

Abstract
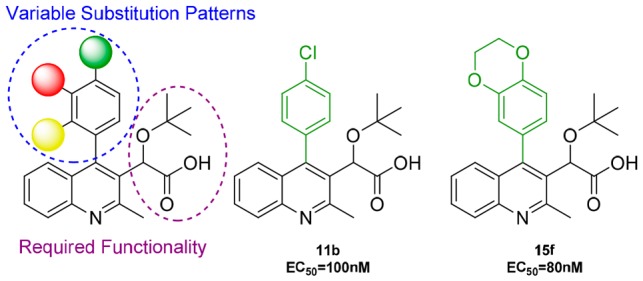
HIV-1 integrase multimerization inhibitors have recently been established as an effective class of antiretroviral agents due to their potent ability to inhibit viral replication. Specifically, quinoline-based inhibitors have been shown to effectively impair HIV-1 replication, highlighting the importance of these heterocyclic scaffolds. Pursuant of our endeavors to further develop a library of quinoline-based candidates, we have implemented a structure–activity relationship study of trisubstituted 4-arylquinoline scaffolds that examined the integrase multimerization properties of substitution patterns at the 4-position of the quinoline. Compounds consisting of substituted phenyl rings, heteroaromatics, or polycyclic moieties were examined utilizing an integrase aberrant multimerization in vitro assay. para-Chloro-4-phenylquinoline 11b and 2,3-benzo[b][1,4]dioxine 15f showed noteworthy EC50 values of 0.10 and 0.08 μM, respectively.
Keywords: HIV integrase, multimerization, quinoline, structure−activity relationship (SAR)
Within the life cycle of HIV-1 there are multiple targets that have been investigated for inhibition of viral replication. These targets have spanned across the full spectrum of the replication cycle from host-cell entry to release and maturation of new virions. Specific stages currently exploited include: (1) cell wall fusion/entry, (2) RNA reverse transcription, (3) DNA integration, and (4) maturation.1
HIV-1 integrase (IN), the enzyme responsible for integration of viral DNA (vDNA) into the host DNA is essential for virus replication and represents an important multifunctional therapeutic target.2 IN contains three main sections: the N-terminus domain (NTD), C-terminus domain (CTD), and the catalytic core domain (CCD).3,4 These three domains work in conjunction to form a tetramer structure where vDNA and host chromosomal DNA are bound to IN.5−8 An additional necessary component for tetramer association and vDNA integration is the cellular chromatin-associated protein lens epithelium-derived growth factor (LEDGF/p75)9 that functions as a tether wherein it binds to both HIV and target chromatin to facilitate integration.10−12
The integration of vDNA occurs in two stages at the active site located within the CCD. The first stage forms the target capture complex, which involves trapping of the target followed by subsequent cleavage of the vDNA 3′ ends. Completion of this leads to a strand transfer complex (STC) that cleaves the target DNA and covalently links the 3′ ends.13 The three amino acid residues required for the strand transfer include aspartates 64 and 116 and glutamate 152. In addition, these residues coordinate with two divalent magnesium cofactors to perform 3′ processing.14
There are currently three FDA approved compounds available for IN via strand transfer inhibition: raltegravir (RAL), elvitegravir (EVG), and dolutegravir (DVG). RAL was developed by Merck and approved in 2007 making it the first treatment available. Following this, Gilead produced EVG and received approval in 2012 with GlaxoSmithKline close thereafter with approval of DVG in 2013. Although these treatments are effective, drug resistant strains have emerged against RAL and EVG due to HIV’s high turnover and mutation rates.15
Development of mutations within the active site of HIV IN makes it pertinent to pursue alternative inhibition sites on the enzyme. HIV IN functions as a tetramer wherein a dimer is formed at the CCD interface for vDNA interaction. Furthermore, LEDGF/p75 interacts at this CCD dimer within an allosteric binding pocket via the C-terminal integrase binding domain.16−18 This binding pocket has become of significant interest as an IN alternative target option.19 Previous studies20−22 have demonstrated that the binding of inhibitors “to this binding pocket” rapidly triggers an aberrant multimerization of the enzyme. A recent crystallographic study23 has suggested the contribution of a third monomer through its CTD. The acetic acid side chain, tert-butyl group, and aromatic properties of the scaffold contribute to the formation of a strong network of hydrogen bonds and electrostatic interactions. This leads to the genesis of large inhibitor-IN multimer complexes that evolves into a nonfunctional open polymer of IN. While the presence of the LEDGF/p75 in the infected cell decreases the efficacy of these inhibitors during the early stage of viral replication,24 there is minimal to no detectable competition from LEDGF/p75 during viral production. Thus, these compounds effectively trigger aberrant IN multimerization within the progeny virions causing severe morphological defect.25 Once multimerized, essential IN interactions with viral RNA are irreversibly compromised, which lead to the release of noninfectious virions.26 Three reported IN multimerization inhibitors include quinoline scaffolds 1,272,25 and 3 (BI 224436)28 (Figure 1).
Figure 1.

Known investigational quinolines utilized for HIV-1 IN inhibition whose EC50 values were previously evaluated utilizing the multimerization assay.
Examination of the crystal structure of 1 within the binding pocket reveals the amino acids of interest in the northern region of the pocket (Figure 2).29 Specifically, the northern aromatic component of the substrate resides in proximity to tryptophan 132 and leucine 102. Therefore, we commenced an investigation to identify relevant stereoelectronic interactions within this pocket. The inhibition data for compounds 1 and 2 led our research group to further investigate a library of 4-aryl quinolines through systematic structure–activity relationship studies using a validated in vitro assay known to accurately predict the antiviral potency for this class of inhibitors.27,30−33 Thus, to survey the quinoline library generated in the present study, we have performed dose response measurements for each synthesized compound using a previously described homogeneous time-resolved fluorescence-based IN multimerization in vitro assay.27,34
Figure 2.
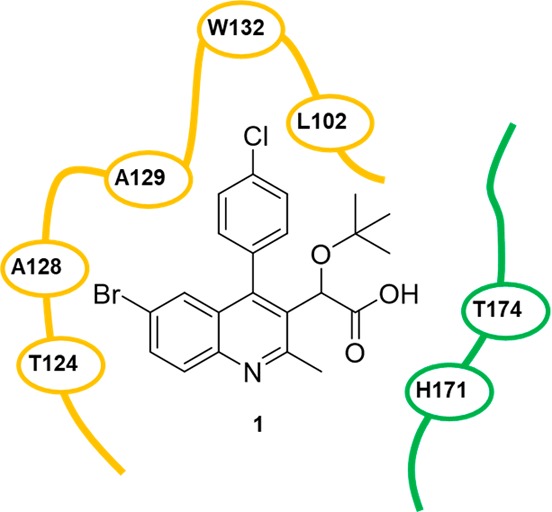
Two-dimensional representation of 1 (PDB: 4gw6) within the target integrase binding pocket. The pocket space is defined with the green and yellow segments, which denote the individual monomer units of the integrase dimer.
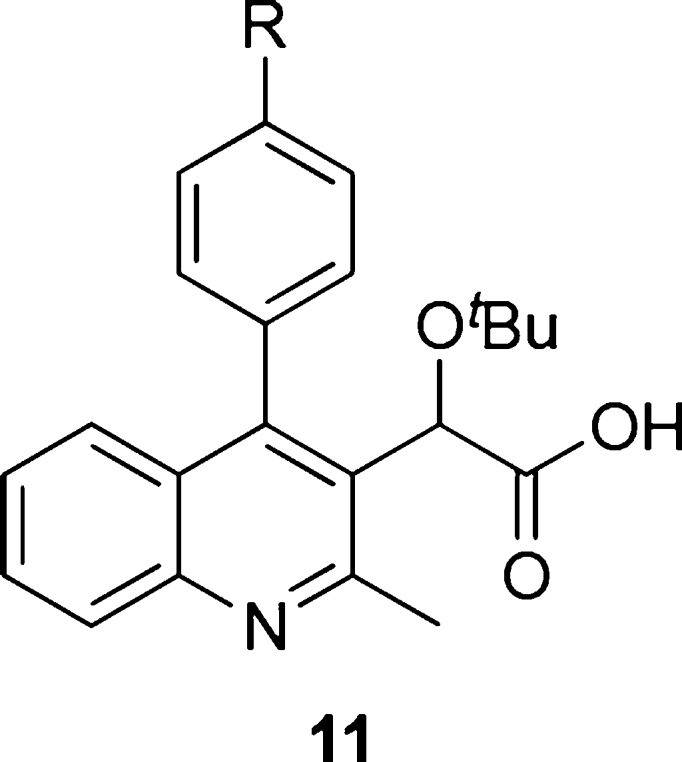
In the present study, the C2 methyl group and C3 acetic acid moiety bearing the tert-butyl ether group of the quinoline scaffold were conserved from original lead structures in the literature (Figure 1). It has been established that the acetic acid moiety participates in crucial hydrogen bond interactions with threonine 174 and histidine 171 (Figure 3). To probe the steric and electronic interactions within the A129-W132-L102 pocket residues, substitution of the C4 aryl group via Suzuki coupling was implemented as the key derivatizing step. Compound 4 was prepared in 65% yield over two steps starting from commercially available 4-hydroxy-2-methylquinoline via ortho-bromination with NBS followed by chloride formation in neat phosphorus oxychloride. Selective halogen-metal exchange of bromide 4 with isopropyl magnesium chloride lithium chloride complex followed by quenching with ethyl chlorooxalate furnished the desired ethyl oxalate (Scheme 1).35 Reduction of the ketone to benzylic alcohol 5 using sodium borohydride in ethanol followed by Finkelstein halide exchange of chlorine for iodine supplied iodoquinoline 6. Installation of the tert-butyl group was accomplished using tert-butyl acetate and perchloric acid to furnish 7, which served as the common scaffold prior to derivatization. Suzuki coupling of 7 with various ortho-, meta-, and para-substituted boronic acids furnished the 4-aryl quinolines 8–10, respectively. Subsequent saponification of the ethyl ester provided the desired substrates 11–13.
Figure 3.
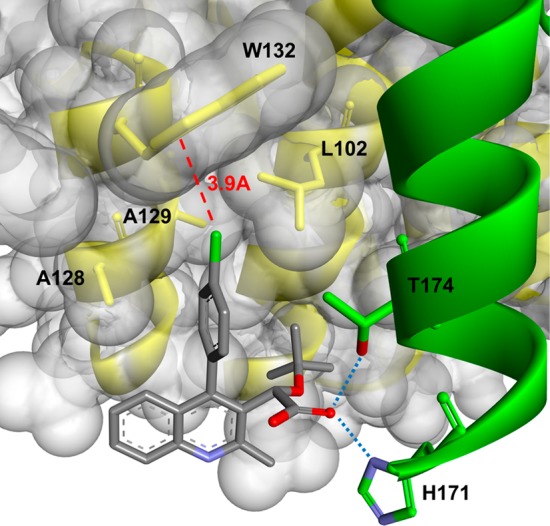
Ribbon representation of 11b within the target integrase binding pocket. The chlorine–W132 distance is marked with a dashed line indicating a favorable distance for Cl−π interactions. Hydrogen bonding interactions are indicated with dotted lines.
Scheme 1. Synthesis of 4-Aryl Quinolines.

Reagents: (i) iPrMgCl·LiCl, CuBr·SMe2, THF, 0 °C, 47%; (ii) NaBH4, THF/EtOH (5:1), 0 °C, 30 min, 77%; (iii) 4 N HCl, THF, rt, 1 h then NaI, MeCN, 90 °C, 16 h, 99%; (iv) HClO4, t-BuOAc, rt, 3 h, 74%; (v) Pd[PPh3]4, K2CO3, Ar–B(OH)2, DMF, 90 °C, 3 h, 38–97%; (vi) 2 N NaOH, EtOH, 60 °C, 3 h, 11–96%.
With the multisubstituted quinolines in hand, we implemented a study to probe for relevant interactions within the hydrophobic pocket of the IN dimer by changing the para substituent (Table 1). The 4-phenylquinoline (11a, R1 = H) was utilized as the reference point for relative comparison of substituted phenyl rings. Substitution for a methyl group (11d, R1 = CH3) resulted in a 6-fold decrease in the EC50 value implying increased ability to cause multimerization. However, substitution of methyl hydrogens with fluorine results in diminished potency as there was only a 2-fold increase in the EC50 value observed for 4-trifluoromethyl 11e. Furthermore, 4-methoxy 11f and 4-trifluoromethoxy 11g followed a similar trend but to a lesser degree. In addition, halogen substitution (entries 2 and 3) showed improvement in potency with para-chloro-4-phenylquinoline 11b having the most effective inhibition among the series with an EC50 of 100 nM. Originally, we postulated that electron density at the aromatic ring was a significant factor, but the presence of chlorine intriguingly interrupted this trend. We examined additional factors including size restrictions, steric interactions, and substrate−π interactions that may contribute significantly to protein affinity. Closer scrutiny of the trends revealed to us that electron density of the ring has minimal, although observable effect, with the size and steric interactions exhibiting the dominating factor for successful protein affinity. For example, 4-cyano 11i had diminished protein binding, which is attributed in part to the rigid, linear geometry of the substituent. We postulate this may result in restricted access to the pocket.
Table 1. IN Aberrant Multimerization Assay of p-Substituted 4-Phenylquinolinesa.
| Entry | Compound | R | EC50 (μM)b |
|---|---|---|---|
| 1 | 11a | H | 1.32 ± 0.53 |
| 2 | 11b | Cl | 0.10 ± 0.02 |
| 3 | 11c | F | 0.49 ± 0.04 |
| 4 | 11d | CH3 | 0.24 ± 0.11 |
| 5 | 11e | CF3 | 0.72 ± 0.06 |
| 6 | 11f | OCH3 | 0.23 ± 0.04 |
| 7 | 11g | OCF3 | 0.35 ± 0.09 |
| 8 | 11h | SCH3 | 0.49 ± 0.01 |
| 9 | 11i | CN | 2.97 ± 0.81 |
| 10 | 11j | COCH3 | 1.39 ± 0.48 |
| 11 | 11k | NHCOCH3 | 8.41 ± 0.73 |
All compounds were demonstrated to have ≥90% chemical purity.
EC50 determined with HTRF-based IN multimerization assay.
Still, steric interactions did not provide sufficient explanation for the increased potency in 11b. Further understanding for diversion from the standard trend can be found in the stabilizing effect of chlorine−π interactions. A theoretical study elaborates on the significance of these interactions wherein chlorine participates in dispersive interactions with the π-systems of phenylalanine, histidine, and tryptophan residues.36 The result of this interaction is a stabilizing effect of −2.01 kcal/mol, attributed to increased protein affinity. As shown in Figure 3, the chlorine resides within 4 Å of tryptophan 132, allowing for this stabilizing Cl−π interaction to occur and providing support for the improved potency.
With the para-substituted phenyl series completed, the ortho- and meta-substituted phenyl coupling partners were examined, and a comparison was made among the candidates (Table 2). Of note is the superiority of para substitution on overall binding of the substrate compared to either ortho or meta. Examination of reported crystal structures provided guidance for the explanation of this trend.23,29,37 Specifically, tryptophan residue 132 resides within 4 Å to para moieties (Figure 3). Movement to the ortho and meta positions places the substituents beyond 4 Å, which attenuates this interaction. For most cases, the meta position results in the most diminished potency with two exceptions to this being methoxy 12f and thiomethyl 12h substituents, both of which have a binding affinity trend of para > meta > ortho. This is attributed to the increased van der Waals radii associated with these substituents. When placed in the ortho position, these moieties are positioned within proximity to alanine 128. It is hypothesized that the increased size of methoxy and thiomethyl displaces the phenyl ring and results in the diminished affinity for the protein. Entries 1–3 have an alternative trend of para > ortho > meta, which supports that electronic contributions do influence protein affinity, but to a lesser degree than steric restrictions and substrate−π interactions.
Table 2. Comparison Series for Phenyl Ring Substitution Patterns.

| Entry | Substituent | para | meta | ortho | |||
|---|---|---|---|---|---|---|---|
| 1 | Cl | 11b | 0.10 ± 0.02 | 12b | 3.79 ± 0.59 | 13b | 0.26 ± 0.04 |
| 2 | F | 11c | 0.49 ± 0.04 | 12c | 2.11 ± 0.59 | 13c | 0.58 ± 0.07 |
| 3 | CH3 | 11d | 0.24 ± 0.11 | 12d | 0.95 ± 0.29 | 13d | 0.51 ± 0.05 |
| 4 | OCH3 | 11f | 0.23 ± 0.04 | 12f | 1.38 ± 0.37 | 13f | 8.39 ± 0.92 |
| 5 | SCH3 | 11h | 0.49 ± 0.01 | 12h | 1.00 ± 0.14 | 13h | 4.51 ± 0.71 |
| 6 | CN | 11i | 2.97 ± 0.81 | 12i | no activity | 13i | 11.14 ± 0.85 |
In addition to the study of phenyl substitution, heterocyclic and bicyclic systems were also examined (Table 3). When testing the biphenyl 15a and carbazole 15b substituents, no activity was observed. This supports the hypothesis that large substrates will exceed the available volume within the binding pocket as defined by tryptophan 132. The most promising candidate from this series was 2,3-benzo[b][1,4]dioxane 15f, which showed an EC50 of 80 nM. This observation led to the hypothesis that larger, multicyclic structures resembling 2 and 3 have the capability to rotate within the pocket allowing for minimization of steric interactions. There are also degrees of freedom available that may further contribute to the increased effectiveness of these molecules. Although 15b contains the same capability, it still occupies a larger three-dimensional space than the other substituents, which may explain the inactivity.
Table 3. Additional 4-Arylquinoline Studies.


To fully confirm that the synthesized compounds induce valid IN multimerization, we have also employed dynamic light scattering (DLS), a laser-based method used for determining the diffusion coefficients of particles in solution. Since the diffusion coefficient depends on the particle size and shape, DLS has been widely used to study protein aggregation.38−41 In previous studies,24,31,34,42−44 we have successfully validated this biophysical technique to observe our inhibitors multimerization properties. Accordingly, in the absence of inhibitor (DMSO control), IN only yielded a background signal centered around 1 nm presumably due to the relatively small size of the fully soluble IN protein (Figure 4A). While the DLS profile of IN with a nonactive compound such as 15a did not significantly differ from the DMSO control (Figure 4B), the addition of two representative active compounds, 11b and 15f (Figure 4C,D, respectively), triggered the appearance of a highly characteristic peak centered at 1000 nm diameter. This peak corresponds to highly multimerized IN, thereby corroborating the validity of the multimerization assay.
Figure 4.
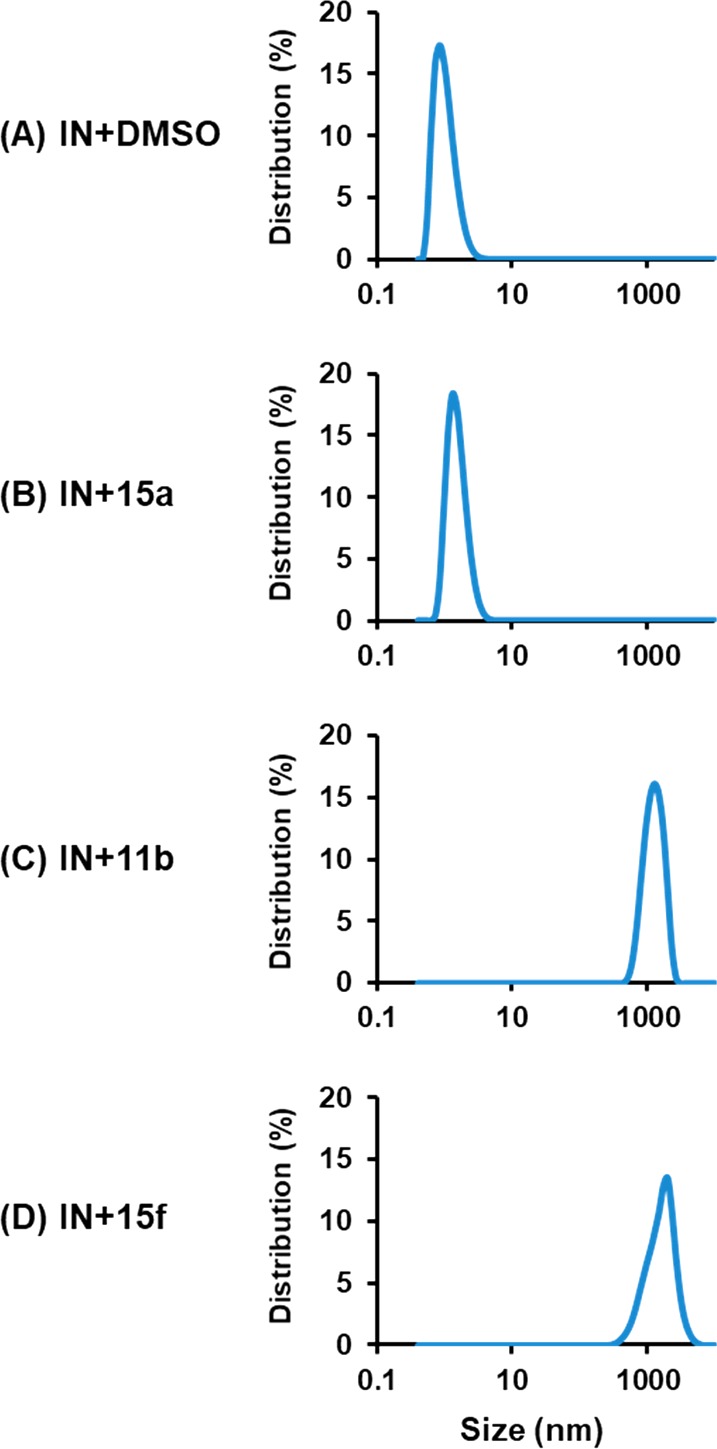
DLS analysis of 15a, 11b, and 15f on multimerization of recombinant HIV-1 IN: (A) IN + DMSO; (B) IN + 15a; (C) IN + 11b; (D) IN + 15f. No detectable signals were recorded in control experiments with the compounds without IN (not shown).
In conclusion, a series of multisubstituted quinolines was prepared in an eight-step synthesis and was examined for their ability to trigger HIV-1 IN multimerization via binding to an allosteric site. The focus was on the substitution pattern of the 4-phenyl moiety and incorporation of heteroaromatics or polycyclics at position 4. In these studies, para-chloro-4-phenylquinoline 11b and 2,3-benzo[b][1,4]dioxine 15f showed the highest potency. This study has provided relevant information regarding defining factors for candidate design within the hydrophobic region of the targeted binding pocket of HIV-1 IN. Based on this data, we are currently investigating substitution at the 5-, 6-, 7-, and 8-positions of the quinoline ring system for further SAR studies.
Acknowledgments
We thank the University of Southern Mississippi Office of the Vice President for Research for financial support, laboratory space, and facilities. M.G.D. is a 2015 and 2018 recipient of the Lucas Award for Faculty Excellence sponsored by the USM Office of the Provost. We thank Dr. Rangachari for access to the DLS instrument.
Glossary
ABBREVIATIONS
- CCD
catalytic core domain
- CTD
carbon terminal domain
- DLS
dynamic light scattering
- DMSO
dimethylsulfoxide
- DVG
dolutegravir
- EC50
effective concentration
- EVG
elvitegravir
- HTRF
homogeneous time-resolved fluorescence
- IN
HIV-1 integrase
- LEDGF
lens epithelium-derived growth factor
- NBS
N-bromosuccinimide
- NTD
N-terminal domain
- RAL
raltegravir
- STC
strand transfer complex
- vDNA
viral DNA
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.8b00269.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
This work was supported by the NIH National Institute of Allergy and Infectious Disease grant number R21AI127282 (J.J.K.) and the USM INTERFACE National Science Foundation National Research Traineeship grant number 1449999 (N.G.J., A.P.H., and J.D.H.). Funding for the Bruker UltraShield Plus 400 MHz NMR and ThermoFinnigan LXQ ESI-LC/MS used in this research was provided by National Science Foundation Major Research Instrumentation under Grant Numbers 0840390 and 0639208, respectively.
The authors declare no competing financial interest.
Supplementary Material
References
- Barre-Sinoussi F.; Ross A. L.; Delfraissy J.-F. Past, present and future: 30 years of HIV research. Nat. Rev. Microbiol. 2013, 11, 877–883. 10.1038/nrmicro3132. [DOI] [PubMed] [Google Scholar]
- Cherepanov P. Integrase illuminated. EMBO Rep. 2010, 11, 328. 10.1038/embor.2010.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng L.; Larue R. C.; Slaughter A.; Kessl J. J.; Kvaratskhelia M. HIV-1 Integrase Multimerization as a Therapeutic Target. Curr. Top. Microbiol. Immunol. 2015, 389, 93–119. 10.1007/82_2015_439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelman A.; Cherepanov P. Retroviral integrase structure and DNA recombination mechanism. Microbiol. Spectrum 2014, 2, 1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnan L.; Engelman A. Retroviral integrase proteins and HIV-1 DNA integration. J. Biol. Chem. 2012, 287, 40858–40866. 10.1074/jbc.R112.397760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherepanov P.; Maertens G. N.; Hare S. Structural insights into the retroviral DNA integration apparatus. Curr. Opin. Struct. Biol. 2011, 21, 249–256. 10.1016/j.sbi.2010.12.005. [DOI] [PubMed] [Google Scholar]
- Li X.; Krishnan L.; Cherepanov P.; Engelman A. Structural biology of retroviral DNA integration. Virology 2011, 411, 194–205. 10.1016/j.virol.2010.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelman A.; Cherepanov P. The structural biology of HIV-1: mechanistic and therapeutic insights. Nat. Rev. Microbiol. 2012, 10, 279–290. 10.1038/nrmicro2747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherepanov P.; Maertens G.; Proost P.; Devreese B.; Van Beeumen J.; Engelborghs Y.; De Clercq E.; Debyser Z. HIV-1 Integrase Forms Stable Tetramers and Associates with LEDGF/p75 Protein in Human Cells. J. Biol. Chem. 2003, 278, 372–381. 10.1074/jbc.M209278200. [DOI] [PubMed] [Google Scholar]
- Marshall H. M.; Ronen K.; Berry C.; Llano M.; Sutherland H.; Saenz D.; Bickmore W.; Poeschla E.; Bushman F. D. Role of PSIP1/LEDGF/p75 in lentiviral infectivity and integration targeting. PLoS One 2007, 2, e1340. 10.1371/journal.pone.0001340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shun M.-C.; Raghavendra N. K.; Vandegraaff N.; Daigle J. E.; Hughes S.; Kellam P.; Cherepanov P.; Engelman A. LEDGF/p75 functions downstream from preintegration complex formation to effect gene-specific HIV-1 integration. Genes Dev. 2007, 21, 1767–1778. 10.1101/gad.1565107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciuffi A.; Llano M.; Poeschla E.; Hoffmann C.; Leipzig J.; Shinn P.; Ecker J. R.; Bushman F. A role for LEDGF/p75 in targeting HIV DNA integration. Nat. Med. 2005, 11, 1287–1289. 10.1038/nm1329. [DOI] [PubMed] [Google Scholar]
- Lesbats P.; Engelman A. N.; Cherepanov P. Retroviral DNA Integration. Chem. Rev. 2016, 116, 12730–12757. 10.1021/acs.chemrev.6b00125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldgur Y.; Dyda F.; Hickman A. B.; Jenkins T. M.; Craigie R.; Davies D. R. Three new structures of the core domain of HIV-1 integrase: an active site that binds magnesium. Proc. Natl. Acad. Sci. U. S. A. 1998, 95, 9150–9154. 10.1073/pnas.95.16.9150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quashie P. K.; Mesplede T.; Wainberg M. A. Evolution of HIV integrase resistance mutations. Curr. Opin. Infect. Dis. 2013, 26, 43–49. 10.1097/QCO.0b013e32835ba81c. [DOI] [PubMed] [Google Scholar]
- Cherepanov P.; Devroe E.; Silver P. A.; Engelman A. Identification of an Evolutionarily Conserved Domain in Human Lens Epithelium-derived Growth Factor/Transcriptional Co-activator p75 (LEDGF/p75) That Binds HIV-1 Integrase. J. Biol. Chem. 2004, 279, 48883–48892. 10.1074/jbc.M406307200. [DOI] [PubMed] [Google Scholar]
- Cherepanov P.; Ambrosio A. L. B.; Rahman S.; Ellenberger T.; Engelman A. Structural basis for the recognition between HIV-1 integrase and transcriptional coactivator p75. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 17308–17313. 10.1073/pnas.0506924102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKee C. J.; Kessl J. J.; Shkriabai N.; Dar M. J.; Engelman A.; Kvaratskhelia M. Dynamic Modulation of HIV-1 Integrase Structure and Function by Cellular Lens Epithelium-derived Growth Factor (LEDGF) Protein. J. Biol. Chem. 2008, 283, 31802–31812. 10.1074/jbc.M805843200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Santo R. Inhibiting the HIV Integration Process: Past, Present, and the Future. J. Med. Chem. 2014, 57, 539–566. 10.1021/jm400674a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelman A.; Kessl J. J.; Kvaratskhelia M. Allosteric inhibition of HIV-1 integrase activity. Curr. Opin. Chem. Biol. 2013, 17, 339–345. 10.1016/j.cbpa.2013.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christ F.; Shaw S.; Demeulemeester J.; Desimmie B. A.; Marchand A.; Butler S.; Smets W.; Chaltin P.; Westby M.; Debyser Z.; Pickford C. Small-molecule inhibitors of the LEDGF/p75 binding site of integrase block HIV replication and modulate integrase multimerization. Antimicrob. Agents Chemother. 2012, 56, 4365–4374. 10.1128/AAC.00717-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsiang M.; Jones G. S.; Niedziela-Majka A.; Kan E.; Lansdon E. B.; Huang W.; Hung M.; Samuel D.; Novikov N.; Xu Y.; Mitchell M.; Guo H.; Babaoglu K.; Liu X.; Geleziunas R.; Sakowicz R. New Class of HIV-1 Integrase (IN) Inhibitors with a Dual Mode of Action. J. Biol. Chem. 2012, 287, 21189–21203. 10.1074/jbc.M112.347534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta K.; Turkki V.; Sherrill-Mix S.; Hwang Y.; Eilers G.; Taylor L.; McDanal C.; Wang P.; Temelkoff D.; Nolte R. T.; Velthuisen E.; Jeffrey J.; Van Duyne G. D.; Bushman F. D. Structural basis for inhibitor-induced aggregation of HIV integrase. PLoS Biol. 2016, 14, e1002584. 10.1371/journal.pbio.1002584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng L.; Dharmarajan V.; Serrao E.; Hoyte A.; Larue R. C.; Slaughter A.; Sharma A.; Plumb M. R.; Kessl J. J.; Fuchs J. R.; Bushman F. D.; Engelman A. N.; Griffin P. R.; Kvaratskhelia M. The Competitive Interplay between Allosteric HIV-1 Integrase Inhibitor BI/D and LEDGF/p75 during the Early Stage of HIV-1 Replication Adversely Affects Inhibitor Potency. ACS Chem. Biol. 2016, 11, 1313–1321. 10.1021/acschembio.6b00167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurado K. A.; Wang H.; Slaughter A.; Feng L.; Kessl J. J.; Koh Y.; Wang W.; Ballandras-Colas A.; Patel P. A.; Fuchs J. R.; Kvaratskhelia M.; Engelman A. Allosteric integrase inhibitor potency is determined through the inhibition of HIV-1 particle maturation. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 8690–8695. 10.1073/pnas.1300703110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessl J. J.; Kutluay S. B.; Townsend D.; Rebensburg S.; Slaughter A.; Larue R. C.; Shkriabai N.; Bakouche N.; Fuchs J. R.; Bieniasz P. D.; Kvaratskhelia M. HIV-1 integrase binds the viral RNA genome and is essential during virion morphogenesis. Cell 2016, 166, 1257–1268. 10.1016/j.cell.2016.07.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessl J. J.; Jena N.; Koh Y.; Taskent-Sezgin H.; Slaughter A.; Feng L.; de Silva S.; Wu L.; Le Grice S. F. J.; Engelman A.; Fuchs J. R.; Kvaratskhelia M. Multimode, Cooperative Mechanism of Action of Allosteric HIV-1 Integrase Inhibitors. J. Biol. Chem. 2012, 287, 16801–16811. 10.1074/jbc.M112.354373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fader L. D.; Malenfant E.; Parisien M.; Carson R.; Bilodeau F.; Landry S.; Pesant M.; Brochu C.; Morin S.; Chabot C.; Halmos T.; Bousquet Y.; Bailey M. D.; Kawai S. H.; Coulombe R.; LaPlante S.; Jakalian A.; Bhardwaj P. K.; Wernic D.; Schroeder P.; Amad M.; Edwards P.; Garneau M.; Duan J.; Cordingley M.; Bethell R.; Mason S. W.; Bös M.; Bonneau P.; Poupart M.-A.; Faucher A.-M.; Simoneau B.; Fenwick C.; Yoakim C.; Tsantrizos Y. Discovery of BI 224436, a Noncatalytic Site Integrase Inhibitor (NCINI) of HIV-1. ACS Med. Chem. Lett. 2014, 5, 422–427. 10.1021/ml500002n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng L.; Sharma A.; Slaughter A.; Jena N.; Koh Y.; Shkriabai N.; Larue R. C.; Patel P. A.; Mitsuya H.; Kessl J. J.; Engelman A.; Fuchs J. R.; Kvaratskhelia M. The A128T Resistance Mutation Reveals Aberrant Protein Multimerization as the Primary Mechanism of Action of Allosteric HIV-1 Integrase Inhibitors. J. Biol. Chem. 2013, 288, 15813–15820. 10.1074/jbc.M112.443390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurado K. A.; Wang H.; Slaughter A.; Feng L.; Kessl J. J.; Koh Y.; Wang W.; Ballandras-Colas A.; Patel P. A.; Fuchs J. R.; Kvaratskhelia M.; Engelman A. Allosteric integrase inhibitor potency is determined through the inhibition of HIV-1 particle maturation. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 8690–8695. 10.1073/pnas.1300703110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma A.; Slaughter A.; Jena N.; Feng L.; Kessl J. J.; Fadel H. J.; Malani N.; Male F.; Wu L.; Poeschla E.; Bushman F. D.; Fuchs J. R.; Kvaratskhelia M. A new class of multimerization selective inhibitors of HIV-1 integrase. PLoS Pathog. 2014, 10, e1004171. 10.1371/journal.ppat.1004171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnard D.; Le Rouzic E.; Eiler S.; Amadori C.; Orlov I.; Bruneau J.-M.; Brias J.; Barbion J.; Chevreuil F.; Spehner D.; Chasset S.; Ledoussal B.; Moreau F.; Saib A.; Klaholz B. P.; Emiliani S.; Ruff M.; Zamborlini A.; Benarous R. Structure-function analyses unravel distinct effects of allosteric inhibitors of HIV-1 integrase on viral maturation and integration. J. Biol. Chem. 2018, 293, 6172–6186. 10.1074/jbc.M117.816793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burlein C.; Wang C.; Xu M.; Bhatt T.; Stahlhut M.; Ou Y.; Adam G. C.; Heath J.; Klein D. J.; Sanders J.; Narayan K.; Abeywickrema P.; Heo M. R.; Carroll S. S.; Grobler J. A.; Sharma S.; Diamond T. L.; Converso A.; Krosky D. J. Discovery of a Distinct Chemical and Mechanistic Class of Allosteric HIV-1 Integrase Inhibitors with Antiretroviral Activity. ACS Chem. Biol. 2017, 12, 2858–2865. 10.1021/acschembio.7b00550. [DOI] [PubMed] [Google Scholar]
- Kessl J. J.; Sharma A.; Kvaratskhelia M. Methods for the Analyses of Inhibitor-Induced Aberrant Multimerization of HIV-1 Integrase. Methods Mol. Biol. 2016, 1354, 149–164. 10.1007/978-1-4939-3046-3_10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babudri F.; Fiandanese V.; Marchese G.; Punzi A. A General and Straightforward Approach to α,ω-Ketoesters. Tetrahedron 1996, 52, 13513–13520. 10.1016/0040-4020(96)00805-8. [DOI] [Google Scholar]
- Imai Y. N.; Inoue Y.; Nakanishi I.; Kitaura K. Cl-π interactions in protein-ligand complexes. Protein Sci. 2008, 17, 1129–1137. 10.1110/ps.033910.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel P. A.; Kvaratskhelia N.; Mansour Y.; Antwi J.; Feng L.; Koneru P.; Kobe M. J.; Jena N.; Shi G.; Mohamed M. S.; Li C.; Kessl J. J.; Fuchs J. R. Indole-based allosteric inhibitors of HIV-1 integrase. Bioorg. Med. Chem. Lett. 2016, 26, 4748–4752. 10.1016/j.bmcl.2016.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy R. M. Static and dynamic light scattering of biological macromolecules: what can we learn?. Curr. Opin. Biotechnol. 1997, 8, 25–30. 10.1016/S0958-1669(97)80153-X. [DOI] [PubMed] [Google Scholar]
- Jachimska B.; Wasilewska M.; Adamczyk Z. Characterization of Globular Protein Solutions by Dynamic Light Scattering, Electrophoretic Mobility, and Viscosity Measurements. Langmuir 2008, 24, 6866–6872. 10.1021/la800548p. [DOI] [PubMed] [Google Scholar]
- Serriere J.; Fenel D.; Schoehn G.; Gouet P.; Guillon C. Biophysical characterization of the Feline Immunodeficiency Virus p24 capsid protein conformation and in vitro capsid assembly. PLoS One 2013, 8, e56424. 10.1371/journal.pone.0056424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A.; Pate K. M.; Moss M. A.; Dean D. N.; Rangachari V. Self-propagative replication of Aβ oligomers suggests potential transmissibility in Alzheimer disease. PLoS One 2014, 9, e111492. 10.1371/journal.pone.0111492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shkriabai N.; Dharmarajan V.; Slaughter A.; Kessl J. J.; Larue R. C.; Feng L.; Fuchs J. R.; Griffin P. R.; Kvaratskhelia M. A Critical Role of the C-terminal Segment for Allosteric Inhibitor-induced Aberrant Multimerization of HIV-1 Integrase. J. Biol. Chem. 2014, 289, 26430–26440. 10.1074/jbc.M114.589572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slaughter A.; Jurado K. A.; Deng N.; Feng L.; Kessl J. J.; Shkriabai N.; Larue R. C.; Fadel H. J.; Patel P. A.; Jena N.; Fuchs J. R.; Poeschla E.; Levy R. M.; Engelman A.; Kvaratskhelia M. The mechanism of H171T resistance reveals the importance of Nδ-protonated His171 for the binding of allosteric inhibitor BI-D to HIV-1 integrase. Retrovirology 2014, 11, 100. 10.1186/s12977-014-0100-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessl J. J.; Sharma A.; Kvaratskhelia M. Methods for the Analyses of Inhibitor-Induced Aberrant Multimerization of HIV-1 Integrase. Methods Mol. Biol. 2016, 1354, 149–164. 10.1007/978-1-4939-3046-3_10. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.