

Abstract
Recruitment of naturally occurring suppressive CD4+, CD25+, and FOXP3+ regulatory T cells (Treg) to the tumor microenvironment (TME) has the potential to weaken the antitumor response in patients receiving treatment with immuno-oncology (IO) agents. Human Treg express CCR4 and can be recruited to the TME through the C–C chemokines CCL17 and CCL22. We have recently developed a series of potent, orally bioavailable small molecule antagonists of CCR4 that can block recruitment of Treg into the TME.
Keywords: CCR4, CCR4 antagonist, Treg migration
Several pathways exist by which cancer can evade or negatively affect the immune response to the tumor. The discovery of monoclonal antibodies and small molecules that regulate the immune system’s response to cancer has reshaped the way scientists design cancer therapies, leading to a plethora of research in the field of immuno-oncology.1,2 With the FDA approval of checkpoint inhibitors such as ipilimumab or nivolumab and pembrolizumab, which target CTLA-4 or PD-1 respectively, researchers and patients have been given new hope in the battle against cancer.
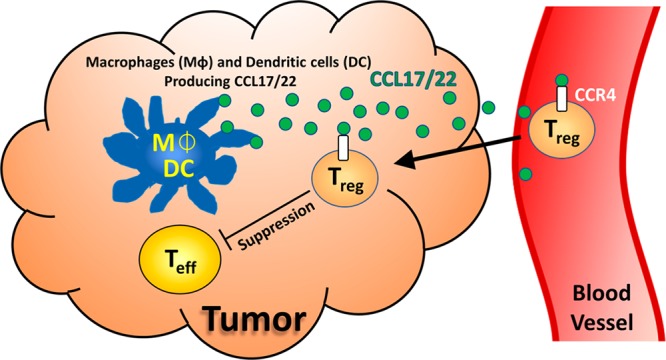
CCR4 is a G protein-coupled receptor (GPCR) that is highly expressed on the most immunosuppressive CD4+, CD25+, and FOXP3+ regulatory T cells (Treg).3 CCR4 is known to play a dominant role in the recruitment of Treg into the tumor microenvironment (TME) via the chemokine ligands CCL17 and CCL22 (Figure 1).4 While Treg can prevent autoimmunity, their recruitment and eventual accumulation in the TME can also cause the functional suppression of CD8+ effector T cells (Teff, Figure 1), leading to a poor patient prognosis.5 Though earlier CCR4 antagonists were developed to suppress Th2 migration for inflammatory disorders,6 there have been few reports investigating their use to affect Treg migration into the tumor microevironment.2
Figure 1.
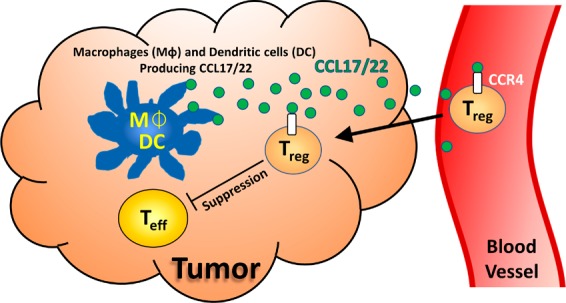
Treg-suppressed tumor microenvironment.
Depending on their binding mode, allosteric CCR4 antagonists can be placed into two distinct classes: Class I allosteric antagonists, which are believed to bind to an extracellular portion of the receptor, and Class II, which bind to an intracellular pocket.7 In general, Class I antagonists comprise a lipophilic arene and a side chain containing a basic amine, which are both linked to a heteroaromatic core in a 1,3-substitution pattern (Figure 2). Class II antagonists contain sulfonamides that are flanked by both a lipophilic arene and a substituted heteroaromatic ring.
Figure 2.

Representative CCR4 antagonists.
Until late 2017, the most advanced CCR4 antagonist to be involved in clinical trials was a Class II antagonist from GlaxoSmithKline, GSK2239633.8 Between 2010 and 2011, GSK2239633 entered healthy volunteer studies with asthma as a possible therapeutic indication. Although no dose limiting toxicity was identified, clinical trials for this compound were discontinued based on its low exposure and target engagement in the blood. Additionally, AstraZeneca has recently disclosed two Class II antagonists as preclinical candidates, AZD-1678 and AZD-2098.9 In 2013, AZD-2098 was licensed to Cancer Research UK for the treatment of kidney cancer; however, no further development has been reported to date.2
In addition to small molecule antagonists, a cell depleting monoclonal antibody recognizing CCR4, mogamulizumab (KY-0761, Kyowa Hakko Kirin Co., Ltd.), has been in several clinical trials.10 In 2014, mogamulizumab received approval for the treatment of hematological malignancies and Cutaneous T-cell Lymphoma (CTCL) in Japan. Data from their recent phase III multicenter study known as MAVORIC showed a statistically significant increase in progression-free survival and overall response rate for CTCL patients when compared to vorinostat, an FDA approved treatment for CTCL. Based on their results, the FDA has approved the use of mogamulizumab for the treatment of adult patients who have received at least one prior systemic therapy for two subtypes of CTCL, mycosis fungoides (MF) or Sézary syndrome (SS). Significantly, mogamulizumab is the first drug approved by the FDA to specifically treat SS and the first approved biologic to target CCR4.
A depleting antibody that causes ADCC (antibody-dependent cell-mediated cytotoxicity) has a vastly different mechanism of action when compared to a small molecule antagonist and may not be ideal for IO. One possible advantage of targeting CCR4 with a small molecule antagonist when compared to a depleting antibody is its ability to block Treg migration into the tumor without depletion of cells from normal tissues or depletion of beneficial immune cells.
Our efforts to design an orally bioavailable small molecule IO therapy have led us to the discovery of FLX475, a potent and selective CCR4 antagonist that blocks Treg migration to the TME in several tumor models.11 Phase I trials for FLX475 are currently in progress. Harnessing data from The Cancer Genome Atlas (TCGA), we have found that several human tumor types present high levels of CCL17 and CCL22 gene expression and show an increased infiltration of Treg.12 These data support the potential for a targeted approach in selecting patient populations.
The field of immuno-oncology has presented the scientific community with new strategies for developing cancer therapies. With several known pathways to target and many more yet to be discovered, our clinical studies could prove pivotal in determining if using small molecule antagonists to inhibit CCR4 will increase patient survival.
Acknowledgments
We would like to acknowledge the contributions from all current and former FLX Bio, Inc. employees. Their hard work has allowed our research to progress to the stage it is at today.
Glossary
ABBREVIATIONS
- CCR4
CC chemokine receptor 4
- CCL17/22
CC chemokine ligand 17/22
- FOXP3
Forkhead box protein 3
- CD4/25
Cluster of differentiation 4/25
Views expressed in this editorial are those of the authors and not necessarily the views of the ACS.
The authors declare the following competing financial interest(s): The authors of this manuscript are employees of FLX Bio, Inc.
References
- Sharpe A. H. Introduction to checkpoint inhibitors and cancer immunotherapy. Immunol. Rev. 2017, 276, 5. 10.1111/imr.12531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toogood P. Small molecule immuno-oncology therapeutic agents. Bioorg. Med. Chem. Lett. 2017, 28, 319. 10.1016/j.bmcl.2017.12.044. [DOI] [PubMed] [Google Scholar]
- Sugiyama D.; Nishikawa H.; Maeda Y.; Nishioka M.; Tanemura A.; Katayama I.; Ezoe S.; Kanakura Y.; Sato E.; Fukumori Y.; Karbach J.; Jäger E.; Sakaguchi S. Anti-CCR4 mAb selectively depletes effector-type FoxP3+CD4+ regulatory T cells, evoking antitumor immune responses in humans. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 17945. 10.1073/pnas.1316796110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishida T.; Ueda R. CCR4 as a novel molecular target for immunotherapy of cancer. Cancer Sci. 2006, 97, 1139. 10.1111/j.1349-7006.2006.00307.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishikawa H.; Sakaguchi S. Regulatory T cells in tumor immunity. Int. J. Cancer 2010, 127, 759. 10.1002/ijc.25429. [DOI] [PubMed] [Google Scholar]
- Solari R.; Pease J. E. Targeting chemokine receptors in disease – a case study of CCR4. Eur. J. Pharmacol. 2015, 763, 169. 10.1016/j.ejphar.2015.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ajram L.; Begg M.; Slack R.; Cryan J.; Hall D.; Hodgson S.; Ford A.; Barnes A.; Swieboda D.; Mousnier A.; Solari R. Internalization of the chemokine receptor CCR4 can be evoked by orthosteric and allosteric receptor antagonists. Eur. J. Pharmacol. 2014, 729, 75. 10.1016/j.ejphar.2014.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahn A.; Hodgson S.; Wilson R.; Robertson J.; Watson J.; Beerahee M.; Hughes S. C.; Young G.; Graves R.; Hall D.; van Marle S.; Solari R. Safety, tolerability, pharmacokinetics and pharmacodynamics of GSK2239633, a CC-chemokine receptor 4 antagonist, in healthy male subjects: results from an open-label and from a randomized study. BMC Pharmacol. Toxicol. 2013, 14, 14. 10.1186/2050-6511-14-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kindon N.; Andrews G.; Baxter A.; Cheshire D.; Hemsley P.; Johnson T.; Liu Y. Z.; McGinnity D.; McHale M.; Mete A.; Reuberson J.; Roberts B.; Steele J.; Teobald B.; Unitt J.; Vaughan D.; Walters I.; Stocks M. Discovery of AZD-2098 and AZD-1678, Two Potent and Bioavailable CCR4 Receptor Antagonists. ACS Med. Chem. Lett. 2017, 8, 981. 10.1021/acsmedchemlett.7b00315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FDA approves treatment for two rare types of non-Hodgkin lymphoma. https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm616176.htm (accessed August 9th, 2018). [Google Scholar]
- Talay O.; Marshall L.; Meleza C.; Reilly M. K.; Robles O.; Zibinsky M.; Okal A.; Seitz L.; McKinnell J.; Jacobson S.; Riegler E.; Karbarz E.; Chian D.; Wadsworth A.; Kassner P.; Wustrow D.; Fridman J. S.. Potent and selective C-C chemokine receptor (CCR4) antagonists potentiate anti-tumor immune responses by inhibiting regulatory T cells (Treg). AACR 2017 Proceedings Abstracts, 107th Annual Meeting of the American Association for Cancer Research, Washington, D. C., April 1–5, 2017. [Google Scholar]
- Talay O.; Jorapur A.; Jacobson S.; Marubayashi S.; Marshall L.; Suthram S.; Robles O.; Ketcham J. M.; Reilly M. K.; Younai A.; Biannic B.; Hu D.; Bui M.; Schwarz J.; Kassner P.; Cutler G.. EBV associated tumors have increased regulatory T cell recruitment and are therefore a potential indication for treatment with potent and selective small molecule CCR4 antagonists. AACR 2018 Proceedings Abstracts, 109th Annual Meeting of the American Association for Cancer Research, Chicago, IL, April 14–18, 2018. [Google Scholar]