

Abstract

Here, we predicted the potential halogen bonding interaction between compound 2 and the 5-hydroxytryptamine 2B (5-HT2B) receptor and systematically assessed this interaction via structure–activity relationship analysis and molecular dynamics simulations. A physics-based computational protocol was then developed to further explore the opportunity of “designing in” halogen bonding interactions in structure-based ligand design for the 5-HT2B receptor, which not only facilitated the identification of previously uncharacterized halogen bonds in known 5-HT2B ligands but also enabled the rational design of halogen bonding interactions for the optimization of 5-HT2B ligands. As a proof-of-concept, a series of halogen-substituted analogues of doxepin was synthesized and evaluated, which showed improved in vitro and in vivo potency.
Keywords: Halogen bond, 5-HT2B receptor, MD simulation, doxepin, irritable bowel syndrome
Halogen bonding is now recognized as an important noncovalent interaction in the protein–ligand binding process.1−3 It is characterized as a directional and electrostatically driven interaction between a crown of positive charge on the outer side of a halogen atom, known as the σ-hole, and an electrically negative site.4 Structural database survey studies have revealed its common occurrence in biological systems.5,6 The concept of halogen bonding is now attractive in drug discovery programs owing to its advantages for improving drug–target binding affinity and for tuning ADME/T properties.7 Although the importance and potential of halogen bond interactions in protein–ligand binding are now widely appreciated by theorists and computational chemistry researchers, it is worth noting that our understanding in this area results largely from serendipitous discoveries or post facto analysis rather than from practical application in effective designs from predictions.8 Recently, however, a few exciting successful utilizations of halogen bonding interactions in lead optimization programs have stimulated efforts to apply this potentially powerful approach in rational drug design.9−11
Our aim in undertaking the present study was to explore the potential of halogen bond interactions in structure-based ligand design by using the 5-hydroxytryptamine 2B (5-HT2B) receptor as a case study. We previously identified a novel series of compounds (Figure 1A) with potent and selective 5-HT2B antagonist activity.12 Here, we carried out structure–activity relationship (SAR) analysis and molecular dynamics (MD) simulations to explore a potential halogen bonding interaction between the bromine-containing compound 2 and the 5-HT2B receptor. We also developed a physics-based computational protocol to systematically utilize halogen bonding interactions for known 5-HT2B ligands. Our protocol successfully led to not only the identification of potential halogen bonding interactions in existing 5-HT2B ligands but also to design ideas to exploit novel halogen bond interactions via specific halogen substitutions. As an example case, we incorporated a bromine substitution on doxepin to design compound 14, which showed a 10-fold increase in binding affinity (Ki = 2.5 nM) and superior in vivo therapeutic potency.
Figure 1.

Predicted halogen bonding interaction between compound 2 and the 5-HT2B receptor. (A) Chemical structures and binding affinities of 1 and 2. (B) Comparison of the predicted binding modes of 1 (blue) and 2 (yellow). (C) MD simulations of compound 2 in complex with the 5-HT2B receptor.
In our previous study, the ethyl benzoate moiety of compound 1 deeply buried into the inner part of the hydrophobic binding pocket was predicted to form a hydrogen bond with residue Thr1403.37 of the 5-HT2B receptor (Figure 1B), where the ethyl formate group of compound 1 appears to be essential for such binding; its removal largely destroyed its activity.12 A SAR study showed that the bromine-containing analogue compound 2, although lacking the ability to hydrogen bond with the aforementioned Thr1403.37, exhibited comparable binding activity with compound 1 (Figure 1A). Here, by examining molecular docking poses, we found that compound 2 could adopt another configuration in which the bromobenzene ring flipped over and pointed toward the exposed backbone carbonyl of Phe2175.38 on the opposite side (Figure 1B). With the concept of halogen bond interactions in mind, we predicted that compound 2 might form a halogen bonding interaction between the bromine atom and the carbonyl oxygen atom of Phe2175.38, which might contribute to its potent binding affinity.
To estimate the potential of the predicted halogen bonding interaction between compound 2 and the 5-HT2B receptor, we synthesized a series of halogen-substituted compounds and evaluated their antagonist activities in cellular function assays (Table 1). Compared with unhalogenated compound 3, introduction of halogen atoms with fluoride, chlorine, bromine, and iodine enhanced the activity by factors of 4, 35, 46, and 70, respectively (compounds 2 and 4–6, respectively), which nicely follows the general trend for the strength order of halogen bonding: F ≪ Cl < Br < I.13 However, replacing the chlorine atom in compound 5 with a methyl group, which is a hydrophobic group but lacks any σ-hole effect, led to a 10-fold decrease in activity for compound 7. In addition, substitution with a hydroxyl group abolished the activity of compound 8, suggesting that it is not likely to introduce a hydrogen bond interaction at this position for targeting the backbone carbonyl of Phe2175.38. Radioligand binding assays were also performed to determine their binding affinities; the results were consistent with the cellular activities (Table 1).
Table 1. Validation of the Halogen Bonding Interaction in Compound 2.

| ID | R | IC50 (nM)a | Ki (nM)b |
|---|---|---|---|
| 3 | -H | 1641.3 ± 221.2 | 151.3 ± 20.7 |
| 4 | -F | 403.4 ± 53.1 | 53.3 + 13.7 |
| 5 | -Cl | 46.9 ± 4.8 | 6.3 ± 0.9 |
| 2 | -Br | 35.4 ± 1.7 | 5.8 ± 1.2 |
| 6 | -I | 23.3 ± 3.7 | 1.7 ± 0.4 |
| 7 | -CH3 | 459.4 ± 118.2 | 19.8 ± 4.8 |
| 8 | –OH | n.a.c | 313.5 ± 43.0 |
The IC50 values were determined for each compound (mean ± SEM) from two independent tests.
The Ki values were determined for each compound (mean ± SEM) from three independent tests.
Not active, <50% inhibition at 10 μM concentration.
MD simulations were carried out to investigate the structural characteristics of this halogen bonding interaction. To account for the contribution of σ-hole effects, we introduced a positively charged extra point (EP) on the bromine atom in compound 2 as described previously.14 A stable Br···O distance of ∼3.08 Å and an almost optimal ∠C–Br···O angle of ∼173.7° were revealed in a 50 ns MD simulation (Figure 1C), suggesting the formation of a halogen bond with the backbone carbonyl of Phe2175.38.
We also synthesized compound 9 in which two bromine substitutions were introduced on both ortho positions in the phenyl ring to further support our prediction that the bromine atom could favor the orientation toward residue Phe2175.38. The IC50 value of compound 9 is 215.0 nM (Table 2). The mildly reduced potency of compound 9 might result from the steric clashes by the bromine substitution on the other ortho position. Clearly, a weakened halogen bond with a deviated distance and angle was observed during the MD simulation (Figure S1). Although single ortho substitution for a large-sized group would be favorable as for the ethyl formate group in compound 1, substitutions on both ortho positions were not additive due to a 27° twist angle between the phenyl-ring planes in their individually optimal configurations in the binding pocket (Figure S2). Thus, large-size substitutions on the other ortho position would twist the phenyl-ring plane, which finally affected the strength of the halogen bond with residue Phe2175.38. This is further supported by compounds 10 and 11 (Table 2). Additionally introducing small groups (a fluorine atom in compound 10) on the other ortho position had almost no effect on the activity (with an IC50 value of 74.9 nM). Instead, substitution with a larger group (the ethyl formate group in compound 11) would bring steric clashes and dramatically reduce the activity (with an IC50 value of 1962.5 nM). MD simulations also indicated that the halogen bonding interaction is well maintained in compound 10 but is disrupted in compound 11 (Figure S1).
Table 2. Validation of the Binding Mode of Compound 2.

| ID | R | IC50 (nM)a |
|---|---|---|
| 2 | -H | 35.4 ± 1.7 |
| 9 | -Br | 215.0 ± 17.2 |
| 10 | -F | 74.9 ± 8.7 |
| 11 | -COOEt | 1962.5 ± 134.1 |
The IC50 values were determined for each compound (mean ± SEM) from two independent tests.
We also developed a physics-based computational protocol to systematically explore the opportunity of utilizing halogen bonding interactions in the design of ligands for the 5-HT2B receptor. From ChEMBL database (version 23, May 2017), 1113 known 5-HT2B ligands were initially docked into the binding pocket, and halogen substitutions were then introduced in silico to identify potential halogen bonding interactions with the 5-HT2B receptor (Figure S3A). A chlorine atom would be added to any unsubstituted aromatic carbon atom of the ligand; note that only the protein backbone carbonyl, which is the most frequently observed binding partner in biomolecular halogen bonds,15 was considered here. Each Car-Cl···O=C interaction pair was evaluated by the defined geometry criteria (Figure S3B). Substitutions that met the relaxed geometry criteria (d < 4 Å and θ > 120°)2,5 and which produced no obvious steric clashes (distance < 2 Å) with surrounding residues were retained for the next optimization, which finally resulted in 1574 modeled halogenated ligands to meet the halogen bonding criteria; note that multiple halogen substitutions could be introduced on different sites in one ligand. These complexes were subjected to minimization and MM-PB/SA calculation with the inclusion of EP on the chlorine atoms. There were 277 chlorine-substituted ligands predicted to form potential halogen bonding interactions with the 5-HT2B receptor by satisfying the precise geometry criteria (d < 3.27 Å and θ > 140°)8 and exhibiting favorable binding free energies (ΔG < 0) related to their parent ligands (Table S1). Among these chlorine-substituted ligands, 11 were found as already existing 5-HT2B ligands with published activities, thereby serving as retrospective validations. Encouragingly, 9 of these 11 ligands have increased activities compared with the parent ligands (Table S2). The improved activities in these cases might result from the halogen bonding, and none of these halogen bonding interactions were characterized previously. Similar results were also obtained for bromine substation (Tables S3 and S4). Moreover, it was notable that our computational protocol could identify the halogen bonds in both agonists and antagonists of the 5-HT2B receptor (as illustrated in Figure S4), suggesting considerable predictive power.
Here, we focused on the predicted potential substitution sites for halogen bonding interactions in 21 FDA approved drugs with known 5-HT2B activities (Table S5). More than half of them belonged to two clusters: ergoline derivatives and tricyclic antidepressants (TCAs) (Table 3). Ergoline derivatives are a class of hemiterpenoid indole alkaloids. Acting as ligands for various serotonin and dopamine receptors, ergolines exert broad pharmacological actions and have been used clinically for vasoconstriction, migraines, and Parkinson’s disease.16 Consistent with the reported crystal structures of the 5-HT2B receptor complexed with ergotamine17 and lysergic acid diethylamide,18 the ergoline scaffold is anchored through the salt-bridge with the carboxylate of Asp1353.32 and buried into the hydrophobic pocket formed by the residues of Val1363.33, Gly2215.42, Phe2175.38, Phe3416.52, and Val3667.39. The indole ring points toward the exposed backbone carbonyl of Phe2175.38, and chlorine substitution at the 3-position was predicted to form a halogen bond with the residue Phe2175.38 (Table 3). TCAs are a class of drugs commonly used to relieve depression. They are often highly promiscuous ligands with nanomolar affinities at various GPCRs. The antagonist activity against the 5-HT2B receptor was also reported for many TCAs. In our predicted binding models, the amine moiety of TCAs forms a salt-bridge with the residue Asp1353.32, and the tricyclic ring sits deeply in the hydrophobic binding pocket and packs against the aromatic side chains of the residues Trp3376.48, Phe3406.51, and Phe3416.52; this resembles the interaction observed in the crystal structure of the doxepin-bound histamine H1 receptor.19 On the basis of this binding configuration, we predicted that a halogen bonding interaction could be formed between residue Phe2175.38 and the substituted chlorine atom on the phenyl ring as illustrated in Table 3. Given emerging opportunities and concerns for the clinical applications of 5-HT2B antagonists,20,21 modification of existing drugs to tune the binding preference against the 5-HT2B receptor may be an effective strategy.
Table 3. Design of Halogen Bonding Interactions with the 5-HT2B Receptor in FDA Approved Drugs.

Interestingly, we found residues Phe2175.38 and Gly2215.42 were the most frequently targeted “hot spots” in the 5-HT2B receptor, accounting for 75.8% of the predicted halogen bonding interactions (Figure 2A). Examining the ligand binding pocket, we found that Phe2175.38, Gly2215.42, and Ala2255.46 are located on three consecutive turns of helix V with side chains orientated toward the binding pocket (Figure 2B). Avoidance of side chain steric hindrance in Gly2215.42 and Ala2255.46 leads to an exposed backbone carbonyl oxygen in residues in the preceding turn (Phe2175.38 and Gly2215.42, respectively) for interaction with the halogen atom. Sequence alignment analysis further suggested that, although Phe5.38 or Tyr5.38 is conserved in most monoamine GPCRs, Gly5.42 is only conserved in the 5-HT2 receptors (Figure S5). Thus, the interaction propensity with Phe5.38 might be unique for three 5-HT2 receptors. Substitution of Gly5.42 to a larger residue with a bulk side chain would block the accessibility of the backbone carbonyl in Phe5.38 (as illustrated in histamine H1 receptor in Figure S6). For Gly5.42, the 5-HT2B and 5-HT2C receptors might have higher propensity of interaction due to the A5.46S substitution in the 5-HT2A receptor (Figure S5). The unique structural features of residues Phe5.38 and Gly5.42 also make possible the rational improvement of binding selectivity against the 5-HT2 receptors. In addition, the designed halogen bonds with the Phe5.38 and Gly5.42 residues tend to be geometrically perpendicular to the shared hydrogen bonds with the backbone amide in the succeeding helix turn, which is quite consistent with the concept that halogen bonds in biomolecular systems tend to be geometrically orthogonal and energetically independent of hydrogen bonds that share the same acceptor.22
Figure 2.

Halogen bonding interaction “hot spots” in the 5-HT2B receptor. (A) Statistics of predicted halogen bond formation with the backbone carbonyl of different residues in the 5-HT2B receptor. (B) Environment of the residues of Phe2175.38, Gly2215.42, and Ala2255.46 in the binding pocket in the 5-HT2B receptor.
As a proof-of-concept for halogen bonding in the design of 5-HT2B ligands, a series of halogen-substituted compounds were synthesized based on doxepin, a widely used TCA (Table 3). Introducing chlorine and bromine substitutions brought 8- and 10-fold improved binding affinities in compounds 13 and 14, respectively (Figure 3A), whereas introducing fluoride substitution in compound 12 had no effect on the binding affinity. MD simulation was carried out for compound 14 complexed with the 5-HT2B receptor, which showed the formation of a halogen bond with the backbone carbonyl of Phe2175.38 (Figure S7). The averaged Br···O distance during 50 ns MD simulation is 3.16 Å, and the averaged ∠C–Br···O angle is 171.0°. We further evaluated the in vivo bioactivity of compound 14 using a castor oil-induced diarrhea model in mice.23 The 5-HT2B receptor was reported to regulate the contraction of smooth muscle to mediate GI motility,24 and its inhibition was shown to produce a benefit for abnormal defecation.25,26 At the same oral dosage (10 mg/kg), we found compound 14 exhibited superior therapeutic efficiency compared with doxepin, significantly improving stool consistency (Figure 3B) and normalizing fecal output (Figure 3C). Moreover, decreases in body weight were also strongly attenuated by compound 14 (Figure S8).
Figure 3.
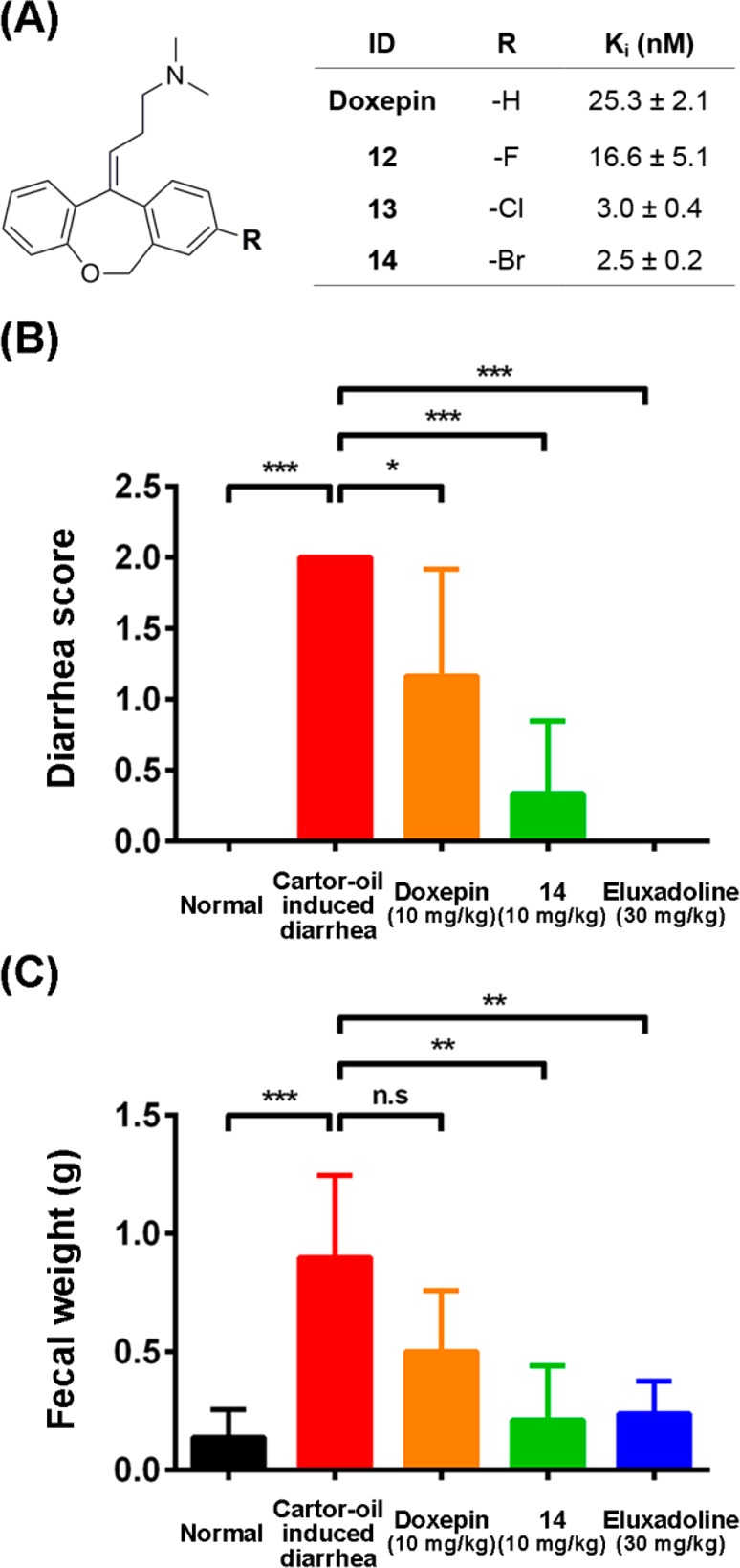
Halogen-substituted doxepin exhibited improved potency both in vitro and in vivo. (A) Binding affinities of doxepin and compounds 12–14 against the 5-HT2B receptor. The Ki values were determined for each compound (mean ± SEM) from three independent tests. (B) Diarrhea score in castor oil-induced diarrhea model. (C) Fecal weight in castor oil-induced diarrhea model.
In this study, we evaluated halogen bonding interactions in the 5-HT2B receptor and used the information we gathered to design new ligands with increased binding activity. Previously uncharacterized halogen bonds in known 5-HT2B ligands were identified, and novel halogen bonding interactions were designed for further ligand optimization. Halogen atoms have been classically incorporated in drug development to increase membrane permeability and metabolic stability. Recent studies have highlighted the importance of halogen bond interactions contributing to 5-HT ligand binding.27,28 Moreover, the potential role of halogen atoms as hydrogen bond acceptors has also been noted,29 which deserves further investigation both computationally and experimentally. Nevertheless, there is great potential to utilize halogen atoms in structure-based drug design in general if those interactions can be accurately predicted and incorporated. Our developed protocol has exhibited encouraging prediction of the binding energies between the parent ligands and their halogenated substitutes (Figure S9), thus providing the opportunity to “design in” halogen bond interaction in the lead optimization process. Doxepin is a commonly used TCA with moderate 5-HT2B antagonist activity.30 Recent studies have suggested that it also may be effective for patients with diarrhea-predominant irritable bowel syndrome (IBS-D) and promoted it into clinical trials.31 Although the pathogenic mechanism of IBS-D is not fully understood, the 5-HT2B receptor was believed to play an important role and could be regulated to improve both visceral hypersensitivity and abnormal defecation in IBS-D models.24−26,32 Our design of compound 14, by successfully exploiting halogen bonding interactions, brings a 10-fold increased binding affinity with the 5-HT2B receptor. Moreover, compound 14 had superior in vivo therapeutic efficiency in a mouse model of diarrhea, making it a promising preclinical candidate. Certainly, further investigation on the PK/ADME properties is required as halogen substitution may significantly alter these properties.
Glossary
Abbreviations
- 5-HT2B
5-hydroxytryptamine 2B
- EP
extra point
- IBS-D
diarrhea-predominant irritable bowel syndrome.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications Web site. (PDF) The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.8b00300.
Author Contributions
# Y.Z. and Y.W. contributed equally to this work.
N.H. was supported by the “Hundred, Thousand and Ten Thousand Talent Project” by the Beijing municipal government (2017A01) and the National Natural Science Foundation of China (81470862). Y.D. was supported by the National Natural Science Foundation of China (No. 21472136) and Tianjin Research Program of Application Foundation and Advanced Technology (15JCZDJC32900). X.-P.H. was supported by the National Institute of Mental Health Psychoactive Drug Screening Program (NIMH-PDSP, HHSN-271-2013-00017-C) directed by Bryan L. Roth (M.D., Ph.D.) at the University of North Carolina at Chapel Hill and Program Officer Jamie Driscoll at NIMH (Bethesda, MD, USA). Computational support was provided by the Special Program for Applied Research on Super Computation of the NSFC-Guangdong Joint Fund (the second phase) under Grant No. U1501501.
The authors declare the following competing financial interest(s): N.H. is founder of RXPDs (Suzhou) Co., Ltd, which develops 5-HT2B receptor antagonists for the treatment of IBS-D.
Supplementary Material
References
- Ford M. C.; Ho P. S. Computational tools to model halogen bonds in medicinal chemistry. J. Med. Chem. 2016, 59, 1655–1670. 10.1021/acs.jmedchem.5b00997. [DOI] [PubMed] [Google Scholar]
- Lu Y.; Liu Y.; Xu Z.; Li H.; Liu H.; Zhu W. Halogen bonding for rational drug design and new drug discovery. Expert Opin. Drug Discovery 2012, 7, 375–383. 10.1517/17460441.2012.678829. [DOI] [PubMed] [Google Scholar]
- Wilcken R.; Zimmermann M. O.; Lange A.; Zahn S.; Boeckler F. M. Using halogen bonds to address the protein backbone: a systematic evaluation. J. Comput.-Aided Mol. Des. 2012, 26, 935–945. 10.1007/s10822-012-9592-8. [DOI] [PubMed] [Google Scholar]
- Clark T.; Hennemann M.; Murray J. S.; Politzer P. Halogen bonding: the σ-hole. J. Mol. Model. 2007, 13, 291–296. 10.1007/s00894-006-0130-2. [DOI] [PubMed] [Google Scholar]
- Auffinger P.; Hays F. A.; Westhof E.; Ho P. S. Halogen bonds in biological molecules. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 16789–16794. 10.1073/pnas.0407607101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirimulla S.; Bailey J. B.; Vegesna R.; Narayan M. Halogen interactions in protein-ligand complexes: implications of halogen bonding for rational drug design. J. Chem. Inf. Model. 2013, 53, 2781–2791. 10.1021/ci400257k. [DOI] [PubMed] [Google Scholar]
- Xu Z.; Yang Z.; Liu Y.; Lu Y.; Chen K.; Zhu W. Halogen bond: its role beyond drug-target binding affinity for drug discovery and development. J. Chem. Inf. Model. 2014, 54, 69–78. 10.1021/ci400539q. [DOI] [PubMed] [Google Scholar]
- Wilcken R.; Zimmermann M. O.; Lange A.; Joerger A. C.; Boeckler F. M. Principles and applications of halogen bonding in medicinal chemistry and chemical biology. J. Med. Chem. 2013, 56, 1363–1388. 10.1021/jm3012068. [DOI] [PubMed] [Google Scholar]
- Xu Z.; Liu Z.; Chen T.; Chen T.; Wang Z.; Tian G.; Shi J.; Wang X.; Lu Y.; Yan X.; Wang G.; Jiang H.; Chen K.; Wang S.; Xu Y.; Shen J.; Zhu W. Utilization of halogen bond in lead optimization: a case study of rational design of potent phosphodiesterase type 5 (PDE5) inhibitors. J. Med. Chem. 2011, 54, 5607–5611. 10.1021/jm200644r. [DOI] [PubMed] [Google Scholar]
- Lange A.; Gunther M.; Buttner F. M.; Zimmermann M. O.; Heidrich J.; Hennig S.; Zahn S.; Schall C.; Sievers-Engler A.; Ansideri F.; Koch P.; Laemmerhofer M.; Stehle T.; Laufer S. A.; Boeckler F. M. Targeting the gatekeeper MET146 of C-Jun N-Terminal Kinase 3 induces a bivalent halogen/chalcogen bond. J. Am. Chem. Soc. 2015, 137, 14640–14652. 10.1021/jacs.5b07090. [DOI] [PubMed] [Google Scholar]
- Li Y.; Guo B.; Xu Z.; Li B.; Cai T.; Zhang X.; Yu Y.; Wang H.; Shi J.; Zhu W. Repositioning organohalogen drugs: a case study for identification of potent B-Raf V600E inhibitors via docking and bioassay. Sci. Rep. 2016, 6, 31074. 10.1038/srep31074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y.; Ma J.; Lin X.; Huang X. P.; Wu K.; Huang N. Structure-based discovery of novel and selective 5-hydroxytryptamine 2B receptor antagonists for the treatment of irritable bowel syndrome. J. Med. Chem. 2016, 59, 707–720. 10.1021/acs.jmedchem.5b01631. [DOI] [PubMed] [Google Scholar]
- Politzer P.; Lane P.; Concha M. C.; Ma Y.; Murray J. S. An overview of halogen bonding. J. Mol. Model. 2007, 13, 305–311. 10.1007/s00894-006-0154-7. [DOI] [PubMed] [Google Scholar]
- Ibrahim M. A. Molecular mechanical study of halogen bonding in drug discovery. J. Comput. Chem. 2011, 32, 2564–2574. 10.1002/jcc.21836. [DOI] [PubMed] [Google Scholar]
- Lu Y.; Wang Y.; Zhu W. Nonbonding interactions of organic halogens in biological systems: implications for drug discovery and biomolecular design. Phys. Chem. Chem. Phys. 2010, 12, 4543–4551. 10.1039/b926326h. [DOI] [PubMed] [Google Scholar]
- Schiff P. L. Ergot and its alkaloids. Am. J. Pharm. Educ. 2006, 70, 98. 10.5688/aj700598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wacker D.; Wang C.; Katritch V.; Han G. W.; Huang X. P.; Vardy E.; McCorvy J. D.; Jiang Y.; Chu M.; Siu F.; et al. Structural features for functional selectivity at serotonin receptors. Science 2013, 340, 615–619. 10.1126/science.1232808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wacker D.; Wang S.; McCorvy J. D.; Betz R. M.; Venkatakrishnan A. J.; Levit A.; Lansu K.; Schools Z. L.; Che T.; Nichols D. E.; Shoichet B. K.; Dror R. O.; Roth B. L. Crystal structure of an LSD-bound human serotonin receptor. Cell 2017, 168, 377–389 e312. 10.1016/j.cell.2016.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimamura T.; Shiroishi M.; Weyand S.; Tsujimoto H.; Winter G.; Katritch V.; Abagyan R.; Cherezov V.; Liu W.; Han G. W.; Kobayashi T.; Stevens R. C.; Iwata S. Structure of the human histamine H1 receptor complex with doxepin. Nature 2011, 475, 65–70. 10.1038/nature10236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poissonnet G.; Parmentier J. G.; Boutin J. A.; Goldstein S. The emergence of selective 5-HT2B antagonists structures, activities and potential therapeutic applications. Mini-Rev. Med. Chem. 2004, 4, 325–330. 10.2174/1389557043487312. [DOI] [PubMed] [Google Scholar]
- Brea J.; Castro-Palomino J.; Yeste S.; Cubero E.; Parraga A.; Dominguez E.; Loza M. I. Emerging opportunities and concerns for drug discovery at serotonin 5-HT2B receptors. Curr. Top. Med. Chem. 2010, 10, 493–503. 10.2174/156802610791111524. [DOI] [PubMed] [Google Scholar]
- Voth A. R.; Khuu P.; Oishi K.; Ho P. S. Halogen bonds as orthogonal molecular interactions to hydrogen bonds. Nat. Chem. 2009, 1, 74–79. 10.1038/nchem.112. [DOI] [PubMed] [Google Scholar]
- Fujita W.; Gomes I.; Dove L. S.; Prohaska D.; McIntyre G.; Devi L. A. Molecular characterization of eluxadoline as a potential ligand targeting mu-delta opioid receptor heteromers. Biochem. Pharmacol. 2014, 92, 448–456. 10.1016/j.bcp.2014.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borman R. A.; Tilford N. S.; Harmer D. W.; Day N.; Ellis E. S.; Sheldrick R. L.; Carey J.; Coleman R. A.; Baxter G. S. 5-HT(2B) receptors play a key role in mediating the excitatory effects of 5-HT in human colon in vitro. Br. J. Pharmacol. 2002, 135, 1144–1151. 10.1038/sj.bjp.0704571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassil A. K.; Taylor C. M.; Bolton V. J. N.; Gray K. M.; Brown J. D.; Cutler L.; Summerfield S. G.; Bruton G.; Winchester W. J.; Lee K.; Sanger G. J. Inhibition of colonic motility and defecation by RS-127445 suggests an involvement of the 5-HT2B receptor in rodent large bowel physiology. Br. J. Pharmacol. 2009, 158, 252–258. 10.1111/j.1476-5381.2009.00155.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi N.; Inagaki K.; Taniguchi K.; Sakaguchi Y.; Kawamura K. The novel 5-HT2B receptor antagonist, RQ-00310941, attenuates visceral hypersensitivity and abnormal defecation in rat models. Gastroenterology 2011, 140, S-607. 10.1016/S0016-5085(11)62513-4. [DOI] [Google Scholar]
- Kapadia N.; Ahmed S.; Harding W. W. New halogenated tris-(phenylalkyl) amines as h5-HT2B receptor ligands. Bioorg. Med. Chem. Lett. 2016, 26, 3216–3219. 10.1016/j.bmcl.2016.05.079. [DOI] [PubMed] [Google Scholar]
- González-Vera J. A.; Medina R. A.; Martín-Fontecha M.; Gonzalez A.; de La Fuente T.; Vázquez-Villa H.; García-Cárceles J.; Botta J.; McCormick P. J.; Benhamú B.; Pardo L.; Lopez-Rodriguez M. L. A new serotonin 5-HT 6 receptor antagonist with procognitive activity–Importance of a halogen bond interaction to stabilize the binding. Sci. Rep. 2017, 7, 41293. 10.1038/srep41293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin F.-Y.; MacKerell A. D. Jr Do Halogen–Hydrogen Bond Donor Interactions Dominate the Favorable Contribution of Halogens to Ligand–Protein Binding?. J. Phys. Chem. B 2017, 121, 6813–6821. 10.1021/acs.jpcb.7b04198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cusack B.; Nelson A.; Richelson E. Binding of antidepressants to human brain receptors: focus on newer generation compounds. Psychopharmacology 1994, 114, 559–565. 10.1007/BF02244985. [DOI] [PubMed] [Google Scholar]
- Kulak-Bejda A.; Bejda G.; Waszkiewicz N. Antidepressants for irritable bowel syndrome-A systematic review. Pharmacol. Rep. 2017, 69, 1366–1379. 10.1016/j.pharep.2017.05.014. [DOI] [PubMed] [Google Scholar]
- Wouters M. M.; Roeder J. L.; Tharayil V. S.; Stanich J. E.; Strege P. R.; Lei S.; Bardsley M. R.; Ordog T.; Gibbons S. J.; Farrugia G. Protein kinase Cγ mediates regulation of proliferation by the serotonin 5-hydroxytryptamine receptor 2B. J. Biol. Chem. 2009, 284, 21177–21184. 10.1074/jbc.M109.015859. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.