

Abstract

XJP-L (8), a derivative of the natural product (±)-7,8-dihydroxy-3-methylisochroman-4-one isolated from the peel of Musa sapien tum L., was found to exhibit weak inhibitory activity of tubulin polymerization (IC50 = 10.6 μM) in our previous studies. Thus, a series of 4-arylisochromene derivatives were prepared by incorporating the trimethoxyphenyl moiety into 8, among which compound (±)-19b was identified as the most potent compound with IC50 values ranging from 10 to 25 nM against a panel of cancer cell lines. Further mechanism studies demonstrated that (±)-19b disrupted the intracellular microtubule network, caused G2/M phase arrest, induced cell apoptosis, and depolarized mitochondria of K562 cells. Moreover, (±)-19b exhibited potent in vitro antivascular and in vivo antitumor activities. Notably, the R-configured enantiomer of (±)-19b, which was prepared by chiral separation, was slightly more potent than (±)-19b and was much more potent than the S-configured enantiomer in both antiproliferative and antitubulin assays. Our findings suggest that (±)-19b deserves further research as a potential antitubulin agent for the treatment of cancers.
Keywords: Combretastatin A-4; (±)-7,8-Dihydroxy-3-methylisochroman-4-one; Tubulin inhibitors; Enantioseparation; Antitumor; Vascular disrupting
Combretastatin A-4 (1, CA-4) (Figure 1), a natural cis- stilbene derivative isolated from the bark of the African willow tree Combretum caffrum,1 was found to be a powerful inhibitor of tubulin polymerization targeting the colchicine binding site.2 CA-4 has strong cytotoxicity against a variety of tumor cells with a broad therapeutic window. Besides, CA-4 was reported to show vascular disrupting properties at a tolerated dose.3 The potent antimitotic and vascular disrupting profiles of CA-4 made it a promising therapy for the treatment of cancers.4−7 However, CA-4P (2, Fosbretabulin), the phosphate prodrug of CA-4, had been discontinued in clinical trials8 due to the lack of a meaningful improvement in progression-free survival (PFS) and unfavorable partial response data.9 In the presence of light, heat, or acid media, but also after in vivo administration, the Z-isomer of CA-4 easily isomerizes to the E-isomer that is significantly less potent at inhibiting tubulin polymerization and cancer cell growth.10 Strategies to surmount this drawback have been taken by modification on the bridge structure of CA-4, which lead to the discovery of various moieties replacing the cis-double bond.11−13 Fusing cis-olefin into the ring B represents an important tactic to lock the cis double bond; compounds 3–6 with highly potent cytotoxicity were thus discovered (Figure 1).14−21
Figure 1.
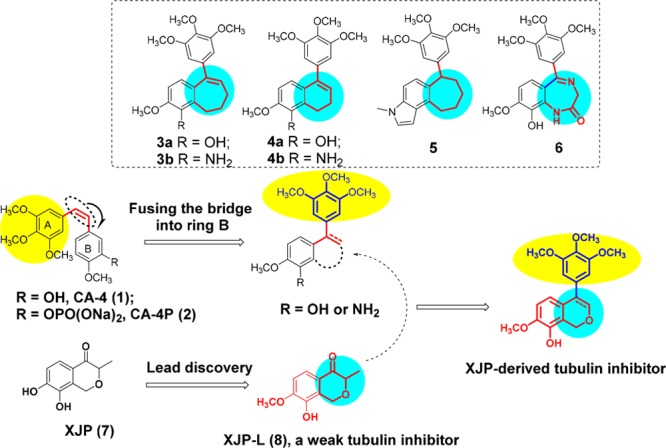
Strategy of fusing the bridge into ring B of CA-4 to surmount its instability and the design of XJP derived tubulin inhibitor.
It is known to all that natural products with structurally diverse frameworks have always been and continue to be an important source for new drug discovery.22 (±)-7,8-Dihydroxy-3-methylisochroman-4-one [7, (±)-XJP] was a structurally unique 4-isochromanone compound which was isolated from the peel of Musa sapientum L. and totally synthesized by our group,23,24 and it exhibited a wide range of favorable pharmacological properties.25,26 In our previous research for discovering new antitubulin agents with novel skeletons, a series of XJP derivatives was screened for their inhibitory activity of tubulin polymerization. Interestingly, XJP-L (8) was found to exhibit weak microtubule polymerization inhibitory activity (IC50 = 10.6 μM).
Considering the vital roles of 3,4,5-trimethoxyphenyl on the activity of inhibitors of tubulin polymerization targeting the colchicine binding site,27−298 was further hybridized with a 3,4,5-trimethoxyphenyl moiety for improving both its antiproliferative and inhibitory activities of tubulin polymerization using the cis-double bond locking strategy. Thus, a series of novel 4-arylisochromenes were designed and synthesized (Figure 1). Herein, we wish to report their synthesis and antitumor activities as antitubulin agents.
The synthetic route for the preparation of 4-arylisochromenes is outlined in Scheme 1. Various substituted XJP scaffolds 12a–e were synthesized according to the method utilizing a Parham-type cyclization with the tert-butyllithium reagent reported by our group.30 Sulfur-containing XJP scaffolds 12f–g were further synthesized via a Friedel–Crafts cyclic reaction.
Scheme 1. Synthetic Route to 4-Arylisochromenes.

Reagents and conditions: (a) NaH, DMF, 2 h, 55–70%; (b) t-BuLi, THF, −78 °C, 15 min, 72–90%; (c) (i) ethyl thioglycolate, K2CO3, CH3CN, 2 h, 75–88%; (ii) 10% NaOH aqueous, CH3OH, 80 °C, 85–90%; (d) (i) oxalyl chloride, DMF(cat.), DCM; (ii) SnCl4, chlorobenzene, 55–65% over two steps; (e) p-Toluenesulfonhydrazide, EtOH, 90 °C, 75–90%; (f) 5-bromo-1,2,3-trimethoxybenzene (16), PdCl2(CH3CN)2, Xphos, t-BuOLi, 90 °C, 45–70%; (g) Pd/C, H2, THF, 65–92%; (h) Pd/C, H2, CH3OH, 72–95%.
Then, 12a–g were transformed into their corresponding N-tosylhydrazones 15a–g in refluxing EtOH with good to excellent yields, followed by the coupling of 15a–g with 5-bromo-1,2,3-trimethoxybenzene (16) via a Pd-catalyzed cross-coupling to afford 17a–g in moderate to good yields. Utilizing the different reducing capacities of Pd–C/H2 in THF and CH3OH, the benzyl groups of 17b and 17d were selectively deprotected using THF as the solvent to give compounds 18a and 18b, respectively, while double bonds were reduced to afford (±)-19a–d with CH3OH as the solvent.
All target compounds 17a, 17c, 17e–g, 18a–b, and (±)-19a–d were initially evaluated for their antiproliferative activities against human hepatocellular carcinoma cells (HepG2) by the MTT assays. Most of the synthesized compounds displayed potent activity against HepG2 cells, and the preliminary structure activity relationships (SARs) were obtained. The position of the hydroxyl group on the isochroman moiety was crucial for antiproliferative activity. Compounds 18b and (±)-19b, which contain hydroxyl at the C-5 position (R2), exhibited the most potent activity with IC50 values of 26 and 15 nM, respectively. Moving the OH group from the C-5 position to the C-3 position led to the loss of antiproliferative activity (18a and 19d, IC50 > 10 μM). When R2 was substituted with other groups such as OCH3 [17c and (±)-19c], Br (17e), or no substitution [17a and (±)-19a], the activity significantly decreased when compared with 18b and (±)-19b. Besides, the activity was maintained when sulfur (X = S) was introduced into the isochroman scaffold (17f and 17g). The reduction of double bonds led to a slight improvement of activity [17a vs 19a, 17c vs (±)-19c, and 18b vs (±)-19b]. Especially, compound (±)-19b displayed the most potent antiproliferative activity (IC50 = 15 nM), which exhibited nearly 1000-fold improvement of activity toward 8 (IC50 = 23.7 μM) (See Supporting Information Table S1).
Subsequently, compounds 18b and (±)-19b were further evaluated for their antiproliferative activity against another five cancer cell lines including KB, HCT-8, MDA-MB-231, K562, and H22 cells. As shown in Table 1, both 18b and (±)-19b showed potent antiproliferative activities. Especially, (±)-19b exhibited the most active against K562 cell lines with the IC50 value of 10 nM, which is more potent than the positive control CA-4 (IC50 = 15 nM); human normal hepatocytes LO2 cells were also used to determine their selectivity toward cancer cells and normal cells, which showed that 18b and (±)-19b selectively inhibited the growth of cancer cells.
Table 1. Cytotoxicity against Five Cancer Cell Lines, Human Normal Hepatocyte LO2 Cells, and ITP of Compounds 18b, (±)-19b, (R)-(+)-19b, and (S)-(−)-19ba.
| IC50 values
(μM) |
|||||||
|---|---|---|---|---|---|---|---|
| Compd. | KB | HCT-8 | MDA-MB-231 | K562 | H22 | LO2 | ITPb |
| 18b | 0.016 ± 0.002 | 0.025 ± 0.004 | 0.028 ± 0.004 | 0.019 ± 0.002 | 0.010 ± 0.001 | 0.138 ± 0.012 | 4.2 ± 0.3 |
| (±)-19b | 0.025 ± 0.002 | 0.030 ± 0.002 | 0.022 ± 0.002 | 0.010 ± 0.001 | 0.011 ± 0.001 | 0.095 ± 0.010 | 3.1 ± 0.2 |
| (R)-(+)-19b | 0.012 ± 0.002 | 0.012 ± 0.001 | 0.011 ± 0.002 | 0.008 ± 0.001 | 0.008 ± 0.002 | 0.058 ± 0.005 | 2.5 ± 0.1 |
| (S)-(−)-19b | 0.87 ± 0.08 | 0.94 ± 0.12 | 0.90 ± 0.09 | 0.46 ± 0.06 | 0.80 ± 0.12 | 0.90 ± 0.08 | 4.3 ± 0.1 |
| CA-4 | 0.012 ± 0.001 | 0.015 ± 0.002 | 0.015 ± 0.002 | 0.015 ± 0.002 | 0.008 ± 0.001 | 0.095 ± 0.002 | 2.5 ± 0.2 |
| 8 | 25.1 ± 0.9 | 35.2 ± 1.3 | 30.2 ± 2.0 | 20.5 ± 1.2 | 18.2 ± 2.1 | 22.2 ± 1.2 | 10.6 ± 0.2 |
MTT methods; cells were incubated with indicated compounds for 72 h (means ± SD, n = 3).
Inhibition of tubulin polymerization activity.
The further in vitro assay for the inhibition of tubulin polymerization demonstrated that compounds 18b and (±)-19b were potent inhibitors of tubulin polymerization. As shown in Table 1, compound (±)-19b (IC50 = 3.1 μM) was more potent than 18b (IC50 = 4.2 μM) and was slightly less potent than CA-4 (IC50 = 2.5 μM). Furthermore, (±)-19b exhibited nearly 3-fold improvement of inhibitory activity of tubulin polymerization compared with 8 (IC50 = 10.6 μM), which suggested that the trimethoxyphenyl moiety has significant roles in binding with tubulin. In addition, in assay of the colchicine competitive experiment, the binding potency of (±)- 19b to the colchicine binding site was comparable to that of CA-4 with the inhibition rates of 76.4% and 89.6% at 1 μM and 5 μM, respectively (see Supporting Information Table S2), indicating that (±)-19b binds to the colchicine binding site.
Moreover, immunofluorescent assays were performed to investigate the effect of (±)-19b on microtubule networks. As shown in Figure 2, K562 cells exhibited normal filamentous microtubules arrays without drug treatment. However, after exposure to (±)-19b at three different concentrations (5 nM, 10 nM, 20 nM) for 24 h, the microtubule networks in cytosol were disrupted, indicating that (±)-19b induced a dose-dependent collapse of the microtubule networks.
Figure 2.
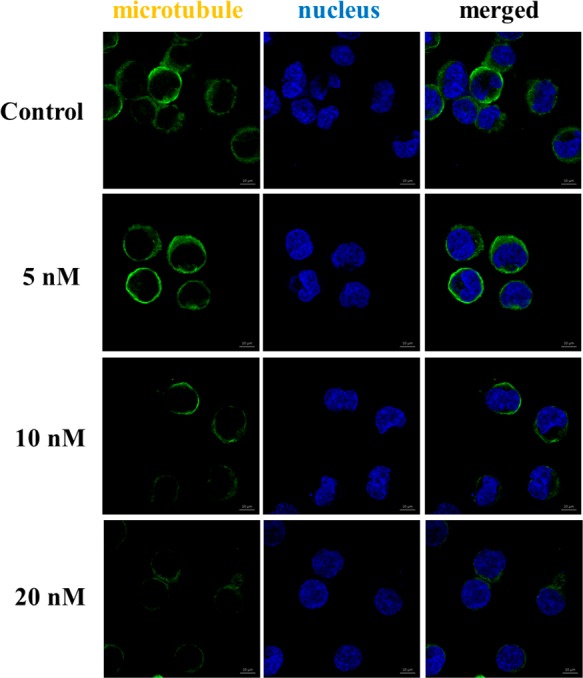
Effects of (±)-19b on the cellular microtubule network visualized by immunofluorescence.
Since most microtubule polymerization inhibitors disrupt cell mitosis and exert cell cycle arrest effects,31 the effect of (±)-19b on cell cycle progression using propidiumiodide (PI) staining in K562 cells was examined. When treated with (±)-19b at 5 nM, 10 nM, and 20 nM for 48 h, the percentages of cells arrested at the G2/M phase were 17.5%, 19.5%, and 22.4%, respectively, indicating that (±)-19b can disrupt the dynamic balance of the tubulin-microtubule system and further induced the cell cycle arrest at the G2/M phase (see Supporting Information Figure S1). Next, an Annexin V-APC/7-AAD binding assay was carried out to assess whether (±)-19b would induce cell apoptosis. The percentage of apoptotic cells after the 48 h treatment was only 5.7% in the control group. The total numbers of early (Annexin-V+/PI−) and late (Annexin-V+/PI+) apoptotic cells increased to 13.7%, 29.0%, and 54.1% after treatment with (±)-19b at 5, 10, and 20 nM for 48 h, respectively (see Supporting Information Figure S2). These results confirmed that (±)-19b effectively induced cell apoptosis in K562 cells in a dose-dependent manner.
In order to determine whether (±)-19b-induced apoptosis was involved in a disruption of mitochondrial membrane integrity, the fluorescent probe JC-1 was employed to measure the mitochondrial membrane potential (MMP). When treated with (±)-19b at concentrations of 0, 5, 10, and 20 nM for 48 h, the number of K562 cells with collapsed MMP increased to 0.6%, 12.7%, 26.7%, and 55.1%, respectively (see Supporting Information Figure S3), suggesting that (±)-19b caused mitochondrial depolarization of K562 cells in the process of apoptosis.
Most microtubule binding agents have antiangiogenic or vascular-disrupting activities or both, which are antivascular effects.32 Angiogenesis inhibiting agents (AIAs) interfere with new vessel formation, require chronic administration, and are likely to be of benefit in early stage or asymptomatic metastatic disease, while vascular-disrupting agents (VDAs) target the established tumor blood vessels, which are often given acutely and have particular efficacy against advanced disease.33 Considering that invasion and tube formation are highly relevant properties in the process of tumor vasculature, the HUVEC culture assay was used to assess the ability of (±)-19b to inhibit HUVEC migration. As a result, the untreated cells migrated to fill the area that was initially scraped after 24 h. In contrast, (±)-19b significantly inhibited the HUVEC migration in a dose-dependent manner (see Supporting Information Figure S4A). Then we further evaluated the antivascular ability of (±)-19b in a tube formation assay. After being seeded on matrigel, HUVECs form the capillary-like tubules with multicentric junctions. After exposure to (±)-19b at doses of 0, 5, 10, and 20 nM for 6 h, the capillary-like tubes were interrupted in different levels (Figure S4B). These results showed that (±)-19b effectively inhibited the tube formation of HUVECs.
Furthermore, we tested the in vivo antitumor activity of (±)-19b based on the in vitro antiproliferative activity and mechanistic studies. Mouse liver cancer xenograft model was established by subcutaneous inoculation of H22 cells into the right flank of mice. The tumor size and body weights of the mice were monitored and recorded every 2 days. Paclitaxel (PTX) was dosed as 8 mg/kg per 2 days (i.v.) due to its severe toxicity. To compare the antitumor efficacy of (±)-19b and CA-4, (±)-19b and CA-4 were dosed at 15 and 30 mg/kg per day (i.v.). None of the mice died in all groups after 21 days treatments. As shown in Figure 3A, both (±)-19b at the dose of 30 mg/kg per day and PTX at the dose of 8 mg/kg per 2 days significantly decreased the tumor volume. The reduction in tumor weight of the PTX group reached 72.1% at 21 days after initiation of treatment as compared to vehicle, while (±)-19b reduced tumor weight by 46.5% and 62.3% at doses of 15 and 30 mg/kg per day (i.v.), respectively, which are more potent than CA-4 treatment groups (inhibitory rates of 44.8% and 55.3% at doses of 15 and 30 mg/kg per day, respectively) (Figure 3C). Importantly, (±)-19b did not significantly affect body weight even at the dose up to 30 mg/kg, while treatment with PTX at a dose of 8 mg/kg per 2 days led to a significant decrease of body weight (Figure 3B). Thus, (±)-19b is worthy of further investigation for the treatment of cancers.
Figure 3.
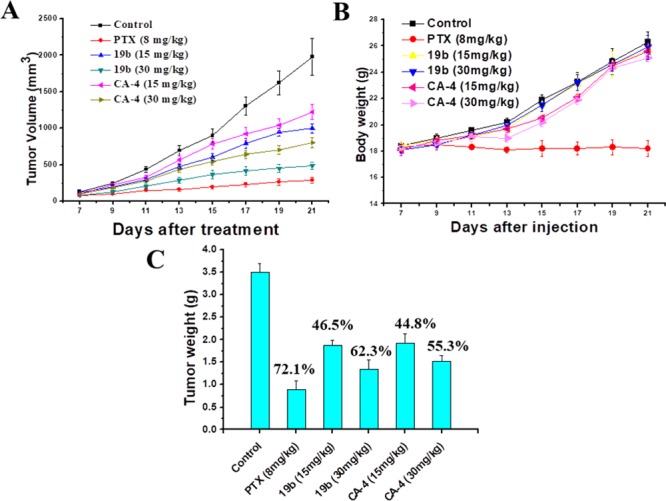
(A) Tumor growth curves after injection with different formulations in H22 tumor bearing mice. (B) Body weight changes of mice during treatment. (C) (±)-19b treatment resulted in significantly lower tumor weight compared with controls.
Finally, in order to investigate the effects of C-4 chirality on activity, the racemic mixture of (±)-19b was enantioseparated on a chiral column (see Supporting Information Figure S5) to afford (+)-19b and (−)-19b. Circular dichroism (CD) was performed to determine the absolute configuration, and the results indicated that the absolute configuration of (+)-19b is R-configured and the absolute configuration of (−)-19b is S-configured (see Supporting Information Figure S6).
Furthermore, (R)-(+)-19b and (S)-(−)-19b were evaluated for their antiproliferative and inhibitory activities of tubulin polymerization. As shown in Table 1, the R-configured 19b still exhibited very potent activity against five cancer cell lines, which were slightly more potent than (±)-19b, whereas (S)-(−)-19b displayed a significant decrease of activity. As regards the antitubulin assays, (R)-(+)-19b (IC50 = 2.5 μM) was about 2-fold more potent than (S)-(−)-19b (IC50 = 4.3 μM). Moreover, in assay of the colchicine competitive experiment, the binding ability of (R)-(+)-19b to the colchicine binding site was more potent than (±)-19b and was comparable to that of CA-4 with the inhibition rates of 79.4% and 92.6% at 1 μM and 5 μM (Table S2).
To explain the significant difference in activity for two enantiomers of (±)-19b, molecular modeling studies were performed by using the DOCK program in Discovery Studio 3.0 software with the tubulin crystal structure (PDB: 5lyj). As a result, (R)-(+)-19b adopted a very similar positioning with that of CA-4. The phenolic hydroxyl and 4-methoyl of (R)-(+)-19b and CA-4 formed hydrogen bonds with Thr179 and Cys241 residues, respectively. The oxygen atom in the isochromene ring interacted with residue Asn258 by a weak hydrogen bond (see Supporting Information Figure S7A). However, the binding pose of (S)-(−)-19b was flipped over 180° compared to that of CA-4, which may explain why both the antitubulin and antiproliferative activity of (S)-(−)-19b decreased dramatically (Figure S7B).
In summary, a series of novel 4-arylisochromenes have been synthesized based on the structure of XJP-L (8). Antiproliferative screening of these new synthesized compounds validated the representative compound (±)-19b as a high cytotoxic compound with IC50 ranging from 10 to 25 nM against a panel of cancer cell lines, which displayed a 1000-fold increase in activity compared with the lead 8. It was found that (±)-19b also displayed potent inhibitory activity in tubulin polymerization assay (IC50 = 3.1 μM). Further mechanism studies demonstrated that (±)-19b caused cell cycle arrest in the G2/M phase, induced cell apoptosis, and depolarized mitochondria of K562 cells. And the immunofluorescent assay indicated that (±)-19b can effectively disrupt microtubule networks in a dose-dependent manner. The wound healing and tube formation assays also identified (±)-19b as a novel inhibitor of tubulin polymerization with potent vascular disrupting activity. Finally, the in vivo antitumor activity of (±)-19b was validated in the H22 liver cancer xenograft mouse model, which is more potent than CA-4. Besides, (R)-(+)-19b and (S)-(−)-19b were obtained by chiral separation of (±)-19b and were evaluated for their antiproliferative and antitubulin activities, respectively. (R)-(+)-19b was slightly more potent than (±)-19b whereas (S)-(−)-19b displayed a significant decrease of activity, which was further elucidated by molecular modeling studies. Altogether, (±)-19b may represent a novel class of antitubulin agent with potent antivascular and antitumor activities and deserves further investigation.
Acknowledgments
This study was supported by the National Natural Science Foundation of China (No. 81673306, 81703348), The Open Project of State Key Laboratory of Natural Medicines, China Pharmaceutical University (No. SKLNMKF 201710), and China Postdoctoral Science Foundation (No. 2017100424). The authors thank Dr. Dahong Li (Key Laboratory of Structure-Based Drug Design and Discovery of Ministry of Education and School of Traditional Chinese Materia Medica, Shenyang Pharmaceutical University, Shenyang, China) for the CD calculations.
Glossary
Abbreviations
- DMF
N, N-dimethylformamide
- Xphos
2-dicyclohexylphosphino-2′,4′,6′-triisopropylbiphenyl
- DCM
dichloromethane
- THF
tetrahydrofuran
- HUVECs
human umbilical vein endothelial cell
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.8b00217.
Synthetic methods and characterization of target compounds; procedures for pharmacological activities (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Pettit G. R.; Singh S. B.; Hamel E.; Lin C. M.; Alberts D. S.; Garcia-Kendal D. Isolation and structure of the strong cell growth and tubulin inhibitor combretastatin A-4. Experientia 1989, 45, 209–211. 10.1007/BF01954881. [DOI] [PubMed] [Google Scholar]
- Lin C. M.; Singh S. B.; Chu P. S.; Dempcy R. O.; Schmidt J. M.; Pettit G. R.; Hamel E. Interactions of tubulin with potent natural and synthetic analogs of the antimitotic agent combretastatin: a structure-activity study. Mol. Pharmacol. 1988, 34, 200–208. [PubMed] [Google Scholar]
- Dark G. G.; Hill S. A.; Prise V. E.; Tozer G. M.; Pettit G. R.; Chaplin D. J. Combretastatin A-4, an agent that displays potent and selective toxicity toward tumor vasculature. Cancer Res. 1997, 57, 1829–1834. [PubMed] [Google Scholar]
- Pérez-Pérez M. J.; Priego E. M.; Bueno O.; Martins M. S.; Canela M. D.; Liekens S. Blocking blood flow to solid tumors by destabilizing tubulin: An approach to targeting tumor growth. J. Med. Chem. 2016, 59, 8685–8711. 10.1021/acs.jmedchem.6b00463. [DOI] [PubMed] [Google Scholar]
- Porcù E.; Bortolozzi R.; Basso G.; Viola G. Recent advances in vascular disrupting agents in cancer therapy. Future Med. Chem. 2014, 6, 1485–1498. 10.4155/fmc.14.104. [DOI] [PubMed] [Google Scholar]
- Jaroch K.; Karolak M.; Górski P.; Jaroch A.; Krajewski A.; Ilnicka A.; Sloderbach A.; Stefański T.; Sobiak S. Combretastatins: In vitro structure-activity relationship, mode of action and current clinical status. Pharmacol. Rep. 2016, 68, 1266–1275. 10.1016/j.pharep.2016.08.007. [DOI] [PubMed] [Google Scholar]
- Ji Y.; Liu Y.; Liu Z. Tubulin colchicine binding site inhibitors as vascular disrupting agents in clinical developments. Curr. Med. Chem. 2015, 22, 1348–1360. 10.2174/0929867322666150114163732. [DOI] [PubMed] [Google Scholar]
- https://www.clinicaltrials.gov/ ; a Focus: PCC + Bevacizumab + CA4P Versus PCC + Bevacizumab + Placebo for subjects with platinum resistant Ovarian Cancer;; b Safety and effectiveness of Combretastatin A-4 phosphate combined with chemotherapy in advanced solid tumors;; c Fosbretabulin or Placebo in combination with Carboplatin/Paclitaxel in Anaplastic Thyroid Cancer.
- http://investor.mateon.com/releasedetail.cfm?ReleaseID=1041745.
- Aprile S.; Del Grosso E.; Tron G. C.; Grosa G. In vitro metabolism study of combretastatin A-4 in rat and human liver microsomes. Drug Metab. Dispos. 2007, 35, 2252–2261. 10.1124/dmd.107.016998. [DOI] [PubMed] [Google Scholar]
- Li W.; Sun H.; Xu S.; Zhu Z.; Xu J. Tubulin inhibitors targeting the colchicine binding site: a perspective of privileged structures. Future Med. Chem. 2017, 9, 1765–1794. 10.4155/fmc-2017-0100. [DOI] [PubMed] [Google Scholar]
- Kaur R.; Kaur G.; Gill R. G.; Soni R.; Bariwal J. Recent developments in tubulin polymerization inhibitors: An overview. Eur. J. Med. Chem. 2014, 87, 89–124. 10.1016/j.ejmech.2014.09.051. [DOI] [PubMed] [Google Scholar]
- Lu Y.; Chen J.; Xiao M.; Li W.; Miller D. An overview of tubulin inhibitors that interact with the colchicine binding site. Pharm. Res. 2012, 29, 2943–2971. 10.1007/s11095-012-0828-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sriram M.; Hall J. J.; Grohmann N. C.; Strecker T. E.; Wootton T.; Franken A.; Trawick M. L.; Pinney K. G. Design, synthesis and biological evaluation of dihydronaphthalene and benzosuberene analogs of the combretastatins as inhibitors of tubulin polymerization in cancer chemotherapy. Bioorg. Med. Chem. 2008, 16, 8161–8171. 10.1016/j.bmc.2008.07.050. [DOI] [PubMed] [Google Scholar]
- Herdman C. A.; Strecker T. E.; Tanpure R. P.; Chen Z.; Winters A.; Gerberich J.; Liu L.; Hamel E.; Mason R. P.; Chaplin D. J.; Trawick M. L.; Pinney K. G. Synthesis and biological evaluation of benzocyclooctene-based and indene-based anticancer agents that function as inhibitors of tubulin polymerization. MedChemComm 2016, 7, 2418–2427. 10.1039/C6MD00459H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinney K. G.; Mocharla V. P.; Chen Z.; Garner C. M.; Ghatak A.; Hadimani M.; Kessler J.; Dorsey J. M.; Edvardsen K.; Chaplin D. J.; Prezioso J.; Ghatak U. R.. Tubulin binding agents and corresponding prodrug constructs. US20040043969-A1, 2004.
- Pinney K. G.; Mocharla V. P.; Chen Z.; Garner C. M.; Ghatak A.; Hadimani M.; Kessler J.; Dorsey J. M.; Edvardsen K.; Chaplin D. J.; Prezioso J.; Ghatak U. R.. Tubulin binding agents and corresponding prodrug constructs: U.S. Patent 7001926, Feb 21, 2006.
- Devkota L.; Lin C.; Strecker T. E.; Wang Y.; Tidmore J. K.; Chen Z.; Guddneppanavar R.; Jelinek C. J.; Lopez R.; Liu L.; Hamel E.; Mason R.; Chaplin D. J.; Trawick M. L.; Pinney K. G. Design, synthesis, and biological evaluation of water-soluble amino acid prodrug conjugates derived from combretastatin, dihydronaphthalene, and benzosuberene-based parent vascular disrupting agents. Bioorg. Med. Chem. 2016, 24, 938–956. 10.1016/j.bmc.2016.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasolofonjatovo E.; Provot O.; Hamze A.; Rodrigo J.; Bignon J.; Wdzieczak-Bakala J.; Desravines D.; Dubois J.; Brion J.; Alami M. Conformationnally restricted naphthalene derivatives type isocombretastatin A-4 and isoerianin analogues: synthesis, cytotoxicity and antitubulin activity. Eur. J. Med. Chem. 2012, 52, 22–32. 10.1016/j.ejmech.2012.03.001. [DOI] [PubMed] [Google Scholar]
- Yan J.; Hu J.; An B.; Huang L.; Li X. Design, synthesis, and biological evaluation of cyclic-indole derivatives as antitumor agents via the inhibition of tubulin polymerization. Eur. J. Med. Chem. 2017, 125, 663–675. 10.1016/j.ejmech.2016.09.056. [DOI] [PubMed] [Google Scholar]
- Galli U.; Travelli C.; Aprile S.; Arrigoni E.; Torretta S.; Grosa G.; Massarotti A.; Sorba G.; Canonico P. L.; Genazzani A. A.; Tron G. C. Design, synthesis, and biological evaluation of combretabenzodiazepines: a novel class of anti-tubulin agents. J. Med. Chem. 2015, 58, 1345–1357. 10.1021/jm5016389. [DOI] [PubMed] [Google Scholar]
- Yao H.; Liu J.; Xu S.; Zhu Z.; Xu J. The structural modification of natural products for novel drug discovery. Expert Opin. Drug Discovery 2017, 12, 121–140. 10.1080/17460441.2016.1272757. [DOI] [PubMed] [Google Scholar]
- Qian H.; Huang W. L.; Wu X. M.; Zhang H. B.; Zhou J. P.; Ye W. C. A new isochroman-4-one derivative from the peel of Musa sapientum L. and its total synthesis. Chin. Chem. Lett. 2007, 18, 1227–1230. 10.1016/j.cclet.2007.07.020. [DOI] [Google Scholar]
- Liu J.; Ren H.; Xu J.; Bai R.; Yan Q.; Huang W.; Wu X.; Fu J.; Wang Q.; Wu Q.; Fu R. Total synthesis and antihypertensive activity of (±) 7, 8-dihydroxy-3-methyl-isochro- -man-4-one. Bioorg. Med. Chem. Lett. 2009, 19, 1822–1824. 10.1016/j.bmcl.2008.12.102. [DOI] [PubMed] [Google Scholar]
- Fu R.; Chen Z.; Wang Q.; Guo Q.; Xu J.; Wu X. XJP-1, a novel ACEI, with anti-inflammatory properties in HUVECs. Atherosclerosis 2011, 219, 40–48. 10.1016/j.atherosclerosis.2011.07.010. [DOI] [PubMed] [Google Scholar]
- Fu R.; Wang Q.; Guo Q.; Xu J.; Wu X. XJP-1 protects endothelial cells from oxidized low-density lipoprotein-induced apoptosis by inhibiting NADPH oxidase subunit expression and modulating the PI3K/Akt/eNOS pathway. Vasc. Pharmacol. 2013, 58, 78–86. 10.1016/j.vph.2012.08.004. [DOI] [PubMed] [Google Scholar]
- Negi A. S.; Gautam Y.; Alam S.; Chanda D.; Luqman S.; Sarkar J.; Khan F.; Konwar R. Natural antitubulin agents: Importance of 3,4,5-trimethoxyphenyl fragment. Bioorg. Med. Chem. 2015, 23, 373–3891. 10.1016/j.bmc.2014.12.027. [DOI] [PubMed] [Google Scholar]
- Dong M.; Liu F.; Zhou H.; Zhai S.; Yan B. Novel natural product and privileged scaffold-based tubulin inhibitors targeting the colchicine binding site. Molecules 2016, 21, 1375. 10.3390/molecules21101375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L.; Jiang S.; Li X.; Liu Y.; Su J.; Chen J. Recent advances in trimethoxyphenyl (TMP) based tubulin inhibitors targeting the colchicine binding site. Eur. J. Med. Chem. 2018, 151, 482–494. 10.1016/j.ejmech.2018.04.011. [DOI] [PubMed] [Google Scholar]
- Wang C.; Wu Z.; Wang J.; Liu J.; Yao H.; Lin A.; Xu J. An efficient synthesis of 4-isochromanones via parham-type cyclization with weinreb amide. Tetrahedron 2015, 71, 8172–8177. 10.1016/j.tet.2015.08.028. [DOI] [Google Scholar]
- Dumontet C.; Jordan M. A. Microtubule-binding agents: a dynamic field of cancer therapeutics. Nat. Rev. Drug Discovery 2010, 9, 790–803. 10.1038/nrd3253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz E. L. Antivascular actions of microtubule-binding drugs. Clin. Cancer Res. 2009, 15, 2594–2601. 10.1158/1078-0432.CCR-08-2710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siemann D. W.; Bibby M. C.; Dark G. G.; Dicker A. P.; Eskens F. A.L.M.; Horsman M. R.; Marme D.; LoRusso P. M. Differentiation and definition of vascular-targeted therapies. Clin. Cancer Res. 2005, 11, 416–420. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.