

Abstract
Protein arginine deiminase 4 (PAD4) is a calcium-dependent enzyme that catalyzes the conversion of arginine to citrulline within target proteins. Dysregulation of PAD4 has been implicated in a number of human diseases, including rheumatoid arthritis and other inflammatory diseases as well as cancer. In this study, we report on the design, synthesis, and evaluation of a new class of haloacetamidine-based compounds as potential PAD4 inhibitors. Specifically, we describe the identification of 4,5,6-trichloroindazole 24 as a highly potent PAD4 inhibitor that displays >10-fold selectivity for PAD4 over PAD3 and >50-fold over PAD1 and PAD2. The efficacy of this compound in cells was determined by measuring the inhibition of PAD4-mediated H4 citrullination in HL-60 granulocytes.
Keywords: Protein arginine deiminase, mechanism-based inhibitor, rheumatoid arthritis, inflammatory disease, citrullination
Protein arginine deiminases (PADs) are a family of Ca2+-dependent enzymes that catalyze the conversion of arginines to citrullines within proteins, a phenomenon termed citrullination (Figure 1). Citrullination is a post-translational event involved in many cellular processes such as gene regulation and cell differentiation.1,2 This post-translational modification results in the loss of the net positive charge on the target protein, which leads to structural changes of the protein and consequently alters the protein function and its interaction with other biomolecules. Five human PADs (1, 2, 3, 4, and 6) have been identified and characterized.3−5 PAD1 to PAD4 are catalytically active, but PAD6 has no detectable activity.2,5,6 Although these isozymes share greater than 50% interisozyme sequence identity,1,5 they display unique tissue expression patterns.4,7 Significantly, dysregulation of PAD activity has been linked to the pathogenesis of many diseases.4,7,8
Figure 1.

PAD-catalyzed conversion of arginine to citrulline.
Among the four PAD isozymes, PAD4 is of great interest due to its relevance to the progression of rheumatoid arthritis (RA).9 In the sera of RA patients, anticitrullinated protein antibodies (ACPA) that specifically recognize PAD4-mediated citrullination are found at high levels.4,9 Because PAD4 is primarily expressed in granulocytes, overexpression of PAD4 also facilitates the formation of NETs (Neutrophil Extracellular Traps), and the resultant aberrant level of NETs is associated with inflammatory disorders such as sepsis,10 lupus,11 vasculitis,12 and thrombosis.13 In addition to promoting NETosis, citrullination by PAD4 critically regulates the expression of inflammatory cytokines in neutrophils such as TNF and IL-1β, which also actively contribute to the pathogenesis of RA.14 In cancer, PAD4 not only acts as a transcriptional corepressor of the tumor suppressor protein p5315 but also is involved in mediating malignancy.16 Due to its regulatory roles in cell signaling pathways and disease pathogenesis, PAD4 has emerged as a potential therapeutic target for a range of disease states, and the development of inhibitors toward PAD4 as biological tools has become of high importance.
In recent years, numerous attempts have been carried out to identify potent and selective inactivators of PAD4. GSK implemented DNA encoded library screening to identify reversible inhibitors of PAD4.17 The Thompson group has also developed a fluorescence polarization-activity based protein-profiling assay that enabled the discovery of streptonigrin, a natural product, as a potent and selective inhibitor of PAD4.18 Unfortunately, off-target effects exhibited by streptonigrin complicate its use as a PAD4 pharmacological tool. Haloacetamidine mechanism-based inhibitors such as Cl-amidine (Figure 2) and BB-Cl-amidine, a second generation derivative of Cl-amidine with enhanced cell permeability and similar potency, have provided many key insights into PAD biology.14,19,21 To date, selective inhibitors containing the haloacetamidine warhead have been developed for PAD1,20 PAD2,21 and PAD322,23 with representative inhibitors shown in Figure 2.
Figure 2.

Potent and selective haloacetamidine-based inhibitors of PAD1, PAD2, and PAD3.
Potent and selective mechanism-based haloacetamidine inhibitors against PAD4 have not yet been developed. Here, we report a new class of selective haloacetamidine-based nonpeptidic inhibitors against PAD4 bearing an indazole core, a heterocycle motif that has been incorporated into many drugs.24 Moreover, we demonstrate that our most potent and selective indazole inhibitor prevents PAD4-mediated histone citrullination in cell culture.
We previously reported a substrate-based fragment screening approach to identify low molecular weight nonpeptidic guanidine substrates of PADs and identified an N-substituted indole appended to guanidine as an effective PAD4 substrate.23 To capitalize on this nonpeptidic substrate, we prepared a series of analogous indole-derived chloracetamidine mechanism-based inhibitors 7a–7d (Scheme 1). Commercially available amide 4 was reduced followed by Boc protection of the resulting free amine to give intermediate 5.25 Indole N-alkylation with several different alkyl halides generated 6a–6d. Cleavage of the Boc group followed by addition of ethyl-2-chloroacetimidate provided inhibitors 7a–7d. We evaluated these inhibitors against PAD4 and observed that increasing the size of the N-alkyl group significantly improved the inhibitory potency against PAD4 with the cyclopentylmethyl substituent in 7d providing the greatest inhibitory activity (Table 1). A number of additional N-alkyl derivatives were prepared and evaluated, but none gave any improvement in inhibitory activity over the N-cyclopentylmethyl derivative 7d (data not shown).
Scheme 1. Synthesis of Alkylated Indole Inhibitors.

Reagents and conditions: (a) LiAlH4, THF, reflux, 2 h; (b) Et3N, Boc2O, CH2Cl2, 0 °C to rt, 1 h; (c) NaH, DMF, 0 °C, 30 min to 1 h; (d) RX, DMF, 0 °C to rt, 1 to 2 h; (e) 33% CF3COOH in CH2Cl2, 1 h; (f) ethyl-2-chloroacetamidate·HCl, Et3N, CH3OH, rt, 1 h.
Table 1. IC50 Values for Compounds 7a–d and 12aa.

IC50 values are reported as mean ± SD and were run in duplicate. Cyp = cyclopentyl. See Supporting Information for further assay details.
Indazole is isosteric with indole, and therefore, the indazole analog 12a (R = H) corresponding to indole inhibitor 7d was synthesized (Scheme 2). Lewis acid-catalyzed addition of aniline (8a) to 4-chlorobutanenitrile provided the 2-aminophenyl ketone 9a.26 Diazotization and then reduction to the hydrazine with in situ cyclization to give the indazole was followed by displacement of the bromide with azide to afford indazole 10a. N-Alkylation of the indazole with cyclopentylmethyl bromide yielded intermediate 11a, which, after reduction to the amine followed by addition of ethyl-2-chloroacetimidate, furnished inhibitor 12a. Evaluation of 12a against PAD4 established that replacing the indole heterocycle core in 7d with indazole resulted in a modest boost in potency (Table 1).
Scheme 2. Synthesis of Alkylated Indazole Inhibitors.

Reagents and conditions: (a) Me2S·BCl3, 1,2-dichloroethane (DCE), 0 °C; (b) 4-chlorobutanenitrile, 0 °C to rt; (c) AlCl3, reflux, overnight; (d) 2 M HCl, reflux, 30 min; (e) NaNO2, H2O, HCl, 0 °C, 1 h; (f) SnCl2, HCl, 0 °C, 1 h; (g) NaN3, DMF, 50 °C, overnight; (h) NaH, DMF, 0 °C, 30 min to 1 h; (i) cyclopentylmethyl bromide, DMF, 0 °C to rt, 1 to 2 h; (j) SnCl2, CH3OH, 2–4 h; (k) ethyl-2-chloroacetamidate·HCl, Et3N, CH3OH, rt, 1–2 h.
Given that indazole 12a is slightly more potent than indole 7d, we next proceeded to functionalize the ring to further enhance PAD4 potency and selectivity. We decided to install chloro substituents around the indole ring because the chloro substituent is relatively small and metabolically stable and also could provide a convenient handle for further synthetic elaboration. The preparation of the chloro substituted inhibitors 12b–12d and the dichloro-substituted inhibitor 12e was accomplished according to the same sequence used to prepare the parent inhibitor 12a employing chloro-substituted anilines 8b–8e starting materials (Scheme 2).
To complete the SAR of all possible monochloro derivatives, we also sought to install the chloro group at position 4 of the indazole ring. However, the route used to prepare the other chloro derivatives is not appropriate for this substitution pattern because Friedel–Crafts acylation does not occur at the more sterically hindered ortho-site of 3-chloroaniline. We therefore developed a different route (Scheme 3). Directed ortho-metalation of commercially available aryl halide 13 resulted in lithiation at the most hindered 2-position, which after addition of trimethylsilyl chloride (TMSCl) afforded 14. The silyl substituent in 14 then served to both activate and direct Lewis acid-catalyzed acylation with 4-chlorobutanenitrile at the silyl-substituted site to give ketone 15.27 Azide displacement of the alkyl chloride followed by hydrazone formation with concomitant SNAr-mediated cyclization then generated indazole 16. Next, N-alkylation with cyclopentylmethyl bromide provided 17. Finally, reduction of the azide and subsequent addition of ethyl chloroacetimidate furnished inhibitor 18.
Scheme 3. Synthesis of 4-Chloroindazole Inhibitor 18.

Reagents and conditions: (a) tetramethylethylenediamine (TMEDA), n-BuLi, THF, −75 °C, 2 h; (b) Me3SiCl, THF, −75 °C, overnight; (c) AlCl3, CH2Cl2, 0 °C; (d) 4-chlorobutanoyl chloride, −80 to −10 °C, 4 h; (e) NaN3, DMF, 50 °C, 16 h; (f) hydrazine monohydrate, dimethoxyethane (DME), reflux, 17 h; (g) NaH, DMF, 0 °C, 30 min to 1 h; (h) cyclopentylmethyl bromide, DMF, 0 °C to rt, 1 to 2 h; (i) SnCl2, CH3OH, 2.5 h; (j) ethyl-2-chloroacetamidate·HCl, Et3N, CH3OH, rt, 1 to 2 h.
Because we desired both potency against PAD4 and selectivity over all of the other PAD isozymes, we carried out more rigorous determination of kinact/KI for the indazole inhibitors (Table 2). The parent indazole inhibitor 12a had excellent selectivity over PAD1 and PAD2. However, selectivity for PAD4 over PAD3 was not obtained. Therefore, all of the monochloro-substituted derivatives, 12b–d and 18, were evaluated for inhibitory activity against both PAD4 and PAD3 (Table 2). Inhibitor 12b, with the chloro substituent installed on position 7 of the indazole ring, resulted in modestly greater inhibitory activity against both PAD4 and PAD3. In contrast, inhibitor 12c with the chloro substitution at position 6 resulted in both increased potency against PAD4 and 2-fold selectivity for PAD4 over PAD3. Inhibitors 18 and 12d with the chloro substituent at positions 4 and 5 of the ring, respectively, both provided significant improvements in inhibitory activity against PAD4 and with relatively modest improvements in selectivity over PAD3. We next explored the summation of the effects of the chloro substitution in 12c and 18 by evaluating the dichloro indazole inhibitor 12e. Interestingly, inhibitor 12e resulted in considerable improvement in PAD4 inhibitory activity while at the same time providing 7-fold selectivity over PAD3. Moreover, a high level of selectivity over PAD1 and PAD2 was maintained.
Table 2. kinact/KI Values for Compounds 12a–e, 18, and 24a.

kinact/KI was determined using six concentrations of inhibitor at five different time points. kinact/KI was calculated from linear fit of kobs obtained from two replicates.
kinact/KI was calculated from nonlinear regression of kobs obtained from two replicates. kobs = kinact/KI because [I] ≪ KI. Cl-amidine under the assay conditions reported here gave the following kinact/KI values: PAD1, 4550 ± 860; PAD2, 520 ± 50; PAD3, 2340 ± 80; PAD4, 1770 ± 470.23 See Supporting Information for further assay details.
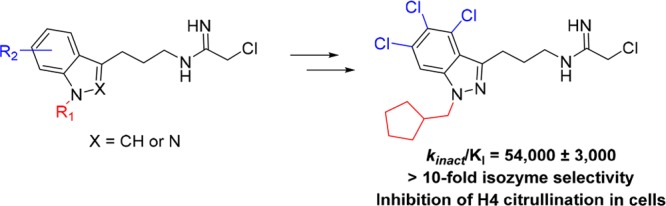
Based on the observation that placement of a chloro substituent at position 5 in inhibitor 12c resulted in the greatest improvement in PAD4 potency and also improved selectivity, we hypothesized that the trichloro analog 24 might result in further enhancement (Scheme 4). The preparation of 24 was synthetically more challenging and lengthier than the other chloro derivatives. Aniline 19 was first converted to aryl fluoride 20 by diazotization and treatment with HBF4.28 Ortho-directed metalation followed by addition of 4-chlorobutanenitrile then provided ketone 21.29 Azide displacement of the alkyl chloride followed by hydrazone condensation and SNAr-mediated cyclization generated azido 4,5,6-trichloroindazole intermediate 22. N-Alkylation with cyclopentylmethyl bromide provided 23. Finally, reduction of the azide to the amine group followed by addition of ethyl-2-chloroacetimidate furnished inhibitor 24. Indeed, the trichloroindazole inhibitor 24 further enhanced PAD4 potency and selectivity, making it the best inhibitor in this series (Table 2).
Scheme 4. Synthesis of 4,5,6-Trichloroindole Inhibitor 24.

Reagents and conditions: (a) NaNO2, H2O, HCl, 30 min; (b) 48% aq. HBF4, 1.5 h; (c) lithium diisopropylamide (LDA), THF, −78 °C, 2 h; (d) ZnCl2, THF, −78 to −10 °C, 4 h; (e) CuCl, THF; (f) 4-chlorobutanoyl chloride, overnight; (g) NaN3, DMF, 50 °C, overnight; (h) hydrazine monohydrate, DME, reflux, overnight; (i) NaH, DMF, 0 °C, 30 min to 1 h; (j) cyclopentylmethyl bromide, DMF, 0 °C to rt, 1 to 2 h; (k) SnCl2, CH3OH, overnight; (l) ethyl-2-chloroacetamidate·HCl, Et3N, CH3OH, rt, 1 to 2 h.
We next evaluated the ability of 24 to inhibit PAD4 activity in cells by monitoring its effect on the level of histone H4 citrullination in differentiated HL-60 cells. PAD4 is naturally overexpressed in mature HL-60 derived granulocytes and is also known to citrullinate H4 upon stimulation with calcium ionophores.30 Western blots of cell lysates visualized with an anticitrulline histone H4 antibody showed that citrullination of H4 was significantly attenuated in cells treated with as little as 5 μM 24. The reduced levels of citrullinated H4 induced by 10 μM or 5 μM 24 were also compared to those induced by Cl-amidine, the most extensively used pan-PAD inhibitor (Figure 3). Qualitative analysis of the Western blot indicated that the PAD4 selective inhibitor 24 is more potent than pan-PAD inhibitor Cl-amidine.
Figure 3.

Evaluation of Cl-amidine and 24 in HL-60 granulocytes after stimulation with the calcium ionophore A23187.
In summary, we synthesized and evaluated a series of N-alkylated indole chloroacetamidine inhibitors as potential PAD4 inactivators. Replacement of the indole heterocycle core with indazole and derivatization around the indazole ring with chloro substituents then led to the identification of trichloroindazole inhibitor 24 with high inhibitory activity and greater than 10-fold selectivity for PAD4 over the other three PAD isozymes. Inhibitor 24 was also evaluated for activity in cells and inhibited PAD4-mediated H4 citrullination at low micromolar concentrations. Exploration of other substituents about the indazole ring might be expected to result in further increases in inhibitor potency and PAD selectivity.
Glossary
ABBREVIATIONS
- PAD
protein arginine deiminase
- RA
rheumatoid arthritis
- ACPA
anticitrullinated protein antibodies
- NET
neutrophil extracellular trap
- TNF
tumor necrosis factor alpha
- IL-1β
interleukin 1 beta
- H4
histone 4
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.8b00283.
Complete experimental procedures, characterization data for all compounds, IC50 and kinact/KI data for inhibitors, HL-60 cell culture and differentiation, and PAD4 inhibition assays and analysis (PDF)
The authors are grateful to the NIH (R35GM122473 to J.A.E and R01CA170741 to A.S.) for support of this work.
The authors declare no competing financial interest.
Supplementary Material
References
- Fuhrmann J.; Thompson P. R. Protein arginine methylation and citrullination in epigenetic regulation. ACS Chem. Biol. 2016, 11, 654–668. 10.1021/acschembio.5b00942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuhrmann J.; Clancy K. W.; Thompson P. R. Chemical biology of protein arginine modifications in epigenetic regulation. Chem. Rev. 2015, 115, 5413–5461. 10.1021/acs.chemrev.5b00003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearney P. L.; Bhatia M.; Jones N. G.; Yuan L.; Glascock M. C.; Catchings K. L.; Yamada M.; Thompson P. R. Kinetic characterization of protein arginine deiminase 4: a transcriptional corepressor Implicated in the onset and progression of rheumatoid arthritis. Biochemistry 2005, 44, 10570–10582. 10.1021/bi050292m. [DOI] [PubMed] [Google Scholar]
- Jones J. E.; Causey C. P.; Knuckley B.; Slack-Noyes J. L.; Thompson P. R. Protein arginine deiminase 4 (PAD4): current understanding and future therapeutic potential. Curr. Opin. Drug Discov. Devel. 2009, 12, 616–627. [PMC free article] [PubMed] [Google Scholar]
- Bicker K. L.; Thompson P. R. The protein arginine deiminases: structure, function, inhibition, and disease. Biopolymers 2013, 99, 155–163. 10.1002/bip.22127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taki H.; Tomoharu G.; Bryan K.; Paul R. T.; Oliver V.; Kazuya H.; Tatsurou M.; Kouichiro S.; Hiroyuki H.; Eiji S. Purification of enzymatically inactive peptidylarginine deiminase type 6 from mouse ovary that reveals hexameric structure different from other dimeric isoforms. Adv. Biosci. Biotechnol. 2011, 2, 304–310. 10.4236/abb.2011.24044. [DOI] [Google Scholar]
- Arita K.; Hashimoto H.; Shimizu T.; Nakashima K.; Yamada M.; Sato M. Structural basis for Ca2+-induced activation of human PAD4. Nat. Struct. Mol. Biol. 2004, 11, 777–783. 10.1038/nsmb799. [DOI] [PubMed] [Google Scholar]
- Witalison E. E.; Thompson P. R.; Hofseth L. J. Protein arginine deiminases and associated citrullination: physiological functions and diseases associated with dysregulation. Curr. Drug Targets 2015, 16, 700–710. 10.2174/1389450116666150202160954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koushik S.; Joshi N.; Nagaraju S.; Mahmood S.; Mudeenahally K.; Padmavathy R.; Jegatheesan S. K.; Mullangi R.; Rajagopal S. PAD4: pathophysiology, current therapeutics and future perspective in rheumatoid arthritis. Expert Opin. Ther. Targets 2017, 21, 433–447. 10.1080/14728222.2017.1294160. [DOI] [PubMed] [Google Scholar]
- Clark S. R.; Ma A. C.; Tavener S. A.; McDonald B.; Goodarzi Z.; Kelly M. M.; Patel K. D.; Chakrabarti S.; McAvoy E.; Sinclair G. D.; Keys E. M.; Allen-Vercoe E.; DeVinney R.; Doig C. J.; Green F. H. Y.; Kubes P. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat. Med. 2007, 13, 463–469. 10.1038/nm1565. [DOI] [PubMed] [Google Scholar]
- Villanueva E.; Yalavarthi S.; Berthier C. C.; Hodgin J. B.; Khandpur R.; Lin A. M.; Rubin C. J.; Zhao W.; Olsen S. H.; Klinker M.; Shealy D.; Denny M. F.; Plumas J.; Chaperot L.; Kretzler M.; Bruce A. T.; Kaplan M. J. Netting neutrophils induce endothelial damage, infiltrate tissues, and expose immunostimulatory molecules in systemic lupus erythematosus. J. Immunol. 2011, 187, 538–552. 10.4049/jimmunol.1100450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohlsson S. M.; Ohlsson S.; Soderberg D.; Gunnarsson L.; Pettersson A.; Segelmark M.; Hellmark T. Neutrophils from vasculitis patients exhibit an increased propensity for activation by anti-neutrophil cytoplasmic antibodies. Clin. Exp. Immunol. 2014, 176, 363–372. 10.1111/cei.12301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinod K.; Demers M.; Fuchs T. A.; Wong S. L.; Brill A.; Gallant M.; Hu J.; Wang Y. M.; Wagner D. D. Neutrophil histone modification by peptidylarginine deiminase 4 is critical for deep vein thrombosis in mice. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 8674–8679. 10.1073/pnas.1301059110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun B.; Dwivedi N.; Bechtel T. J.; Paulsen J. L.; Muth A.; Bawadekar M.; Li G.; Thompson P. R.; Shelef M. A.; Schiffer C. A.; Weerapana E.; Ho I. C. Citrullination of NF- κB p65 promotes its nuclear localization and TLR-induced expression of IL-1β and TNFα. Sci. Immunol. 2017, 2, eaal3062. 10.1126/sciimmunol.aal3062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P.; Yao H.; Zhang Z.; Li M.; Luo Y.; Thompson P. R.; Gilmour D. S.; Wang Y. M. Regulation of p53 target gene expression by peptidylarginine deiminase 4. Mol. Cell. Biol. 2008, 28, 4745–4758. 10.1128/MCB.01747-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang X.; Han J.; Pang L.; Zhao Y.; Yang Y.; Shen Z. Increased PADI4 expression in blood and tissues of patients with malignant tumors. BMC Cancer 2009, 9, 40. 10.1186/1471-2407-9-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis H. D.; Liddle J.; Coote J. E.; Atkinson S. J.; Barker M. D.; Bax B. D.; Bicker K. L.; Bingham R. P.; Campbell M.; Chen Y. H.; Chung C. W.; Craggs P. D.; Davis R. P.; Eberhard D.; Joberty G.; Lind K. E.; Locke K.; Maller C.; Martinod K.; Patten C.; Polyakova O.; Rise C. E.; Ruediger M.; Sheppard R. J.; Slade D. J.; Thomas P.; Thorpe J.; Yao G.; Drewes G.; Wagner D. D.; Thompson P. R.; Prinjha R. K.; Wilson D. M. Inhibition of PAD4 activity is sufficient to disrupt mouse and human NET formation. Nat. Chem. Biol. 2015, 11, 189–191. 10.1038/nchembio.1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knuckley B.; Jones J. E.; Bachovchin D. A.; Slack J.; Causey C. P.; Brown S. J.; Rosen H.; Cravatt B. F.; Thompson P. R. A fluopol-ABPP HTS assay to identify PAD inhibitors. Chem. Commun. 2010, 46, 7175–7177. 10.1039/c0cc02634d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willis V. C.; Gizinski A. M.; Banda N. K.; Causey C. P.; Knuckley B.; Cordova K. N.; Luo Y.; Levitt B.; Glogowska M.; Chandra P.; Kulik L.; Robinson W. H.; Arend W. P.; Thompson P. R.; Holers V. M. N-α-benzoyl-N5-(2-chloro-1-iminoethyl)-L-ornithine amide, a protein arginine deiminase inhibitor, reduces the severity of murine collagen-induced arthritis. J. Immunol. 2011, 186, 4396–4404. 10.4049/jimmunol.1001620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian V.; Knight J. S.; Parelkar S.; Anguish L.; Coonrod S. A.; Kaplan M. J.; Thompson P. R. Design, synthesis, and biological evaluation of tetrazole analogs of CI-Amidine as protein arginine deiminase inhibitors. J. Med. Chem. 2015, 58, 1337–1344. 10.1021/jm501636x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muth A.; Subramanian V.; Beaumont E.; Nagar M.; Kerry P.; McEwan P.; Srinath H.; Clancy K.; Parelkar S.; Thompson P. R. Development of a selective inhibitor of protein arginine deiminase 2. J. Med. Chem. 2017, 60, 3198–3211. 10.1021/acs.jmedchem.7b00274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamali H.; Khan H. A.; Tjin C. C.; Ellman J. A. Cellular activity of new small molecule protein arginine deiminase 3 (PAD3) inhibitors. ACS Med. Chem. Lett. 2016, 7, 847–851. 10.1021/acsmedchemlett.6b00215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamali H.; Khan H. A.; Stringer J. R.; Chowdhury S.; Ellman J. A. Identification of multiple structurally distinct, nonpeptidic small molecule inhibitors of protein arginine deiminase 3 using a substrate-based fragment method. J. Am. Chem. Soc. 2015, 137, 3616–3621. 10.1021/jacs.5b00095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bendazac, niraparib, granisetron, pazopanib, axitinib, lonidamine, and lificiguat are approved drugs and drug candidates containing the indazole heterocycle. Detailed information on these drugs including compound structure, bioactivity, published studies, clinical trials, applications, and usage can be obtained at PubChem (http://pubchem.ncbi.nlm.nih.gov/).
- Mewshaw R. E.; Zhou D.; Zhou P.; Shi X. J.; Hornby G.; Spangler T.; Scerni R.; Smith D.; Schechter L. E.; Andree T. H. Studies toward the discovery of the next generation of antidepressants. 3. Dual 5-HT1A and serotonin transporter affinity within a Class of N-aryloxyethylindolylalkylamines. J. Med. Chem. 2004, 47, 3823–3842. 10.1021/jm0304010. [DOI] [PubMed] [Google Scholar]
- Cinelli M. A.; Morrell A.; Dexheimer T. S.; Scher E. S.; Pommier Y.; Cushman M. Design, synthesis, and biological evaluation of 14-substituted aromathecins as topoisomerase I inhibitors. J. Med. Chem. 2008, 51, 4609–4619. 10.1021/jm800259e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennetau B.; Rajarison F.; Dunoguès J.; Babin P. Fonctionnalisation régiosélective en position 2 de benzènes 1,3-disubstitués. Tetrahedron 1993, 49, 10843–10854. 10.1016/S0040-4020(01)80238-6. [DOI] [Google Scholar]
- Mahiou B.; Deinzer M. L. Synthetic strategies in the preparation of regiospecifically chlorine-37 labeled polychlorinated dibenzo-p-diozins. J. Labelled Compd. Radiopharm. 1991, 31, 261–287. 10.1002/jlcr.2580310403. [DOI] [Google Scholar]
- Rosen J.; Steinhuebel D.; Palucki M.; Davies I. One-step synthesis of α-chloro acetophenones from acid chlorides and aryl precursors. Org. Lett. 2007, 9, 667–9. 10.1021/ol0629667. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Wysocka J.; Sayegh J.; Lee Y.; Perlin J. R.; Leonelli L.; Sonbuchner L. S.; McDonald C. H.; Cook R. G.; Dou Y.; Roeder R. G.; Clarke S.; Stallcup M. R.; Allis C. D.; Coonrod S. A. Human PAD4 regulates histone arginine methylation levels via demethylimination. Science 2004, 306, 279–283. 10.1126/science.1101400. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.