

Abstract
Famotidine, an antiulcer drug incorporating a sulfamide motif, was investigated as carbonic anhydrase inhibitor (CAI). It acts as a nanomolar inhibitor of several human (hCA II, VI, VII and XII) and Helicobacter pylori CAs. The high resolution X-ray structures of famotidine bound to hCA I and II revealed interesting aspects related to its CA inhibition mechanism, offering the possibility to develop antibacterials with a novel mechanism of action.
Keywords: Carbonic anhydrase, famotidine, Helicobacter pylori, inhibitor, metalloenzymes
The third generation histamine H2 receptor antagonists such as cimetidine, ranitidine, and famotidine (Figure 1) are widely used for the treatment of duodenal and gastric ulcers, gastroesophageal reflux disease, and pathological hypersecretory conditions such as Zollinger–Ellison syndrome.1−3 Famotidine incorporates a sulfamide moiety, a bioisostere of the sulfonamide group, a well-known zinc binding group present in carbonic anhydrase (CA, EC 4.2.1.1) inhibitors (CAIs).4 However, the effects of famotidine on these zinc metallo-enzymes was scarcely investigated until now.5 CAs are ubiquitous metallo-enzymes that catalyze the reversible hydration of carbon dioxide to bicarbonate and protons.6,7 In humans, 15 different α-isoforms are present, with a very different subcellular localization and tissue distribution.8 There are several cytosolic isoforms (hCA I–III, VII, XIII), four membrane-bound isozymes (CA IV, IX, XII, and XIV), two mitochondrial ones (CA VA, VB), and a secreted CA isozyme, CA VI, apart from three acatalytic ones (CA VIII, X and XI).8
Figure 1.

Third generation of H2 antagonists in clinical use.
These enzymes play crucial physiological/pathological roles in acid–base homeostasis, secretion of electrolytes, transport of ions, by providing bicarbonate and by regulating the pH in the gastric acid production, within the stomach parietal cells, as well as bile production, pancreatic juice production, intestinal ion transport, and protection of gastrointestinal tract from extreme pH conditions (too acidic or too basic).1,9,10 In this particular context, we decided to investigated whether famotidine shows CA inhibitory effects. All catalytically active human CA isoforms (hCA I–XIV), as well as the two CAs present in the bacterial pathogen Helicobacter pylori (α-HpCA, β-HpCA), the etiologic agent responsible of gastric ulcers, were included in the study. Furthermore, we report the X-ray structure of famotidine in complex with hCA I and hCA II, allowing us to unravel interesting aspects related to its CA inhibition mechanism. The inhibition profiles of famotidine and acetazolamide (AAZ), as standard sulfonamide inhibitor against all catalytically active CA isoforms is shown in Table 1. The cytosolic isoforms had a very different inhibition profile with famotidine. hCA VII was the most susceptible isoform to inhibition with this drug, with an inhibition constant Ki of 3.0 nM. However, the two widespread cytosolic isoforms hCA I and hCA II showed a very different inhibition with this compound: hCA II was effectively inhibited (Ki 57.9 nM), whereas hCA I was more than ten times less sensitive to famotidine inhibition compared to hCA II, with a Ki of 922.4 nM. Famotidine was a moderate inhibitor of isoform hCA XIII (Ki 171.5 mM) and lost any activity against hCA III, with a Ki > 10 μM. The two mitochondrial isoforms (hCA VA and VB) were poorly inhibited by this sulfamide drug, in the micromolar range (Kis of 1.4 and 5.3 μM, respectively). The membrane-bound isoforms, similar to the cytosolic ones discussed above, showed a heterogeneous inhibition pattern. hCA XII, a tumor associated isoform and recently validated as antitumor target , was effectively inhibited by famotidine with a Ki of 45.3 nM. The second enzyme overexpressed in hypoxic tumors, hCA IX, was, however, less inhibited by famotidine, with a Ki of 126.3 nM (Table 1). The last two membrane-associated isoforms hCA IV and XIV, instead, were weakly inhibited by this drug, in the high nanomolar range (Ki 938.8 and 677.2 nM).
Table 1. hCA I-XIV Inhibition Data with Famotidine and Acetazolamide (AAZ) as a Standard Inhibitor, by a CO2 Hydration, Stopped-Flow Technique11.
|
Ki (nM)a | ||
|---|---|---|
| cmpd | famotidine | AAZ |
| hCA I | 922.4 | 250.0 |
| hCA II | 57.9 | 12.1 |
| hCA III | >10000 | >10000 |
| hCA IV | 938.8 | 74.0 |
| hCA VA | 1429.5 | 63.0 |
| hCA VB | 5328.2 | 54.0 |
| hCA VI | 98.2 | 11.0 |
| hCA VII | 3.0 | 2.5 |
| hCA IX | 126.3 | 25.8 |
| hCA XII | 45.3 | 5.7 |
| hCA XIII | 171.5 | 17.0 |
| hCA XIV | 677.2 | 41.0 |
Mean from three different assays, by a stopped-flow technique (errors were in the range of ±5–10% of the reported values).
H. pylori, a Gram-negative neutralophile, was shown to be the cause of chronic gastritis, peptic ulcers, and, more recently, gastric cancer, the second most common tumor in humans.12,13 This gastric pathogen encodes for two classes of CAs, an α- and a β-one. Both enzymes are crucial for its survival in the acidic environment within the stomach, and recent studies showed that sulphonamides block the growth of the pathogen in vitro and in vivo.14−16 Thus, in case of interference with these enzymes, famotidine might be used as a new pharmacologic tool in the management of drug-resistant H. pylori.17 For this reason, we performed inhibition studies of the two bacterial CAs (Table 2). The α-class enzyme was effectively inhibited by this compound, with an inhibition constant of 20.7 nM, comparable to clinically used sulphonamide acetazolamide. However, hpβCA was less effectively inhibited by famotidine (Ki of 49.8 nM). However, also for this enzyme the activity was comparable to AAZ. An interesting observation was that famotidine proved to be more active against H. pylori enzymes compared to the two human dominant cytosolic isoforms (hCA I and hCA II), whereas AAZ was an effective inhibitor for both of them (Table 1).
Table 2. hpαCA and hpβCA Inhibition Data with Famotidine and Acetazolamide (AAZ) as a Standard Inhibitor, by a CO2 Hydration, Stopped-Flow Technique11.
|
Ki (nM)a | ||
|---|---|---|
| cmpd | famotidine | AAZ |
| hpαCA | 20.7 | 21.4 |
| hpβCA | 49.8 | 40.0 |
Mean from three different assays, by a stopped-flow technique (errors were in the range of ±5–10% of the reported values).
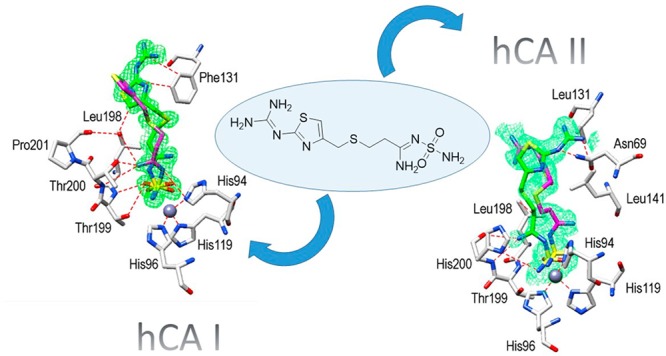
Considering the abundant localization in the gastrointestinal tract of the cytosolic hCA I and II, as well as their crucial roles in pH regulation, we obtained the high-resolution X-ray crystal structures of the adducts of famotidine with these enzymes, in order to understand in detail the binding modes of the drug. Inspection of the initial Fo–Fc electron density maps of both active sites showed well-defined electron density, fully compatible with the presence of famotidine, and, surprisingly, revealed two possible conformation of the drug for both adducts (Figure 2A,B). Famotidine coordinates the catalytic zinc ion of the two CA isoforms by means of one nitrogen atom of the sulfamide group, displacing the zinc bound water molecule/hydroxide ion, resulting in a tetrahedral coordination geometry of the metal ion, similarly to other sulfamide and sulfonamide compounds.4,18
Figure 2.
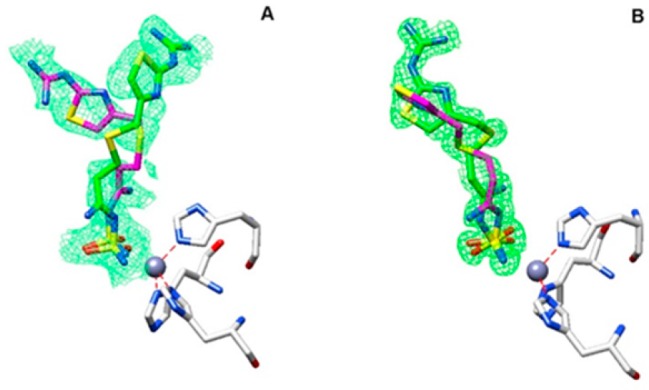
Active site region of hCA I/famotidine (A) and hCA II/famotidine (B) complexes. Inhibitors shown as σA-weighted |Fo–Fc| density maps at 2.0σ. The zinc ion (gray sphere) and its three His ligands are shown in CPK color.
The hCA I/famotidine complex showed two different orientations of the inhibitor (Figure 3). The first one, labeled in green, exhibited different hydrogen bonds stabilizing the enzyme–inhibitor adduct, such as the ones involving the guanidine tail and Asn69 or the nitrogen atom near the sulfamide moiety and Thr199 and His200. However, hydrophobic interactions were almost nonexistent in this complex. We found only one such interaction among Leu198 and the methylene chain of famotidine. The opposite orientation of the second structure (labeled in magenta), surprisingly, did not show any interaction with side chains of the protein (Figure 3). A completely different situation was observed when the hCA II active site was analyzed. Again, two possible orientations of famotidine were observed, both located in the hydrophobic region delimited by a small pocket aligned by the side chains of Phe131, Leu198, and Pro201 (Figure 4). The thiazole ring of one famotidine orientation (labeled in green) formed strong hydrophobic interactions with these residues and a water bridge with Pro201 by means of the azomethine nitrogen atom. This portion of the molecule was oriented differently in two conformations: in one, the nitrogen atom participates in several hydrogen bonds with Thr199 and Thr200. In the second one, a reverse orientation of the nitrogen was seen, with no interactions with the protein. Finally, the sulfamide oxygen is involved in a bifurcated hydrogen bond with Thr200 (Figure 4). Electron density was absent for the guanidine tail of the conformation labeled in magenta in Figure 4 and was not introduced in the complex model.
Figure 3.
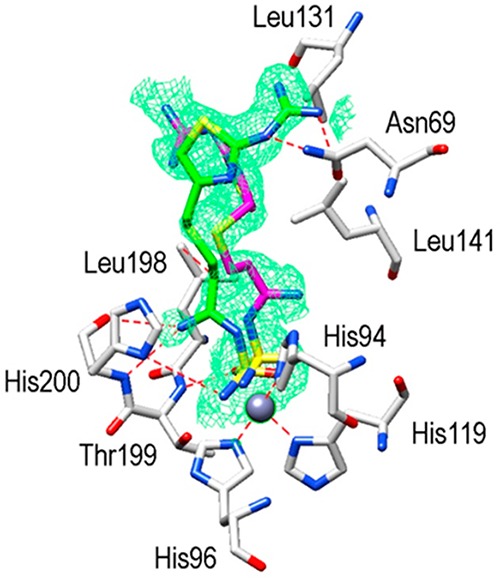
Active site region of hCA I/famotidine adduct. Inhibitor shown as σA-weighted |Fo–Fc| density map at 2.0σ. Hydrogen bonds, van der Waals interactions, and the active site Zn2+-ion coordination are also shown.
Figure 4.
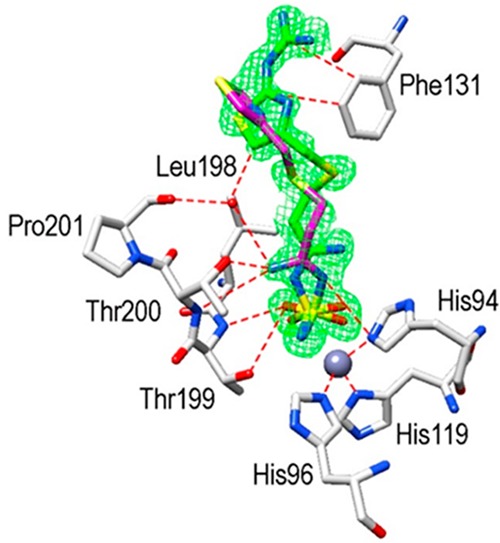
Active site region of hCA II/famotidine adduct. Inhibitor shown as σA-weighted |Fo–Fc| density map at 2.0σ. Hydrogen bonds, van der Waals interactions, and the active site Zn2+-ion coordination are also shown.
The structural superimposition between the hCA I and hCA II complexes is reported in Figure 5, which shows that, while the sulfamide moieties of famotidine are rather well superimposable, the two aliphatic tails present different orientations and, as a consequence, diverse interactions with amino acid side chains from the two active sites. Phe131 in hCA II proved to be essential to collocate famotidine within the lipophilic side of the active site cavity, which strongly correlates with the inhibition potency of this drug against hCA II. However, the presence of Leu131 in hCA I led to a loose van der Waals interaction, which did not force famotidine toward the lipophilic side of the active site, leading thus to two opposite orientations of the scaffold and few hydrophobic interactions with the active site. These characteristics reflect the loss of inhibitory potency against this isoform compared to hCA II.
Figure 5.
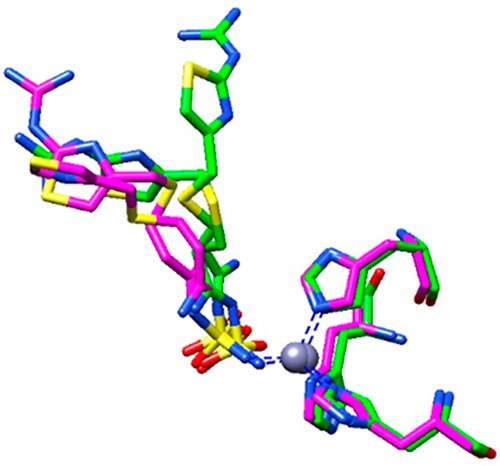
Structural superimposition of famotidine/hCA I and famotidine/hCA II complexes. hCA I and hCA II adducts are colored in green and magenta, respectively.
In summary, we have analyzed the inhibition pattern of famotidine against all human active CAs. The drug shows particular efficacy for the cytosolic isoforms hCA II and hCA VII. Furthermore, the two class bacterial enzymes from H. pylori (hpαCA and hpβCA) were also highly inhibited by this drug, raising thus the possibility that the effective antiulcer effects of famotidine may be due not only to its action of H2-receptor antagonist but also due to the fact that, by inhibiting the bacterial CAs, the survival of the pathogen within the gastric niche is compromised, as demonstrated earlier for acetazolamide.19 Indeed, this compound was used as an antiulcer agent with some success, but its side effects due to inhibition of CAs in other tissues than the stomach precluded its large clinical use.20 Famotidine is a much more isoform-selective CAI compared to acetazolamide and effectively inhibits the bacterial CAs too, allowing us to propose it as a new lead for the design of anti-infectives with a multifactorial mechanism of action.
Glossary
ABBREVIATIONS
- CAs
carbonic anhydrases
- AAZ
acetazolamide
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.8b00334.
Synthetic procedures, characterization of compounds, in vitro kinetic procedure, X-ray crystallography, and biological assay (PDF)
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Sakurai K.; Nagahara A.; Inoue K.; Akiyama J.; Mabe K.; Suzuki J.; Habu Y.; Araki A.; Suzuki T.; Satoh K.; Nagami H.; Harada R.; Tano N.; Kusaka M.; Fujioka Y.; Fujimura T.; Shigeto N.; Oumi T.; Miwa J.; Miwa H.; Fujimoto K.; Kinoshita Y.; Haruma K. Efficacy of omeprazole, famotidine, mosapride and teprenone in patients with upper gastrointestinal symptoms: an omeprazole-controlled randomized study. BMC Gastroenterol. 2012, 12, 42. 10.1186/1471-230X-12-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howden C. W. Clinical pharmacology of omeprazole. Clin. Pharmacokinet. 1991, 20, 38. 10.2165/00003088-199120010-00003. [DOI] [PubMed] [Google Scholar]
- Ahmadi A.; Ebrahimzadeh M. A.; Ahmad-Ashrafi S.; Karami M.; Mahdavi M. R.; Saravi S. S. Hepatoprotective, antinociceptive and antioxidant activities of cimetidine, ranitidine and famotidine as histamine H2 receptor antagonists. Fundam. Clin. Pharmacol. 2011, 25, 72. 10.1111/j.1472-8206.2009.00810.x. [DOI] [PubMed] [Google Scholar]
- De Simone G.; Pizika G.; Monti S. M.; Di Fiore A.; Ivanova J.; Vozny I.; Trapencieris P.; Zalubovskis R.; Supuran C. T.; Alterio V. Hydrophobic substituents of the phenylmethylsulfamide moiety can be used for the development of new selective carbonic anhydrase inhibitors. BioMed Res. Int. 2014, 2014, 523210. 10.1155/2014/523210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demir Y.; Nadaroğlu H.; Demir N. Effects of omeprazole, famotidine, and ranitidine on the enzyme activities of carbonic anhydrase from bovine stomach in vitro and rat erythrocytes in vivo. Biol. Pharm. Bull. 2004, 27, 1730. 10.1248/bpb.27.1730. [DOI] [PubMed] [Google Scholar]
- Supuran C. T. Structure and function of carbonic anhydrases. Biochem. J. 2016, 473, 2023. 10.1042/BCJ20160115. [DOI] [PubMed] [Google Scholar]
- Supuran C. T. Carbonic anhydrases: novel therapeutic applications for inhibitors and activators. Nat. Rev. Drug Discovery 2008, 7, 168. 10.1038/nrd2467. [DOI] [PubMed] [Google Scholar]
- Supuran C. T. Advances in structure-based drug discovery of carbonic anhydrase inhibitors. Expert Opin. Drug Discovery 2017, 12, 61. 10.1080/17460441.2017.1253677. [DOI] [PubMed] [Google Scholar]
- Lönnerholm G.; Selking O.; Wistrand P. J. Amount and distribution of carbonic anhydrases CA I and CA II in the gastrointestinal tract. Gastroenterology 1985, 88, 1151. 10.1016/S0016-5085(85)80074-3. [DOI] [PubMed] [Google Scholar]
- Carter M. J.; Parsons D. S. The isoenzymes of carbonic anhydrase: tissue, subcellular distribution and functional significance, with particular reference to the intestinal tract. J. Physiol. 1971, 215, 71. 10.1113/jphysiol.1971.sp009458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khalifah R. G. The carbon dioxide hydration activity of carbonic anhydrase. I. Stop-flow kinetic studies on the native human isoenzymes B and C. J. Biol. Chem. 1971, 246, 2561. [PubMed] [Google Scholar]
- Nishimori I.; Minakuchi T.; Kohsaki T.; Onishi S.; Takeuchi H.; Vullo D.; Scozzafava A.; Supuran C. T. Carbonic anhydrase inhibitors: the beta-carbonic anhydrase from Helicobacter pylori is a new target for sulfonamide and sulfamate inhibitors. Bioorg. Med. Chem. Lett. 2007, 17, 3585. 10.1016/j.bmcl.2007.04.063. [DOI] [PubMed] [Google Scholar]
- Nishimori I.; Vullo D.; Minakuchi T.; Morimoto K.; Onishi S.; Scozzafava A.; Supuran C. T. Carbonic anhydrase inhibitors: DNA cloning and inhibition studies of the alpha-carbonic anhydrase from Helicobacter pylori, a new target for developing sulfonamide and sulfamate gastric drugs. Bioorg. Med. Chem. Lett. 2006, 16, 2182. 10.1016/j.bmcl.2006.01.044. [DOI] [PubMed] [Google Scholar]
- Maresca A.; Vullo D.; Scozzafava A.; Supuran C. T. Inhibition of the alpha- and beta-carbonic anhydrases from the gastric pathogen Helycobacter pylori with anions. J. Enzyme Inhib. Med. Chem. 2013, 28, 388. 10.3109/14756366.2011.649268. [DOI] [PubMed] [Google Scholar]
- Capasso C.; Supuran C. T. Bacterial, fungal and protozoan carbonic anhydrases as drug targets. Expert Opin. Ther. Targets 2015, 19, 1689. 10.1517/14728222.2015.1067685. [DOI] [PubMed] [Google Scholar]
- Modak J. K.; Liu Y. C.; Supuran C. T.; Roujeinikova A. Structure-Activity Relationship for Sulfonamide Inhibition of Helicobacter pylori α-Carbonic Anhydrase. J. Med. Chem. 2016, 59, 11098. 10.1021/acs.jmedchem.6b01333. [DOI] [PubMed] [Google Scholar]
- Amin M.; Iqbal M. S.; Hughes R. W.; Khan S. A.; Reynolds P. A.; Enne V. I.; Mirza A. S. Mechanochemical synthesis and in vitro anti-Helicobacter pylori and uresase inhibitory activities of novel zinc(II)-famotidine complex. J. Enzyme Inhib. Med. Chem. 2010, 25, 383. 10.3109/14756360903179518. [DOI] [PubMed] [Google Scholar]
- Di Fiore A.; De Simone G.; Alterio V.; Riccio V.; Winum J. Y.; Carta F.; Supuran C. T. The anticonvulsant sulfamide JNJ-26990990 and its S,S-dioxide analog strongly inhibit carbonic anhydrases: solution and X-ray crystallographic studies. Org. Biomol. Chem. 2016, 14, 4853. 10.1039/C6OB00803H. [DOI] [PubMed] [Google Scholar]
- Marcus E. A.; Moshfegh A. P.; Sachs G.; Scott D. R. The periplasmic alpha-carbonic anhydrase activity of Helicobacter pylori is essential for acid acclimation. J. Bacteriol. 2005, 187, 729. 10.1128/JB.187.2.729-738.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buzás G. M.; Supuran C. T. The history and rationale of using carbonic anhydrase inhibitors in the treatment of peptic ulcers. In memoriam Ioan Puşcaş (1932–2015). J. Enzyme Inhib. Med. Chem. 2016, 31, 527. 10.3109/14756366.2015.1051042. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.