

Abstract

Schistosomiasis is a major human parasitic disease afflicting more than 250 million people, historically treated with chemotherapies praziquantel or oxamniquine. Since oxamniquine is species-specific, killing Schistosoma mansoni but not other schistosome species (S. haematobium or S. japonicum) and evidence for drug resistant strains is growing, research efforts have focused on identifying novel approaches. Guided by data from X-ray crystallographic studies and Schistosoma worm killing assays on oxamniquine, our structure-based drug design approach produced a robust structure–activity relationship (SAR) program that identified several new lead compounds with effective worm killing. These studies culminated in the discovery of compound 12a, which demonstrated broad-species activity in killing S. mansoni (75%), S. haematobium (40%), and S. japonicum (83%).
Keywords: Schistosomiasis, oxamniquine, structure−activity relationships, X-ray crystallographic studies, aminopyrrolidine, aminopiperidine
Schistosomiasis is a neglected tropical disease caused by flatworms of the genus Schistosoma. After malaria, it is the second most endemic parasitic disease, estimated to affect over 250 million people worldwide and is responsible for almost 200,000 deaths each year.1 There are three major pathogenic species of Schistosoma: S. haematobium (Africa, 119 million cases), S. mansoni, (South America and Africa, 67 million cases), and S. japonicum (South-East Asia, 1 million cases). Although some small therapeutics have been employed (Figure 1) to combat the disease, broad range efficacy and effectiveness to drug-resistant strains of Schistosoma still represent a significant unmet medical need.2 The general antiparasitic drug Praziquantel 1 is the only treatment on the market and is active against all species of Schistosoma. Oxamniquine 3 was discovered through an optimization study on Mirasan 2, a lead found by Kikuth and Gönnert at Bayer. Compound 2 was found to have schistosomicidal activity in mice while completely inactive in a monkey model. It was later discovered that an active metabolite isolated from the urine of treated mice proved to have high potency on other species. The Mirasan series was revived at Pfizer in 1968, in which they found that an active metabolite, the hydroxymethyl derivative of 2, UK-3883, was three times as potent as 2. Through structure–activity relationships (SARs) and lead optimization, 3 was developed and used as first-line treatment in Brazil until the late 1990s and remained in use until 2010.3 It has a robust safety record, but unlike 1, its treatment efficacy is limited to S. mansoni.4
Figure 1.

Structure of Praziquantel 1, Mirasan 2, and Oxamniquine 3. The hydroxymethyl moiety of 3 is sulfated by SmSULT-OR.
In 2013, Valentim et al. discovered the mechanism of action of 3 by a genetic approach and comparison of gene sequences of oxamniquine-sensitive and resistant S. mansoni. Compound 3 is a prodrug, which, through sulfation of the hydroxymethyl moiety (Figure 1), is converted to the active species upon exposure to a sulfotransferase present in S. mansoni (SmSULT-OR). The active drug is then released from the enzyme and alkylates the parasite’s DNA [in a SN2 reaction] resulting in the death of the parasite.5,6 In order to understand the molecular basis of resistance, SmSULT-OR was cocrystallized with 3 and the sulfate-depleted version of its cofactor, 3′-phosphoadenosine 5′-phosphonate (PAP). From these X-ray structures shown in Figure 2, it was determined that 3 makes 98 contacts with the central cavity of SmSULT-OR, with the most important interactions being van der Waals interactions with F39, F153, and M233 as well as three hydrogen bonds with D91, T157, and D144. Specifically, D91 forms an H-bond with the benzyl alcohol, T157 with the nitro group, and D144 with N12. As shown in Figure 2B, lipophilic pockets above and below the central binding cavity of 3 were identified. Additionally, phylogenetic analysis found homologous sulfotransferases for S. haematobium and S. japonicum with SmSULT-OR.
Figure 2.
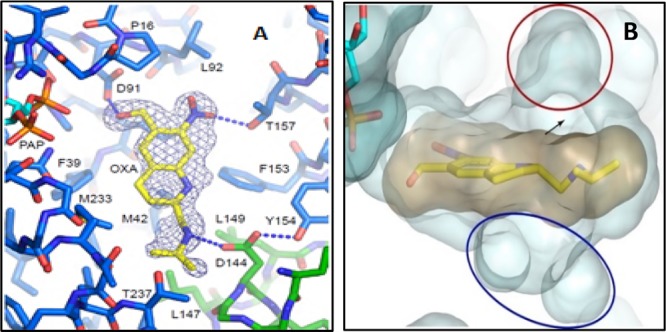
(A) Oxamniquine 3 bound to SmSULT-OR with key contacts. (B) Lipophilic pockets (blue and red) above and below OXA structure.
Despite significant sequence identity shared between SmSULT-OR and the sulfotransferases in S. haematobium (71%) and S. japonicum (58%), 3 is effective only against S. mansoni. Alignment of the crystal structures indicates that the three enzymes have the same catalytic mechanism, but sequence variations in the active site such as a phenylalanine in SmSULT-OR (F39) to tyrosine in the others and a glycine in SmSULT-OR (G143) to valine in S. japonicum were predicted to negatively impact compound 3’s binding and efficacy for the resistant species.5,6 Using a genetic, molecular biology, and biochemistry approach, we demonstrated that a schistosome sulfotransferase was responsible for the mode of action of oxamniquine.5 Comparison of the DNA sequence of the sulfotransferase from a susceptible worm with the sequence from a resistant worm identified the deletion of a glutamate at position 142 as responsible for drug resistance. This was confirmed by a functional assay and in the crystal structure of the sulfotransferase,5 which showed that deletion of glutamate 142 would likely disrupt an α-helix containing aspartate 144 that forms a hydrogen bond to oxamniquine. Current treatment for schistosomiasis uses a praziquantel monotherapy, and a recent review article provides evidence for schistosomes with decreased praziquantel sensitivity n the field in Africa.7 Thus, a structure-based design strategy (Figure 3) of oxamniquine derivatives, to be used alongside praziquantel as a combination therapy, could possibly surmount developing praziquantel resistance as the two drugs act on different targets.
Figure 3.
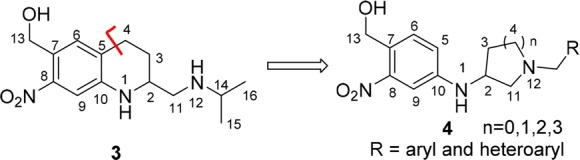
Design of novel analog subtype 4.
To date, the development of novel antischistosomal agents has focused on a variety of small molecule approaches,8,9 including statins,10 cysteine proteases,11 anticancer and kinase targets,12−14 and natural products,15 as examples. Additional analog design approaches have focused on FDA registered drugs, such as 1 or 3, as either starting points for further derivatization and/or as the basis for SAR studies.16−21 More recently, ruthenocenyl- and ferrocenyl-based organometallic oxamniquine conjugates have also been described.22 Given the high production costs and diminishing supply of oxamniquine, partially due to a biotransformation hydroxylation process,23,24 our approach focused on developing a novel small molecule that had efficacious broad-range antischistosomal activity and favorable “drug-like” physicochemical properties and ultimately provided an opportunity for a simplified and efficient synthesis approach. Thus, the goal of our research program was to initiate a structure-based drug design approach based on the X-ray structural data of SmSULT-OR and compound 3, as well as the structural information on S. haematobium and S. japonicum to identify a novel small molecule that would be capable of effective killing activity across all three species of Schistosoma and show activity against 1 resistant forms. Herein we report the design, synthesis, and in vitro evaluation of novel analogs of compound 3, lead compounds of which have been shown to be efficacious against all three species of Schistosoma.
To identify novel, broad-acting antischistosomal agents of general structure 4, we envisioned structural modifications to 3 to accomplish the following (Figure 3): (1) maintain the required 4-amino-2-nitro-benzyl alcohol moiety based on the pro-drug sulfotransferase mechanism associated with compound 3, (2) remove an element of rigidity by removing the tetrahydroquinoline ring of 3 and introduce two rotatable bonds between C10–N1 and N1–C2, (3) install various heterocyclic rings between C2 to N12 to explore the structural effects of different ring sizes, and (4) introduce lipophilic groups (R = aryl and heteroaryl) off of the N12 position to possibly access the lipophilic regions highlighted above in Figure 2B.
In addition to these structure-based drug design objectives, we also incorporated in silico drug-like physicochemical property calculations and in silico molecular modeling and docking studies to aid in compound design cycles. Thus, we aimed to maintain favorable “drug-like” physiochemical properties (LogP, tPSA, MW, and number of hydrogen bond donors/acceptors) across all analogs to maintain good solubility and ADME properties.25,26
The analogs prepared for these studies were synthesized as highlighted in Scheme 1. This synthesis strategy was designed to accomplish two goals: (1) to have flexibility to incorporate numerous N-Boc-cyclic amine templates for structural diversity and (2) to allow for late-stage diversification of the R groups through reductive amination, both of which would support evaluation and SAR development across numerous analogs. Starting from commercially available 4-bromo-2-nitrobenzoic acid 5, reduction with BH3-THF27 followed by protection of the resulting alcohol as a TBS ether produced the desired compound 6.28 Compound 6 was then used as a common intermediate for Buchwald–Hartwig amination conditions29,30 with a variety of commercially available N-Boc-protected diamines, producing a diverse set of cyclic amine templates (compound 7) in moderate to good yields (50–80%).
Scheme 1. General Synthesis Route To Prepare Compound 4.

Reagents and conditions: (a) BH3 THF, THF, 93%; (b) TBSCI, imidazole, DCM, 81%; (c) pd(OAc)2, BINAP, Cs2CO3, 1,4-dioxane, N-Boc-heterocyclic amine, 50–80%; (d) TBAF, THF, 90–98%; (e) BF3OEt2, DCM, −10 °C to r.t., 1 h, 71–99%; (f) NaBH(OAc)3, 1,2-DCE, r.t., RCHO, 50–90%.
Simultaneous deprotection of the N-Boc and TBS-ether groups in compound 7 under a variety of conditions screened proved to be problematic and produce complex product mixtures. Thus, a two-step deprotection sequence utilizing TBAF-mediated removal of the TBS group (90–98%), followed by BF3OEt2 deprotection of the N-Boc group31 was employed. This two-step transformation cleanly produced the desired amine 8, which was then further functionalized via reductive amination conditions with various aldehydes to produce the final analogs (template 4) for screening. All analogs were prepared as racemic mixtures.
In order to test our central hypothesis in our design strategy, our first research objective was to explore the effects of various heterocyclic ring cores and a small number of different R-groups had on S. mansoni. Table 1 highlights our initial investigations focused on the 3-aminopyrrolidine (compounds 9a–9g) and 3-aminopiperidine (compounds 10a–10g) templates. We were encouraged with the initial result showing the secondary amine derivative of the 3-aminopiperidine core, compound 10a, displayed moderate activity (50%) in killing S. mansoni. Interestingly, the corresponding structural analog in the 3-aminopyrrolidine series (compound 9a) was found to be significantly less active than 3 or 10a. These initial results prompted us to prepare a number of analogs with substitution on the nitrogen atoms of the piperidine and pyrrolidine rings. Small alkyl side chains (isopropyl) showed a decrease in S. mansoni killing activity relative to compound 3, as highlighted by analogs 9b and 10b. Increasing the size and aromatic nature of the R-group produced some interesting results. Benzyl groups showed a marked increase in S. mansoni killing activity for both series as observed with compounds 9c and 10c. Additionally, heterocyclic groups such as pyridines or imidazoyl (9d–e and 10d–e) were not well tolerated for either core; however, the 3-indolylmethyl substitution on the 3-aminopyrrolidine core (compound 9f) showed equivalent activity to 3. Finally, as a direct comparison to the secondary piperidine amine 10a, increasing the size of the heterocyclic ring to an azepane or decreasing the size of the ring to the corresponding azetidine significantly decreased the activity in killing S. mansoni (see SI Table S1). Increasing the length of the R group to phenethyl also showed a decrease in activity (9g and 10g).
Table 1. SAR Data on Worm Killing of S. mansoni.


Compounds were tested against adult male S. mansoni (S. m.) worms in vitro. All compounds were tested at a final concentration of 143 μM. Percent killing value and sd are reported. All screens were performed in experimental and biological triplicate. Positive control, compound 3 kills 85% ± 15 of S. mansoni parasites in vitro.
Following these results, we expanded our investigations to include 4-aminopiperidines derivatives as well as the 3-aminopiperidines and 3-aminopyrrolidine cores. We explored the effects of benzyl substitution across all three series, testing for activity on S. mansoni. As highlighted in Table 2, across the series, benzyl derivatives substituted with certain polar groups (i.e., −NO2, −CN, −OCH3) or halogens in the 2- or 4-position showed a marked decrease across all three templates when compared to the unsubstituted benzyl derivatives (9c and 10c). However, 2-trifluoromethyl (12a), 3-trifluoromethyl (11h and 12d), 2-trifluoromethoxy (11d), and 4-trifluoromethoxy (11g) derivatives were shown to exhibit high activity against S. mansoni depending on the specific amine core. The trend appears to show preference for lipophilic moieties, which is consistent with the tyrosine and phenylalanine residues occupying the binding pocket for this side chain. To test the limits of these trends, bis-2,4-trifluoromethyl, 2-methyl, and 4-tert-butyl substitutions of the 3-aminopiperidine core were prepared; however, they failed to exhibit any substantial killing activity for S. mansoni (see SI Table S1).
Table 2. SAR Data on Worm Killing of S. mansoni (S. m.), S. haematobium (S. h.), and S. japonicum (S. j.)a.


Percent killing value is reported for S. mansoni, S. haematobium, and S. japonicum worms in vitro. All derivatives were solubilized in 100% DMSO and administered at a final concentration of 143 μM per well for all assays. All screens were performed in experimental and biological triplicate. Positive control, compound 3 kills 85% ± 15 of S. mansoni parasites in vitro. ND, not determined.
Based on the SAR results above, compounds 9f, 11d, 11f, 11h, 12a–12d, and 13b–13c were screened against S. haematobium and if active, S. japonicum (Table 2). Compound 3 is completely inactive against S. haematobium and S. japonicum. The 3-aminopyrrolidine analogs 9f, 11f, and 11h showed no appreciable activity against S. haematobium, while the corresponding p-OCF3 analog, 11g, showed modest activity. Unfortunately, 11g was inactive against S. japonicum, and the 2-OCF3 derivative 11d was inactive against S. haematobium. In the 3-aminopiperdine series, 12a and 12c showed promising activity against S. haematobium, and compound 12a was found to kill >80% of S. japonicum worms at the end of the 12-day study. The most active 4-aminopiperidine analogs in S. mansoni (13b and 13c) were found to be void of any appreciable activity against S. haematobium.
The worm killing SAR differences can be influenced by factors outside of direct SmSULT-OR binding interactions, such as kinetics of the sulfotransferase process, membrane permeability, and metabolic stability. However, given the structural and property similarities between the different chemical series investigated, we rationalized some of the differences observed in S. mansoni SAR through modeling and docking studies. Docking of all 9–13 compounds from Tables 1 and 2 (both S- and R-enantiomers) provided docking scores partially consistent with the observed SAR in S. mansoni, which suggested a preference for the −CF3 substitution (see Table S3 in SI). A selection of some of the best performing compounds in the docking studies are overlaid in Figure 4 (11f, 12a, 12d, and 13b), suggesting crucial π–π stacking interactions of the phenyl groups with the F39 and F43 residues, while the carboxyl group of D144 forms a salt bridge with the pyrrolidine or piperidine nitrogen. Conversely, some compounds with para substitution, namely, 11e and 11j have also received similar scores; however, they performed poorly in the S. mansoni killing assay. The sequence variations in the active site for S. haematobium and S. japonicum discussed earlier might cause preferential binding on specific CF3-phenyl analogs like 12a; however, docking studies on S. haematobium and S. japonicum were inconclusive. This discrepancy might be rationalized by limitations of current force field calculations to account for unique properties of fluorine in small molecule–protein interactions, an observed phenomenon with extensive literature precedent;32 thus, further structural and binding studies are warranted to provide insight into these observed species differences.
Figure 4.
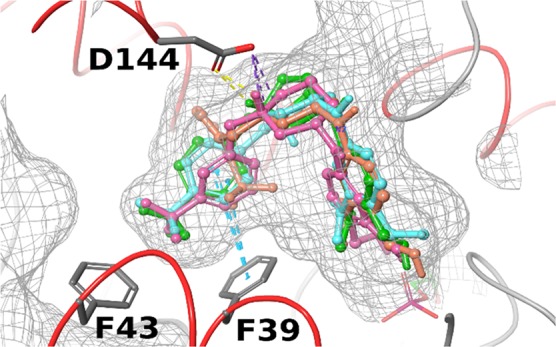
Molecular docking in S. mansoni. Superposition of docking poses of high scoring compounds 11f (green), 12a (orange), 12d (pink), and 13b (cyan) in S. mansoni suggesting −CF3 aryl groups bind to side chains of F39 and F43. Protein surface shown in mesh. D144 forms a salt bridge with protonated nitrogen and hydrogen bond with its hydrogen.
Compound 12a (Figure 5) represents an extremely interesting compound for further follow up studies due to the demonstrated broad-species activity in killing (S. mansoni, 75%; S. haematobium, 40%; and S. japonicum, 83%) and favorable drug-like physicochemical properties (Figure 5).33
Figure 5.

Structure and physiochemical properties of 12a.
Compound 12a is profiled and reported herein as a racemic mixture; however, preparation of the enantiomers and detailed in vitro and in vivo characterization will be reported in due course.
Our most active analogs were soaked in racemic form into native S. mansoni sulfotransferase crystals containing PAP; however, preferential binding of enantiomers (R)-9f and (S)-11f was observed.34 The structures show that compounds were able to enter and bind in the active site without major conformational changes to the unbound enzyme structure. All compounds are observed in the active site overlapping with the oxamniquine position (Figure 6). Interestingly, for compound (S)-11f (Figure 6B), the phenyl ring containing the nitro- and hydroxymethyl groups is rotated 180° such that the nitro moiety is in an alternate location in the active site near R17 and N228. However, this orientation still places the hydroxymethyl moiety in a similar alignment to that observed for (S)-3, consistent with the proposed sulfotransferase mechanism. To clearly depict this observed difference, Figure 6C shows the crystal structure of the SmSULT-OR/compound (S)-11f complex (magenta) overlaid with the (S)-3 (yellow)/SmSULT-OR complex.
Figure 6.
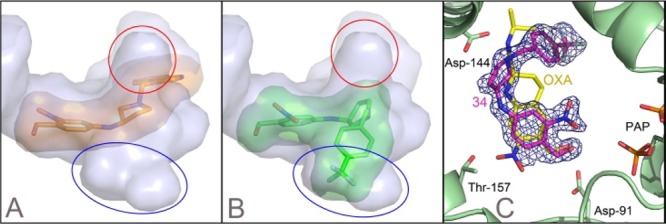
X-ray crystal structures. Derivatives are shown in the SmSULT-OR crystal structure with solvent-accessible surfaces indicated. (A) Compound (R)-9f is shown as orange sticks. The solvent-accessible surface reveals two internal cavities into which additions to the OXA scaffold could be accommodated (red and blue ovals). (B) SmSULT-OR compound (S)-11f. (C) Electron density was calculated as a composite omit map35 for the X-ray crystal structure of the (S)-11f complex (magenta) with SmSULT-OR. Compound (S)-3 (OXA, yellow) from the complex with SmSULT-OR is overlaid for comparison (Protein Data Bank entry 5BYK(36)). Although the (S)-11f nitro group does not overlay the OXA position, the hydroxymethyl group matches the position observed with bound OXA (some atoms have been removed for clarity).
We speculate that the alternate orientation for these two compounds occurs because the crystal structure contains the sulfate-depleted cosubstrate PAP where the absence of the sulfate enlarges the binding pocket. When the active cosubstrate 3′-phosphoadenosine-5′-phosphosulfate (PAPS) is bound, we expect the sulfate group will repel the alternate nitro position and the compounds will adopt the previously observed oxamniquine position. Additional observations in the crystal structures include a partially ordered (S)-11f molecule bound nonspecifically between sulfotransferase molecules at crystal packing interfaces and an oxidized M233 in the active site of the 3 complex structure. The nonspecific binding of compounds at the crystal packing interfaces appears to be an artifact caused by using a saturating concentration of the compounds to soak into the crystals. Oxidation of the methionine likely occurred with crystals stored at 22 °C over several weeks, while the protection from the reducing agent included in the protein preparation decreased over time.
As previously mentioned, the design and SAR of these analogs focused on manipulating the size and electronic nature of the N-benzyl substituent, along with a flexible amine core that allowed rotation of the benzyl moiety into the lipophilic pockets not occupied by 3. Gratifyingly, the crystal structures of SmSULT-OR of analogs (R)-9f and (S)-11f provide evidence to support our design hypothesis focused on accessing the lipophilic regions (red and blue ovals in Figure 2B) not occupied by 3.
In conclusion, a new chemical series of anti-schistosomal agents has been developed. Structure-based drug design guided by X-ray crystal structure analysis and SAR data from worm killing assays produced a number of novel small molecules with killing activity against S. mansoni, while a small number of analogs also showed varying levels of additional activity against S. haematobium and S. japonicum. To our knowledge, compound 12a is the first example of an oxamniquine derivative active against all three species of Schistosoma. Our efforts are ongoing to find an analog that is more active against S. haematobium while maintaining drug-like physicochemical properties. We will, in future reports, continue our exploration of the side chains and central core motifs and examine the SAR of the nitro group. Ideally, an anti-schistosomal agent will be obtained that can be used in combination therapy with 1 to prevent resistance.
Glossary
ABBREVIATIONS
- OXA
oxamniquine;
- N-Boc
N-tert-butylcarbamate
- TBAF
tetrabutylammonium fluoride;
- 1,2-DCE
1,2-dichloroethane
- DCM
dichloromethane
- S. m.
S. mansoni
- S. h.
S. haematobium
- S. j.
S. japonicum
- SmSULT-OR
S. mansoni sulfotransferase
- ND
not determined.
Biographies
Philip T. LoVerde received his Ph.D. in Epidemiologic Science from the University of Michigan under the direction of Profs. John Burch and Henry van der Schalie. His research interests are in host−parasite interactions, especially those that involve the human blood fluke, Schistosoma. His current research involves vaccine development, role of signal transduction in schistosome−host interactions, interplay between male and female parasites that results in female reproductive development, role of host genes in infection outcomes, genomics and genetic approach to identifying drug resistant genes, and a rational approach to novel drug development. He has published over 185 papers.
Stanton F. McHardy graduated from the University of Utah (Ph.D.) and served as a Pfizer Post-Doctoral Fellow with Professor Gary E. Keck. In 1996, Stan joined the Neuroscience group at Pfizer Global Research in Connecticut, focusing on Addiction, Schizophrenia, Cognition, and ADHD. Stan helped advance clinically investigated candidates in multiple programs and, as Associate Director, managed the CNS hit-to-lead efforts. In 2012, Stan became the Director of the Center for Innovative Drug Discovery (CIDD) at UT San Antonio. In the CIDD, Stan’s collaborative programs focus on the discovery of small molecule therapeutics for a variety of disease areas.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.8b00257.
Experimental procedures and data for all new compounds synthesized, schistosomal assay description and additional S. mansoni data, X-ray structural data, molecular modeling and docking studies, and copies of 2D NMR spectra for compound 12a (PDF)
Author Contributions
○ These two Ph.D. student authors contributed equally to this work. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The research was supported by a grant to P.T.L. and P.J.H. from the NIH, NIAID R01 AI115691. P.J.H. (AQ-1399) was funded by The Welch Foundation. The X-ray Crystallography Core Laboratory is a part of the Institutional Research Cores at the University of Texas Health Science Center at San Antonio (UT Health San Antonio) supported by the Office of the Vice President for Research and the Mays Cancer Center (NIH P30 CA054174). This work is based upon research conducted at the Northeastern Collaborative Access Team beamlines, which are funded by the National Institute of General Medical Sciences from the National Institutes of Health (P41 GM103403). This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. Schistosome material was provided by BRI via the NIAID schistosomiasis resource center under NIH-NIAID Contract No. HHSN272201000005I.
The authors declare no competing financial interest.
Supplementary Material
References
- King C. H.; Dickman K.; Tisch D. J. Reassessment of the cost of chronic helmintic infection: a meta-analysis of disability-related outcomes in endemic schistosomiasis. Lancet 2005, 365, 1561–1569. 10.1016/S0140-6736(05)66457-4. [DOI] [PubMed] [Google Scholar]
- Doenhoff M. J.; Cioli D.; Utzinger J. Praziquantel: mechanisms of action, resistance and new derivatives for schistosomiasis. Curr. Opin. Infect. Dis. 2008, 21, 659–667. 10.1097/QCO.0b013e328318978f. [DOI] [PubMed] [Google Scholar]
- Cioli D.; Pica-Mattoccia L.; Archer S. AntiSchistosomal Drugs: Past, Present··· and Future?. Pharmacol. Ther. 1995, 68, 35–85. 10.1016/0163-7258(95)00026-7. [DOI] [PubMed] [Google Scholar]
- Foster R.; Cheetham B. L. Studies with the schistosomicide oxamniquine (UK-4271) I. Activity in rodents and in vitro. Trans. R. Soc. Trop. Med. Hyg. 1973, 67, 674–684. 10.1016/0035-9203(73)90038-2. [DOI] [PubMed] [Google Scholar]
- Valentim C. L. L.; Cioli D.; Chevalier F. D.; Cao X. H.; Taylor A. B.; et al. Genetic and molecular basis of drug resistance and species-specific drug action in schistosome parasites. Science 2013, 342, 1385–1389. 10.1126/science.1243106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor A. B.; Roberts K. M.; Cao X.; Clark N. E.; Holloway S. P.; Donati E.; Polcaro C. M.; Pica-Mattoccia L.; Tarpley R. S.; McHardy S. F.; Cioli D.; LoVerde P. T.; Fitzpatrick P. F.; Hart P. J. Structural and enzymatic insights into species-specific resistance to schistosome parasite drug therapy. J. Biol. Chem. 2017, 292, 11154–11164. 10.1074/jbc.M116.766527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenberg R. M. New approaches for understanding mechanisms of drug resistance in schistosomes. Parasitology 2013, 140, 1534–1546. 10.1017/S0031182013000231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geary T. G.; Sakanari J.; Caffrey C. R. Anthelmintic drug discovery: into the future. J. Parasitol. 2015, 10, 125–33. 10.1645/14-703.1. [DOI] [PubMed] [Google Scholar]
- Galdinoda Rocha Pitta M.; Galdinoda Rocha Pitta M.; de Melo Rêgo M. J. B.; Galdino S. L. The Evolution of Drugs on Schistosoma Treatment: Looking to the Past to Improve the Future. Mini-Rev. Med. Chem. 2013, 13, 493–508. 10.2174/1389557511313040003. [DOI] [PubMed] [Google Scholar]
- Rojo-Arreola L.; Long T.; Asarnow D.; Suzuki B. M.; Singh R.; Caffrey C. R. Chemical and genetic validation of the statin drug target to treat the helminth disease, schistosomiasis. PLoS One 2014, 9, e87594. 10.1371/journal.pone.0087594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonseca N. C.; da Cruz L. F.; da Silva Villela F.; do Nascimento Pereira G. A.; de Siqueira-Neto J. L.; Kellar D.; et al. Synthesis of a Sugar-Based Thiosemicarbazone Series and Structure-Activity Relationship versus the Parasite Cysteine Proteases Rhodesain, Cruzain, and Schistosoma mansoni Cathepsin B1. Antimicrob. Agents Chemother. 2015, 59, 2666–2677. 10.1128/AAC.04601-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morel M.; Vanderstraete M.; Cailliau K.; Lescuyer A.; Lancelot J.; Dissous C. Compound library screening identified Akt/PKB kinase pathway inhibitors as potential key molecules for the development of new chemotherapeutics against schistosomiasis. Int. J. Parasitol.: Drugs Drug Resist. 2014, 4, 256–66. 10.1016/j.ijpddr.2014.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel G.; Roncal N. E.; Lee P. J.; Leed S. E.; Erath J.; Rodriguez A.; et al. Repurposing human Aurora kinase inhibitors as leads for anti-protozoan drug discovery. MedChemComm 2014, 5, 655–658. 10.1039/C4MD00045E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long T.; Neitz R. J.; Beasley R.; Kalyanaraman C.; Suzuki B. M.; Jacobson M. P.; et al. Structure-Bioactivity Relationship for Benzimidazole Thiophene Inhibitors of Polo-Like Kinase 1 (PLK1), a Potential Drug Target in Schistosoma mansoni. PLoS Neglected Trop. Dis. 2016, 10, e0004356. 10.1371/journal.pntd.0004356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neves B. J.; Andrade C. H.; Cravo P. V. Natural products as leads in schistosome drug discovery. Molecules 2015, 20, 1872–903. 10.3390/molecules20021872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- da Silva V. B. R.; Campos B. R. K.; de Oliveira J. F.; Decout J.-L.; do Carmo Alves de Lima M. Medicinal chemistry of antiSchistosomal drugs: Praziquantel and Oxamniquine. Bioorg. Med. Chem. 2017, 25, 3259–3277. 10.1016/j.bmc.2017.04.031. [DOI] [PubMed] [Google Scholar]
- Pellegrino J.; Pereira L. H.; Mello R. T.; Katz N. Activity of Some Tetrahydro- and Pyrazinoquinolines against Early Developing Forms of Schistosoma mansoni. J. Parasitol. 1974, 60, 723–725. 10.2307/3278748. [DOI] [PubMed] [Google Scholar]
- Filho R. P.; Souza Menezes C. M.; Pinto P. L. S.; Paula G. A.; Brandt C. A.; Silveira M. A. B. Design, synthesis, and in vivo evaluation of oxamniquine methacrylate and acrylamide prodrugs. Bioorg. Med. Chem. 2007, 15, 1229–1236. 10.1016/j.bmc.2006.11.027. [DOI] [PubMed] [Google Scholar]
- Filho S. B.; Gargioni C.; Silva Pinto P. L.; Chiodelli S. G.; Gurgel Vellosa S. A.; Silva R. M. da; et al. Synthesis and evaluation of new oxamniquine derivatives. Int. J. Pharm. 2002, 233, 35–41. 10.1016/S0378-5173(01)00917-6. [DOI] [PubMed] [Google Scholar]
- Sadhu P. S.; Kumar S. N.; Chandrasekharam M.; Pica-Mattoccia L.; Cioli D.; Rao V. J. Synthesis of new praziquantel analogues: Potential candidates for the treatment of schistosomiasis, P. S. Bioorg. Med. Chem. Lett. 2012, 22, 1103–1106. 10.1016/j.bmcl.2011.11.108. [DOI] [PubMed] [Google Scholar]
- Kumar S. N.; Sadhu P. S.; Sharma K. K.; Pica-Mattoccia L.; Basso A.; Cioli D.; Rao V. J. Synthesis and antischistosomal activity of new furoxan derivatives of praziquantel. Indian J. Chem., SEC B 2017, 56B, 112–119. [Google Scholar]
- Hess J.; Panic G.; Patra M.; Mastrobuoni L.; Spingler B.; Roy S.; Keiser J.; Gasser G. ACS Infect. Dis. 2017, 3, 645–652. 10.1021/acsinfecdis.7b00054. [DOI] [PubMed] [Google Scholar]
- Straathof A. J. J.; Adlercreutz P.. Applied Biocatalysis; CRC Press, 2003. [Google Scholar]
- Boehm D.; Pryce D. J. Schistosoma haematobium. N. Engl. J. Med. 2001, 344, 1170. 10.1056/NEJM200104123441514. [DOI] [PubMed] [Google Scholar]
- Lu J. J.; Crimin K.; Goodwin J. T.; Crivori P.; Orrenius C.; Xing L.; Tandler P. J.; Vidmar T. J.; Amore B. M.; Wilson A. G. E.; Stouten P. F. W.; Burton P. S. Influence of Molecular Flexibility and Polar Surface Area Metrics on Oral Bioavailability in the Rat. J. Med. Chem. 2004, 47, 6104–6107. 10.1021/jm0306529. [DOI] [PubMed] [Google Scholar]
- Lipinski C. A.; Lombardo F.; Dominy B. W.; Feeney P. J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Delivery Rev. 2001, 46, 3–26. 10.1016/S0169-409X(00)00129-0. [DOI] [PubMed] [Google Scholar]
- Ackermann J.; Bleicher K.; Chomienne O.; Mattei P.; Sander l. O. Novel Bicyclic Sulfonamide Derivativies Which Are L-CPT1 inhibitors. US20100130484A1, 2007.
- Csatayova K.; Davies S. G.; Lee J. A.; Ling K. B.; Roberts P. M.; Russell A. J.; Thomson J. E. Syntheses of trans-SCH-A and cis-SCH-A via a Stereodivergent Cyclopropanation Protocol. Org. Lett. 2010, 12, 3152–3155. 10.1021/ol101295t. [DOI] [PubMed] [Google Scholar]
- Ruiz-Castillo P.; Buchwald S. L. Applications of Palladium-Catalyzed C–N Cross-Coupling Reactions. Chem. Rev. 2016, 116, 12564–12649. 10.1021/acs.chemrev.6b00512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jean L.; Rouden J.; Maddaluno J.; Lasne M. C. Palladium-Mediated Arylations of 3-Aminopiperidines and 3-Aminopyrrolidines. J. Org. Chem. 2004, 69, 8893–8902. 10.1021/jo0487193. [DOI] [PubMed] [Google Scholar]
- Evans E. F.; Lewis N. J.; Kapfer I.; Macdonald G.; Taylor R. J. K. N-tert-Butoxycarbonyl (BOC) Deprotection Using Boron Trifluoride Etherate. Synth. Commun. 1997, 27, 1819–1825. 10.1080/00397919708006783. [DOI] [Google Scholar]
- Gillis E. P.; Eastman K. J.; Hill M. D.; Donnelly D. J.; Meanwell N. A. Applications of Fluorine in Medicinal Chemistry. J. Med. Chem. 2015, 58, 8315–8359. 10.1021/acs.jmedchem.5b00258. [DOI] [PubMed] [Google Scholar]
- Property calculations and data were archived and analyzed using the CDD Vault from Collaborative Drug Discovery (https://www.collaborativedrug.com). Within CDD Vault, ChemAxon’s JChem Base was used for structure searching, chemical database access and management, and chemical property calculations. ChemAxon (http://www.chemaxon.com).
- The evidence for preferentially binding and sulfur transfer of enantiomers of compound 3 was observed in previous work. See refs (5) and (6).
- Terwilliger T. C.; Grosse-Kunstleve R. W.; Afonine P. V.; Moriarty N. W.; Adams P. D.; Read R. J.; Zwart P. H.; Hung L. W. Iterative-build OMIT maps: map improvement by iterative model building and refinement without model bias. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2008, D64, 515–524. 10.1107/S0907444908004319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor A. B.; Pica-Mattoccia L.; Polcaro C. M.; Donati E.; Cao X.; Basso A.; Guidi A.; Rugel A. R.; Holloway S. P.; Anderson T. J.; Hart P. J.; Cioli D.; LoVerde P. T. Structural and functional characterization of the enantiomers of the antiSchistosomal drug oxamniquine. PLoS Neglected Trop. Dis. 2015, 9, e0004132. 10.1371/journal.pntd.0004132. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.