] [Google Scholar]
- Okamoto K, Nagai T, Miyawaki A, Hayashi Y (2004) Rapid and persistent modulation of actin dynamics regulates postsynaptic reorganization underlying bidirectional plasticity. Nat Neurosci 7: 1104–1112 [DOI] [PubMed] [Google Scholar]
- Oliet SH, Malenka RC, Nicoll RA (1997) Two distinct forms of long‐term depression coexist in CA1 hippocampal pyramidal cells. Neuron 18: 969–982 [DOI] [PubMed] [Google Scholar]
- Oliveria SF, Dell'acqua ML, Sather WA (2007) AKAP79/150 anchoring of calcineurin controls neuronal L‐Type Ca(2+) channel activity and nuclear signaling. Neuron 55: 261–275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opazo P, Labrecque S, Tigaret CM, Frouin A, Wiseman PW, De Koninck P, Choquet D (2010) CaMKII triggers the diffusional trapping of surface AMPARs through phosphorylation of stargazin. Neuron 67: 239–252 [DOI] [PubMed] [Google Scholar]
- Opazo P, Choquet D (2011) A three‐step model for the synaptic recruitment of AMPA receptors. Mol Cell Neurosci 46: 1–8 [DOI] [PubMed] [Google Scholar]
- Otmakhov N, Tao‐Cheng JH, Carpenter S, Asrican B, Dosemeci A, Reese TS, Lisman J (2004) Persistent accumulation of calcium/calmodulin‐dependent protein kinase II in dendritic spines after induction of NMDA receptor‐dependent chemical long‐term potentiation. J Neurosci 24: 9324–9331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SY, Avraham HK, Avraham S (2004) RAFTK/Pyk2 activation is mediated by trans‐acting autophosphorylation in a Src‐independent manner. J Biol Chem 279: 33315–33322 [DOI] [PubMed] [Google Scholar]
- Park J, Chavez AE, Mineur YS, Morimoto‐Tomita M, Lutzu S, Kim KS, Picciotto MR, Castillo PE, Tomita S (2016) CaMKII phosphorylation of TARPgamma‐8 Is a mediator of LTP and learning and memory. Neuron 92: 75–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passafaro M, Piech V, Sheng M (2001) Subunit‐specific temporal and spatial patterns of AMPA receptor exocytosis in hippocampal neurons. Nat Neurosci 4: 917–926 [DOI] [PubMed] [Google Scholar]
- Patel N, Stengel F, Aebersold R, Gold MG (2017) Molecular basis of AKAP79 regulation by calmodulin. Nat Commun 8: 1681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patriarchi T, Qian H, Di Biase V, Malik ZA, Chowdhury D, Price JL, Hammes EA, Buonarati OR, Westenbroek RE, Catterall WA, Hofmann F, Xiang YK, Murphy GG, Chen C‐Y, Navedo MF, Hell JW (2016) Phosphorylation of Cav1.2 on S1928 uncouples the L‐type Ca2+ channel from the β2 adrenergic receptor. EMBO J 35: 1330–1345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen SW, Albertsen L, Moran GE, Levesque B, Pedersen SB, Bartels L, Wapenaar H, Ye F, Zhang M, Bowen ME, Stromgaard K (2017) Site‐specific phosphorylation of PSD‐95 PDZ domains reveals fine‐tuned regulation of protein‐protein interactions. ACS Chem Biol 12: 2313–2323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penn AC, Zhang CL, Georges F, Royer L, Breillat C, Hosy E, Petersen JD, Humeau Y, Choquet D (2017) Hippocampal LTP and contextual learning require surface diffusion of AMPA receptors. Nature 549: 384–388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penzes P, Cahill ME, Jones KA, Srivastava DP (2008) Convergent CaMK and RacGEF signals control dendritic structure and function. Trends Cell Biol 18: 405–413 [DOI] [PubMed] [Google Scholar]
- Petralia RS, Zhao HM, Wang YX, Wenthold RJ (1998) Variations in the tangential distribution of postsynaptic glutamate receptors in Purkinje cell parallel and climbing fiber synapses during development. Neuropharmacology 37: 1321–1334 [DOI] [PubMed] [Google Scholar]
- Petrini EM, Lu J, Cognet L, Lounis B, Ehlers MD, Choquet D (2009) Endocytic trafficking and recycling maintain a pool of mobile surface AMPA receptors required for synaptic potentiation. Neuron 63: 92–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plant K, Pelkey KA, Bortolotto ZA, Morita D, Terashima A, McBain CJ, Collingridge GL, Isaac JT (2006) Transient incorporation of native GluR2‐lacking AMPA receptors during hippocampal long‐term potentiation. Nat Neurosci 9: 602–604 [DOI] [PubMed] [Google Scholar]
- Prybylowski K, Chang K, Sans N, Kan L, Vicini S, Wenthold RJ (2005) The Synaptic Localization of NR2B‐Containing NMDA Receptors Is Controlled by Interactions with PDZ Proteins and AP‐2. Neuron 47: 845–857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian H, Matt L, Zhang M, Nguyen M, Patriarchi T, Koval ON, Anderson ME, He K, Lee H‐K, Hell JW (2012) β2 adrenergic receptor supports prolonged theta tetanus – induced LTP. J Neurophysiol 107: 2703–2712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian H, Patriarchi T, Price JL, Matt L, Lee B, Nieves‐Cintron M, Buonarati OR, Chowdhury D, Nanou E, Nystoriak MA, Catterall WA, Poomvanicha M, Hofmann F, Navedo MF, Hell JW (2017) Phosphorylation of Ser1928 mediates the enhanced activity of the L‐type Ca2+ channel Cav1.2 by the beta2‐adrenergic receptor in neurons. Sci Signal 10: eaaf9659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Racca C, Stephenson FA, Streit P, Roberts JD, Somogyi P (2000) NMDA receptor content of synapses in stratum radiatum of the hippocampal CA1 area. J Neurosci 20: 2512–2522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson HR, Gibson ES, Benke TA, Dell'Acqua ML (2009) Regulation of postsynaptic structure and function by an A‐kinase anchoring protein‐membrane‐associated guanylate kinase scaffolding complex. J Neurosci 29: 7929–7943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roche KW, O'Brien RJ, Mammen AL, Bernhardt J, Huganir RL (1996) Characterization of multiple phosphorylation sites on the AMPA receptor GluR1 subunit. Neuron 16: 1179–1188 [DOI] [PubMed] [Google Scholar]
- Roche KW, Standley S, McCallum J, Dune Ly C, Ehlers MD, Wenthold RJ (2001) Molecular determinants of NMDA receptor internalization. Nat Neurosci 4: 794–802 [DOI] [PubMed] [Google Scholar]
- Rose J, Jin SX, Craig AM (2009) Heterosynaptic molecular dynamics: locally induced propagating synaptic accumulation of CaM kinase II. Neuron 61: 351–358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenmund C, Carr DW, Bergeson SE, Nilaver G, Scott JD, Westbrook GL (1994) Anchoring of protein kinase A is required for modulation of AMPA/kainate receptors on hippocampal neurons. Nature 368: 853–856 [DOI] [PubMed] [Google Scholar]
- Sadybekov A, Tian C, Arnesano C, Katritch V, Herring BE (2017) An autism spectrum disorder‐related de novo mutation hotspot discovered in the GEF1 domain of Trio. Nat Commun 8: 601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sainlos M, Tigaret C, Poujol C, Olivier NB, Bard L, Breillat C, Thiolon K, Choquet D, Imperiali B (2011) Biomimetic divalent ligands for the acute disruption of synaptic AMPAR stabilization. Nat Chem Biol 7: 81–91 [DOI] [PubMed] [Google Scholar]
- Sala C, Piech V, Wilson NR, Passafaro M, Liu G, Sheng M (2001) Regulation of dendritic spine morphology and synaptic function by Shank and Homer. Neuron 31: 115–130 [DOI] [PubMed] [Google Scholar]
- Sanderson JL, Dell'Acqua ML (2011) AKAP signaling complexes in regulation of excitatory synaptic plasticity. Neuroscientist 17: 321–336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanderson JL, Gorski JA, Gibson ES, Lam P, Freund RK, Chick WS, Dell'Acqua ML (2012) AKAP150‐anchored calcineurin regulates synaptic plasticity by limiting synaptic incorporation of Ca2+‐permeable AMPA receptors. J Neurosci 32: 15036–15052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanderson JL, Gorski JA, Dell'Acqua ML (2016) NMDA receptor‐dependent LTD requires transient synaptic incorporation of Ca(2+)‐Permeable AMPARs mediated by AKAP150‐Anchored PKA and calcineurin. Neuron 89: 1000–1015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanderson JL, Scott JD, Dell'Acqua ML (2018) Control of Homeostatic Synaptic Plasticity by AKAP‐Anchored Kinase and Phosphatase Regulation of Ca(2+)‐Permeable AMPA Receptors. J Neurosci 38: 2863–2876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheiffele P, Fan J, Choih J, Fetter R, Serafini T (2000) Neuroligin expressed in nonneuronal cells triggers presynaptic development in contacting axons. Cell 101: 657–669 [DOI] [PubMed] [Google Scholar]
- Schikorski T, Stevens CF (1997) Quantitative ultrastructural analysis of hippocampal excitatory synapses. J Neurosci 17: 5858–5867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schluter OM, Xu W, Malenka RC (2006) Alternative N‐terminal domains of PSD‐95 and SAP97 govern activity‐dependent regulation of synaptic AMPA receptor function. Neuron 51: 99–111 [DOI] [PubMed] [Google Scholar]
- Schnell E, Sizemore M, Karimzadegan S, Chen L, Bredt DS, Nicoll RA (2002) Direct interactions between PSD‐95 and stargazin control synaptic AMPA receptor number. Proc Natl Acad Sci USA 99: 13902–13907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwenk J, Harmel N, Zolles G, Bildl W, Kulik A, Heimrich B, Chisaka O, Jonas P, Schulte U, Fakler B, Klocker N (2009) Functional proteomics identify cornichon proteins as auxiliary subunits of AMPA receptors. Science 323: 1313–1319 [DOI] [PubMed] [Google Scholar]
- Sculptoreanu A, Rotman E, Takahashi M, Scheuer T, Catterall WA (1993) Voltage‐dependent potentiation of the activity of cardiac L‐type calcium channel alpha 1 subunits due to phosphorylation by cAMP‐dependent protein kinase. Proc Natl Acad Sci USA 90: 10135–10139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaffer J, Adams JA (1999) Detection of conformational changes along the kinetic pathway of protein kinase A using a catalytic trapping technique. Biochemistry 38: 12072–12079 [DOI] [PubMed] [Google Scholar]
- Shen K, Meyer T (1999) Dynamic control of CaMKII Translocation in hippocampal neurons by NMDA receptor stimulation. Science 284: 162–166 [DOI] [PubMed] [Google Scholar]
- Shen A, Nieves‐Cintron M, Deng Y, Shi Q, Chowdhury D, Qi J, Hell JW, Navedo MF, Xiang YK (2018) Functionally distinct and selectively phosphorylated GPCR subpopulations co‐exist in a single cell. Nat Commun 9: 1050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng M, Hoogenraad CC (2007) The postsynaptic architecture of excitatory synapses: a more quantitative view. Annu Rev Biochem 76: 823–847 [DOI] [PubMed] [Google Scholar]
- Sheng N, Bemben MA, Diaz‐Alonso J, Tao W, Shi YS, Nicoll RA (2018) LTP requires postsynaptic PDZ‐domain interactions with glutamate receptor/auxiliary protein complexes. Proc Natl Acad Sci USA 115: 3948–3954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shenoy SK, Lefkowitz RJ (2011) beta‐Arrestin‐mediated receptor trafficking and signal transduction. Trends Pharmacol Sci 32: 521–533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepherd GM, Harris KM (1998) Three‐dimensional structure and composition of CA3–>CA1 axons in rat hippocampal slices: implications for presynaptic connectivity and compartmentalization. J Neurosci 18: 8300–8310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih M, Lin F, Scott JD, Wang HY, Malbon CC (1999) Dynamic complexes of beta2‐adrenergic receptors with protein kinases and phosphatases and the role of gravin. J Biol Chem 274: 1588–1595 [DOI] [PubMed] [Google Scholar]
- Shinohara Y, Hirase H, Watanabe M, Itakura M, Takahashi M, Shigemoto R (2008) Left‐right asymmetry of the hippocampal synapses with differential subunit allocation of glutamate receptors. Proc Natl Acad Sci USA 105: 19498–19503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shipman SL, Schnell E, Hirai T, Chen BS, Roche KW, Nicoll RA (2011) Functional dependence of neuroligin on a new non‐PDZ intracellular domain. Nat Neurosci 14: 718–726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinnen BL, Bowen AB, Forte JS, Hiester BG, Crosby KC, Gibson ES, Dell'Acqua ML, Kennedy MJ (2017) Optogenetic control of synaptic composition and function. Neuron 93:646–660.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skeberdis VA, Chevaleyre V, Lau CG, Goldberg JH, Pettit DL, Suadicani SO, Lin Y, Bennett MV, Yuste R, Castillo PE, Zukin RS (2006) Protein kinase A regulates calcium permeability of NMDA receptors. Nat Neurosci 9: 501–510 [DOI] [PubMed] [Google Scholar]
- Smith MA, Ellis‐Davies GC, Magee JC (2003) Mechanism of the distance‐dependent scaling of Schaffer collateral synapses in rat CA1 pyramidal neurons. J Physiol 548: 245–258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith KE, Gibson ES, Dell'Acqua ML (2006) cAMP‐dependent protein kinase postsynaptic localization regulated by NMDA receptor activation through translocation of an A‐kinase anchoring protein scaffold protein. J Neurosci 26: 2391–2402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith FD, Esseltine JL, Nygren PJ, Veesler D, Byrne DP, Vonderach M, Strashnov I, Eyers CE, Eyers PA, Langeberg LK, Scott JD (2017) Local protein kinase A action proceeds through intact holoenzymes. Science 356: 1288–1293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soares C, Lee KF, Nassrallah W, Beique JC (2013) Differential subcellular targeting of glutamate receptor subtypes during homeostatic synaptic plasticity. J Neurosci 33: 13547–13559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobolevsky AI, Rosconi MP, Gouaux E (2009) X‐ray structure, symmetry and mechanism of an AMPA‐subtype glutamate receptor. Nature 462: 745–756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somogyi P, Tamas G, Lujan R, Buhl EH (1998) Salient features of synaptic organisation in the cerebral cortex. Brain Res Brain Res Rev 26: 113–135 [DOI] [PubMed] [Google Scholar]
- Spruston N, Jonas P, Sakmann B (1995) Dendritic glutamate receptor channels in rat hippocampal CA3 and CA1 pyramidal neurons. J Physiol 482(Pt 2): 325–352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staus DP, Wingler LM, Choi M, Pani B, Manglik A, Kruse AC, Lefkowitz RJ (2018) Sortase ligation enables homogeneous GPCR phosphorylation to reveal diversity in beta‐arrestin coupling. Proc Natl Acad Sci USA 115: 3834–3839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinberg SF, Brunton LL (2001) Compartmentation of G protein‐coupled signaling pathways in cardiac myocytes. Annu Rev Pharmacol Toxicol 41: 751–773 [DOI] [PubMed] [Google Scholar]
- Steiner P, Higley MJ, Xu W, Czervionke BL, Malenka RC, Sabatini BL (2008) Destabilization of the postsynaptic density by PSD‐95 serine 73 phosphorylation inhibits spine growth and synaptic plasticity. Neuron 60: 788–802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strack S, Colbran RJ (1998) Autophosphorylation‐dependent targeting of calcium/calmodulin‐dependent protein kinase II by the NR2B subunit of the N‐methyl‐D‐aspartate receptor. J Biol Chem 273: 20689–20692 [DOI] [PubMed] [Google Scholar]
- Strack S, Hell JW (2008) Postsynaptic targeting of kinases and phosphatases In Structural and Functional Organization of the Synapse, Hell JW, Ehlers MD. (eds), pp. 459–500. Heidelberg: Springer; [Google Scholar]
- Sudhof TC, Malenka RC (2008) Understanding synapses: past, present, and future. Neuron 60: 469–476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sumioka A, Yan D, Tomita S (2010) TARP phosphorylation regulates synaptic AMPA receptors through lipid bilayers. Neuron 66: 755–767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sumioka A, Brown TE, Kato AS, Bredt DS, Kauer JA, Tomita S (2011) PDZ binding of TARPgamma‐8 controls synaptic transmission but not synaptic plasticity. Nat Neurosci 14: 1410–1412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X, Zhao Y, Wolf ME (2005) Dopamine receptor stimulation modulates AMPA receptor synaptic insertion in prefrontal cortex neurons. J Neurosci 25: 7342–7351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutton MA, Ito HT, Cressy P, Kempf C, Woo JC, Schuman EM (2006) Miniature neurotransmission stabilizes synaptic function via tonic suppression of local dendritic protein synthesis. Cell 125: 785–799 [DOI] [PubMed] [Google Scholar]
- Takumi Y, Ramirez‐Leon V, Laake P, Rinvik E, Ottersen OP (1999) Different modes of expression of AMPA and NMDA receptors in hippocampal synapses. Nat Neurosci 2: 618–624 [DOI] [PubMed] [Google Scholar]
- Tan HL, Queenan BN, Huganir RL (2015) GRIP1 is required for homeostatic regulation of AMPAR trafficking. Proc Natl Acad Sci USA 112: 10026–10031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka J, Matsuzaki M, Tarusawa E, Momiyama A, Molnar E, Kasai H, Shigemoto R (2005) Number and density of AMPA receptors in single synapses in immature cerebellum. J Neurosci 25: 799–807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang AH, Chen H, Li TP, Metzbower SR, MacGillavry HD, Blanpied TA (2016) A trans‐synaptic nanocolumn aligns neurotransmitter release to receptors. Nature 536: 210–214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao J, Wang HY, Malbon CC (2003) Protein kinase A regulates AKAP250 (gravin) scaffold binding to the beta2‐adrenergic receptor. EMBO J 22: 6419–6429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tardin C, Cognet L, Bats C, Lounis B, Choquet D (2003) Direct imaging of lateral movements of AMPA receptors inside synapses. EMBO J 22: 4656–4665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavalin SJ, Colledge M, Hell JW, Langeberg LK, Huganir RL, Scott JD (2002) Regulation of GluR1 by the A‐kinase anchoring protein 79 (AKAP79) signaling complex shares properties with long‐term depression. J Neurosci 22: 3044–3051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavalin SJ (2008) AKAP79 selectively enhances protein kinase C regulation of GluR1 at a Ca2+‐calmodulin‐dependent protein kinase II/Protein Kinase C Site. J Biol Chem 283: 11445–11452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor SS, Ilouz R, Zhang P, Kornev AP (2012) Assembly of allosteric macromolecular switches: lessons from PKA. Nat Rev Mol Cell Biol 13: 646–658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiagarajan TC, Lindskog M, Tsien RW (2005) Adaptation to synaptic inactivity in hippocampal neurons. Neuron 47: 725–737 [DOI] [PubMed] [Google Scholar]
- Thomas MJ, Moody TD, Makhinson M, O'Dell TJ (1996) Activity‐dependent beta‐adrenergic modulation of low frequency stimulation induced LTP in the hippocampal CA1 region. Neuron 17: 475–482 [DOI] [PubMed] [Google Scholar]
- Tomita S, Stein V, Stocker TJ, Nicoll RA, Bredt DS (2005) Bidirectional synaptic plasticity regulated by phosphorylation of stargazin‐like TARPs. Neuron 45: 269–277 [DOI] [PubMed] [Google Scholar]
- Tovar KR, Westbrook GL (2002) Mobile NMDA receptors at hippocampal synapses. Neuron 34: 255–264 [DOI] [PubMed] [Google Scholar]
- Traynelis SF, Wollmuth LP, McBain CJ, Menniti FS, Vance KM, Ogden KK, Hansen KB, Yuan H, Myers SJ, Dingledine R (2010) Glutamate receptor ion channels: structure, regulation, and function. Pharmacol Rev 62: 405–496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsvetanova NG, von Zastrow M (2014) Spatial encoding of cyclic AMP signaling specificity by GPCR endocytosis. Nat Chem Biol 10: 1061–1065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tunquist BJ, Hoshi N, Guire ES, Zhang F, Mullendorff K, Langeberg LK, Raber J, Scott JD (2008) Loss of AKAP150 perturbs distinct neuronal processes in mice. Proc Natl Acad Sci USA 105: 12557–12562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valtschanoff JG, Burette A, Davare MA, Leonard AS, Hell JW, Weinberg RJ (2000) SAP97 concentrates at the postsynaptic density in cerebral cortex. Eur J Neurosci 12: 3605–3614 [DOI] [PubMed] [Google Scholar]
- Valtschanoff JG, Weinberg RJ (2001) Laminar organization of the NMDA receptor complex within the postsynaptic density. J Neurosci 21: 1211–1217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker‐Gray R, Stengel F, Gold MG (2017) Mechanisms for restraining cAMP‐dependent protein kinase revealed by subunit quantitation and cross‐linking approaches. Proc Natl Acad Sci USA 114: 10414–10419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walkup WG, Mastro TL, Schenker LT, Vielmetter J, Hu R, Iancu A, Reghunathan M, Bannon BD, Kennedy MB (2016) A model for regulation by SynGAP‐alpha1 of binding of synaptic proteins to PDZ‐domain ‘Slots’ in the postsynaptic density. eLife 5: e16813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang XB, Yang Y, Zhou Q (2007) Independent expression of synaptic and morphological plasticity associated with long‐term depression. J Neurosci 27: 12419–12429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D, Govindaiah G, Liu R, De Arcangelis V, Cox CL, Xiang YK (2010) Binding of amyloid beta peptide to beta2 adrenergic receptor induces PKA‐dependent AMPA receptor hyperactivity. FASEB J 24: 3511–3521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson JF, Ho H, Greger IH (2017) Synaptic transmission and plasticity require AMPA receptor anchoring via its N‐terminal domain. eLife 6: e23024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisenhaus M, Allen ML, Yang L, Lu Y, Nichols CB, Su T, Hell JW, McKnight GS (2010) Mutations in AKAP5 disrupt dendritic signaling complexes and lead to electrophysiological and behavioral phenotypes in mice. PLoS ONE 5: e10325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenthold RJ, Petralia RS, Blahos J II, Niedzielski AS (1996) Evidence for multiple AMPA receptor complexes in hippocampal CA1/CA2 neurons. J Neurosci 16: 1982–1989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf ME, Tseng KY (2012) Calcium‐permeable AMPA receptors in the VTA and nucleus accumbens after cocaine exposure: when, how, and why? Front Mol Neurosci 5: 72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolfrey KM, Sanderson JL, Dell'Acqua ML (2015) The palmitoyl acyltransferase DHHC2 regulates recycling endosome exocytosis and synaptic potentiation through palmitoylation of AKAP79/150. J Neurosci 35: 442–456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolfrey KM, O'Leary H, Goodell DJ, Robertson HR, Horne EA, Coultrap SJ, Dell'Acqua ML, Bayer KU (2018) CaMKII regulates the depalmitoylation and synaptic removal of the scaffold protein AKAP79/150 to mediate structural long‐term depression. J Biol Chem 293: 1551–1567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu D, Bacaj T, Morishita W, Goswami D, Arendt KL, Xu W, Chen L, Malenka RC, Sudhof TC (2017) Postsynaptic synaptotagmins mediate AMPA receptor exocytosis during LTP. Nature 544: 316–321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Z, Srivastava DP, Photowala H, Kai L, Cahill ME, Woolfrey KM, Shum CY, Surmeier DJ, Penzes P (2007) Kalirin‐7 controls activity‐dependent structural and functional plasticity of dendritic spines. Neuron 56: 640–656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, Schluter OM, Steiner P, Czervionke BL, Sabatini B, Malenka RC (2008) Molecular dissociation of the role of PSD‐95 in regulating synaptic strength and LTD. Neuron 57: 248–262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, Ginsburg KS, Hall DD, Zimmermann M, Stein IS, Zhang M, Tandan S, Hill JA, Horne MC, Bers D, Hell JW (2010) Targeting of protein phosphatases PP2A and PP2B to the C‐terminus of the L‐type calcium channel Ca v1.2. Biochemistry 49: 10298–10307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Kurup P, Bartos JA, Patriarchi T, Hell JW, Lombroso PJ (2012) Striatal‐enriched protein‐tyrosine phosphatase (STEP) regulates Pyk2 kinase activity. J Biol Chem 287: 20942–20956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S, Fletcher WH, Johnson DA (1995) Regulation of cAMP‐dependent protein kinase: Enzyme activation without dissociation. Biochemistry 34: 6267–6271 [DOI] [PubMed] [Google Scholar]
- Yang Y, Wang XB, Frerking M, Zhou Q (2008a) Delivery of AMPA receptors to perisynaptic sites precedes the full expression of long‐term potentiation. Proc Natl Acad Sci USA 105: 11388–11393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Wang XB, Frerking M, Zhou Q (2008b) Spine expansion and stabilization associated with long‐term potentiation. J Neurosci 28: 5740–5751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Wang XB, Zhou Q (2010) Perisynaptic GluR2‐lacking AMPA receptors control the reversibility of synaptic and spines modifications. Proc Natl Acad Sci USA 107: 11999–12004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S, Roselli F, Patchev AV, Yu S, Almeida OF (2013) Non‐receptor‐tyrosine kinases integrate fast glucocorticoid signaling in hippocampal neurons. J Biol Chem 288: 23725–23739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang YP, Holbro N, Oertner TG (2008) Optical induction of plasticity at single synapses reveals input‐specific accumulation of alphaCaMKII. Proc Natl Acad Sci USA 105: 12039–12044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, Patriarchi T, Stein IS, Qian H, Matt L, Nguyen M, Xiang YK, Hell JW (2013) Adenylyl cyclase anchoring by a kinase anchor protein AKAP5 (AKAP79/150) is important for postsynaptic beta‐adrenergic signaling. J Biol Chem 288: 17918–17931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Matt L, Patriarchi T, Malik ZA, Chowdhury D, Park DK, Renieri A, Ames JB, Hell JW (2014) Capping of the N‐terminus of PSD‐95 by calmodulin triggers its postsynaptic release. EMBO J 33: 1341–1353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong H, Sia GM, Sato TR, Gray NW, Mao T, Khuchua Z, Huganir RL, Svoboda K (2009) Subcellular dynamics of type II PKA in neurons. Neuron 62: 363–374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Adams JA (1997) Participation of ADP dissociation in the rate‐determining step in cAMP‐dependent protein kinase. Biochemistry 36: 15733–15738 [DOI] [PubMed] [Google Scholar]
- Zhou Q, Homma KJ, Poo MM (2004) Shrinkage of dendritic spines associated with long‐term depression of hippocampal synapses. Neuron 44: 749–757 [DOI] [PubMed] [Google Scholar]
- Zhu J, Qin Y, Zhao M, Van Aelst L, Malinow R (2002) Ras and Rap Control AMPA Receptor Trafficking during Synaptic Plasticity. Cell 110: 443 [DOI] [PubMed] [Google Scholar]
- Zhu J, Shang Y, Zhang M (2016) Mechanistic basis of MAGUK‐organized complexes in synaptic development and signalling. Nat Rev 17: 209–223 [DOI] [PubMed] [Google Scholar]